American Journal of Analytical Chemistry
Vol. 3 No. 12 (2012) , Article ID: 25699 , 4 pages DOI:10.4236/ajac.2012.312111
A Novel Validated Stability Indicative UP-LC Method for Etravirine for the Determination of Process Related and Degradation Impurities
1Hetero Labs Ltd., Hetero House, Hyderabad, India
2Department of Chemistry, Sri Krishna Devaraya University, Anantapur, India
Email: *mohanchilukuri69@gmail.com
Received October 1, 2012; revised November 11, 2012; accepted November 19, 2012
Keywords: Etravirine; Forced Degradation; Ultra-Performance; Validation; Stability-Indicating
ABSTRACT
A novel stability indicating reverse phase ultra performance liquid chromatographic (UP-LC) method has been developed for Etravirine along with eight impurities (imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8) and validated as per ICH recommendations. Stress degradation conditions were established for Etravirine by subjecting it to stress conditions of acid, base, oxidation, humidity, thermal and photolysis. Significant degradation is observed in base stress condition and the major degradant (RRT at about 0.94) is identified by LC-MS and spectral analysis. The stress samples were assayed against a qualified reference standard and the mass balance was found close to 99.0%. Efficient chromatographic separation was achieved on a Shimpack ODS-II stationary phase with a gradient mobile phase combination. Quantification was carried at 303 nm at a flow rate of 0.6 mL·min−1. The resolution between Etravirine and eight potential impurities is found to be greater than 2.0. Regression analysis shows as r value (correlation coefficient) of greater than 0.999 for Etravirine and eight potential impurities. This method is capable to detect the impurities of Etravirine at a level of 0.003% with respect to test concentration of 1.0 mg·mL−1.
1. Introduction
During the past decade HIV infection has become, at least in developed countries, a largely manageable but incurable disease. This is due to the advent of Highly Active Anti Retroviral Therapy (HAART), in which patients are treated with a cocktail of drugs designed to reduce their viral loads to extremely low levels [1].
Etravirine (4-[[6-amino-5-bromo-2-[(4-cyanophenyl) amino]-4-pyrimidinyl]oxy]-3,5-dimethylben-zonitrile) , a type of organic molecule having some conformational isomerism that can bind the reverse transcriptase enzyme in multiple conformations, allowing for a more robust interaction between etravirine and the enzyme, even in the presence of mutations. Etravirine can be used by patients infected with HIV that is resistant to other NNRTIs [2]. When a CD4 cell in a person’s body effected by HIV, it copies its own genetic code into the cell’s DNA. In this way, the cell is then “programmed” to create new copies of HIV. The genetic material of HIV is in the form of RNA. In order for it to infect CD4 cells, it must first convert its RNA into DNA. HIV’s reverse transcriptase enzyme is needed to perform this process. NNRTIs, also known as “non-nucleosides” or “non-nukes” for short, attach themselves to reverse transcriptase and prevent the enzyme from converting RNA to DNA [3,4]. In turn, HIV’s genetic material cannot be incorporated into the healthy genetic material of the cell, and prevents the cell from producing new virus [5].
A few chromatographic methods have appeared in the literature for the quantification of Etravirine by LCMS and UP-LC [6-8]. The purpose of the present research work is to develop a single stability-indicating UP-LC method for the determination of Etravirine and its related impurities and to establish the degradation conditions for Etravirine. The developed LC method is validated with respect to specificity, LOD, LOQ, linearity, precision, accuracy and robustness as per ICH recommended conditions [9].
2. Experimental
2.1. Chemicals
Samples of Etravirine and its related impurities are received from Hetero labs limited a research foundation of the firm Hetero Labs Ltd, Hyderabad, India (Figure 1).
 (a)
(a) (b)
(b)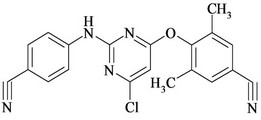 (c)
(c)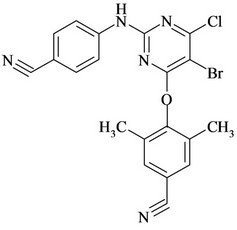 (d)
(d)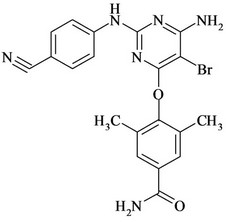 (e)
(e)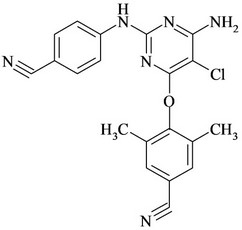 (f)
(f)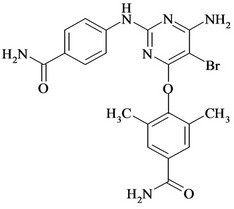 (g)
(g)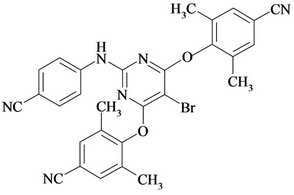 (h)
(h)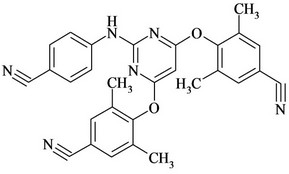 (i)
(i)
Figure 1. Chemical structures and names of Etravirine and its impurities; (a) Etravirine: 4-[[6-amino-5-bromo-2-[(4- cyanophenyl) amino]-4-pyrimidinyl]oxy]-3,5-dimethylbenzonitrile. (molecular weight 435.28) C20H15BrN6O; (b) Imp-1: 4-(6-amino-2-(4-cyanophenylamino) pyrimidin-4- yloxy)-3, 5-dimethyl benzonitrile. (molecular weight 356.38) C20H16N6O; (c) Imp-2: 4-(6-Chloro-2-(4-cyanophenylamino) pyrimidin-4-yloxy)-3, 5-dimethyl benzonitrile. (molecular weight 375.81) C20H1ClN5O; (d) Imp-3: 4-[[6-Chloro-5 -bromo-2-[(4-cyanophenyl) amino]-4-pyrimidinyl] oxy]-3, 5- dimethyl benzonitrile. (molecular weight 454.71) C20H13BrClN5O; (e) Imp-4: 4-(6-amino-5-bromo-2-(4-cyanophenylamino) pyrimidin-4-yloxy)-3, 5-dimethyl benzamide. (molecular weight 453.29) C20H17BrN6O2; (f) Imp-5: 4-(6- amino-5-chloro-2-(4-cyanophenylamino) pyrimidin-4-yloxy) -3, 5-dimethyl benzonitrile. (molecular weight 390.83) C20H15ClN6O; (g) Imp-6: 4-(6-amino-5-bromo-2-[(4-aminocarbonyl) aniline] pyrimidin-4-yloxy)-3, 5-dimethyl benzamide. (molecular weight 471.31) C20H19BrN6O3; (h) Imp- 7: 4-[5-Bromo-2-(4-cynoanilino)-6-(4-cyano-2, 6-dimethyl phenoxy)-4-pyrimidinyl] oxy-3, 5-dimethyl benzonitrile. (molecular weight 565.42) C29H21BrN6O2; (i) Imp-8: 4-[2-(4- cyanoanilini)-6-(4-cyano-2,6-dimethylphenoxy)-4-pyramidinyl]oxy-3,5-dimethylbenzonitrile.(molecular weight 486.52) C29H22N6O2.
All impurities and the Etravirine standard are of >97% purity and as follows: Etravirine (99.6%), imp-1 (97.8%), imp-2 (98.2%), imp-3 (99.3%), imp-4 (99.6%), imp-5 (98.5%), imp-6 (99.0%), imp-7 (97.6%) and imp-8
(97.2%). In addition, HPLC grade acetonitrile and methanol are purchased from Merck (Darmstadt, Germany). Acetic acid are purchased from Merck. Ammonium acetate are purchased from Rankem. Highly pure water was prepared with the Millipore Milli-Q Plus water purification system.
2.2. Equipment
The LC system used for method development (Shimadzu UP-LC), forced degradation studies and method validation consisted of a Waters 2695 binary pump with an auto sampler and a 2996 photo diode array detector (PDA). The output signal is monitored and processed using Empower software on a Pentium computer (Digital equipment Co.). Photo stability studies are carried out in a photo stability chamber (Atlas Suntest CPS+). Thermal stability studies are carried out in a dry hot air oven (Cintex precision hot air oven).
2.3. Chromatographic Conditions
A Shim-pack ODS-II 100 mm × 3.0 mm, 2.2 microns column is used with a mobile phase containing a gradient of solvents A (3.08 g of ammonium acetate in 1000 ml of water and adjusted to pH 6.0 ± 0.05 with acetic acid) and B (mixture of methanol and acetonitrile in the ratio of 50:50 v/v). The flow rate of the mobile phase is 0.6 mL·min–1 with a gradient program of 0/60, 5.10/90, 6.81/90, 7.66/60, 8.51/60 and 10/60 (time (min)/%B). The column temperature is maintained at 40˚C and the detection wavelength is set at 303 nm. The injection volume is 2 µl methanol is used as diluent.
2.4. LC-Mass Spectrum Conditions
The LC-MS system (Agilent 2010 EV series liquid chromatography system triple quadrapole mass spectrometer) was used for the identification of unknown compounds formed during forced degradation. A Symmetry shield RP18 100 × 3.0 mm, 3.5 µm column is used as the stationary phase with a mobile phase containing a gradient of solvents A (1.55 g of ammonium acetate in 1000 ml of water and adjusted to pH 6.0 ± 0.05 with acetic acid) and B (mixture of methanol and acetonitrile in the ratio of 50:50 v/v). The flow rate of the mobile phase is 0.6 mL·min−1 with a gradient program of 0/60, 5.10/90, 6.81/90, 7.66/60, 8.51/60 and 10/60 (time (min)/%B). The injection volume is 20 µl. The analysis is performed in positive and negative electrospray ionization modes. The capillary and cone voltages are 3.50 KV and 25.00 V, respectively. The source and disolvation temperatures are 120˚C and 350˚C, respecttively, and the cone gas flow and disolvation gas flow are 100 L·hr−1 650 L·hr−1 respectively.
2.5. Preparation of Standard Solutions and Sample Solutions
A stock solution of Etravirine (1.0 mg/ml) is prepared by dissolving the appropriate amount of Etravirine solid in the diluent. Working solutions of 0.10% and for impurities 0.10% are prepared from the stock solution for the determinations of related substances respectively. A stock solution of impurities (mixture of imp-1, imp-2, impy-3, imp-4, imp-5, imp-6, imp-7 and imp-8) at 0.02 mg/ml is also prepared in the diluent. The drug substance powder equivalent to 100 mg of sample is transferred into a 100 ml volumetric flask, and 70 ml of diluent is added. The flask is attached to a rotary shaker and shaken for 2 min to disperse the powder completely. The mixture is sonicated for 2 min and then diluted to the appropriate volume with diluent to make a solution containing 1.0 mg/ml. The solution is then filtered through a 0.45-µ Nylon 66 membrane filter.
3. Results and Discussion
3.1. Method Development and Optimization
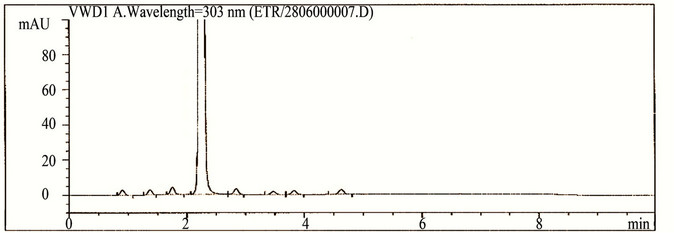
The main objective of the chromatographic method was to separate imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7, imp-8 and Etravirine and the generated degradation products from the analyte peak during stress studies. Impurities and degradation products are co-eluted by using different stationary phases, such as C18 with various mobile phases, and organic modifiers, including acetonitrile and methanol, in the mobile phase. SolutionA and Solution-B ratio in gradient program (50:50, v/v) at a flow rate of 0.5 mL·min−1 is chosen for the initial trail with a BEH Shield RP-18, 100 × 2.1 mm, 1.7 µm stationary phase. When an impurity-spiked solution is injected, two peaks are merging (Imp-5 and Etravirine) (Figure 2). Impurities are almost co-eluted with the analyte. To improve the resolution between the impurities and analyte, mobile phase ratio is slightly changed (60:40, v/v) and injected into the impurity-spiked solution. The resolution between the impurities and analyte is very poor (0.521 between imp-5 and Etravirine). In this case no separation between imp-5 and compound. Therefore, a gradient method is selected using solution-A (0.04M Ammonium acetate buffer) and Solution-B (mixture of acetonitrile and methanol in a ratio of 50:50 (v/v). Different gradient programs are investigated and satisfactory results are obtained when a gradient program with a flow rate of the mobile phase at 0.6 mL·min−1 and a gradient program of 0/60, 5.10/90, 6.81/90, 7.66/60, 8.51/60 and 10/60 (time (min)/%B) is used. The column used with the said satisfactory conditions is Shim-pack ODS-II 100 mm × 3.0 mm, 2.2 microns column. All the impurities are well resolved from each other and Etravirine (resolution is observed above 2 between each other) (Table 1).
 (a)
(a)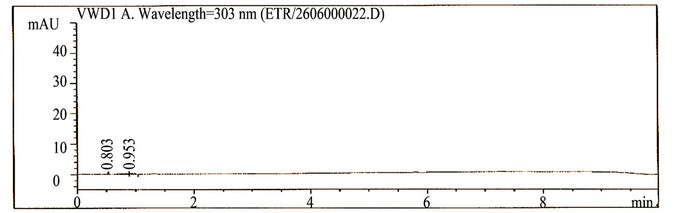 (b)
(b)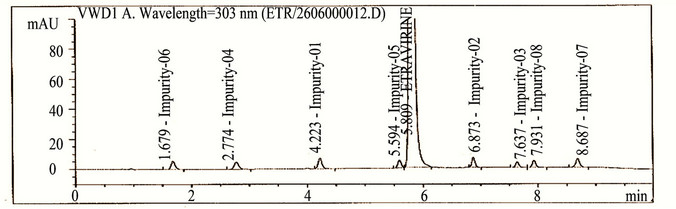 (c)
(c)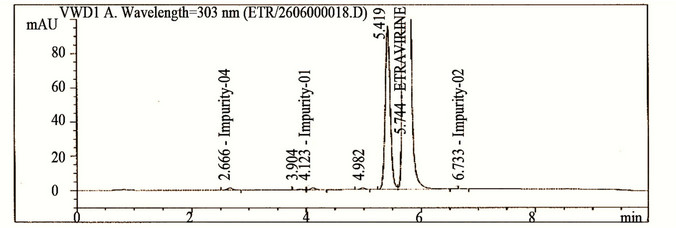 (d)
(d) (e)
(e)
Figure 2. Typical chromatograms etravirine (method development trials, blank, test spiked with impurities and stressed sample chromatograms).
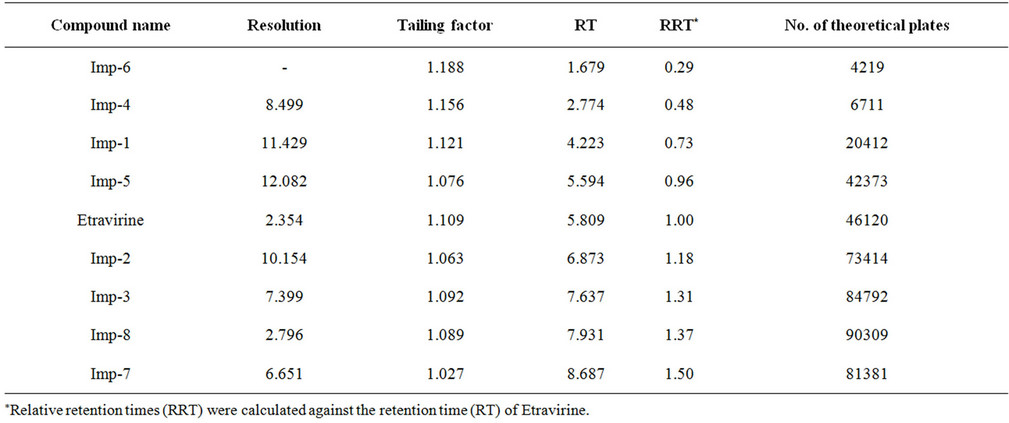
Table 1. System suitability report.
3.2. Analytical Method Validation
The proposed method was validated as per the International Conference on Harmonization (ICH) guidelines [9,13].
3.3. Stress Studies/Specificity
Specificity is the ability of the method to measure the analyte response in the presence of its potential impurities [10-12]. The specificity of the developed UP-LC method for Etravirine is determined in the presence of its eight related impurities (namely imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8) and degradation products. Forced degradation studies are also performed on Etravirine to provide an indication of the stabilityindicating property and specificity of the proposed method [12-17]. The stress conditions employed for the degradation study included light 1.2 million LUX hrs [11] (carried out as per ICH Q1B Option-1), heat (105˚C ), humidity (90% RH), acid hydrolysis (2 M HCl), base hydrolysis (2 M NaOH) and oxidation (30% H2O2). For heat, humidity and light studies, the samples were exposed for 5 days and 1.2 million LUX hrs, 200 WattHours/Sq.mts respectively, whereas the samples are treated for 8 hour for acid, base hydrolysis and oxidation as shows in Table 2. The peak purity of the Etravirine stressed samples is also checked by using a waters 2996 photo diode array detector (PDA). The purity angle is within the purity threshold limit in all of the stressed samples, demonstrating the homogeneity of the analyte peak. The contents of impurities were calculated for the stress samples against a qualified reference standard. The mass balance (% assay + % of impurities + % of degradation products) is calculated for all of the samples.
3.4. Precision
The precision of the method is evaluated by carrying out six independent preparations of a test sample of Etravirine against a qualified reference standard. Etravirine spiked with 0.10% each of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8. The % RSD of the areas of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 is calculated. The intermediate precision of the method is evaluated using a different analyst, different lot column using, different reagents and instrument located within the same laboratory. The % RSD of Etravirine during the method precision study is within 2.0% and the % RSD values of the area of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 in the related substance method precision study are within 5.0%. The %RSD of the results obtained in the intermediate precision study was within 2.0% and the % RSD of the areas of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 are within 5.0%, revealing the high precision of the method (Table 3).
3.5. Limit of Detection (LOD) and Limit of Quantification (LOQ)
The LOD and LOQ for imp-1, imp-2, imp-3, imp-4, imp- 5, imp-6, imp-7 and imp-8 are estimated at a signalto-noise ratio is 3: 1, 10: 1 respectively, by injecting a series of dilute solutions with known concentrations. The precision study is also carried out at the LOQ level by injecting eight individual preparations of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 calculated the %RSD of the areas. The limits of detection and quantification of Etravirine, imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 for a 2-ml injection volume are given in Table 3. The precision at the LOQ

Table 2. Summary of forced degradation results.
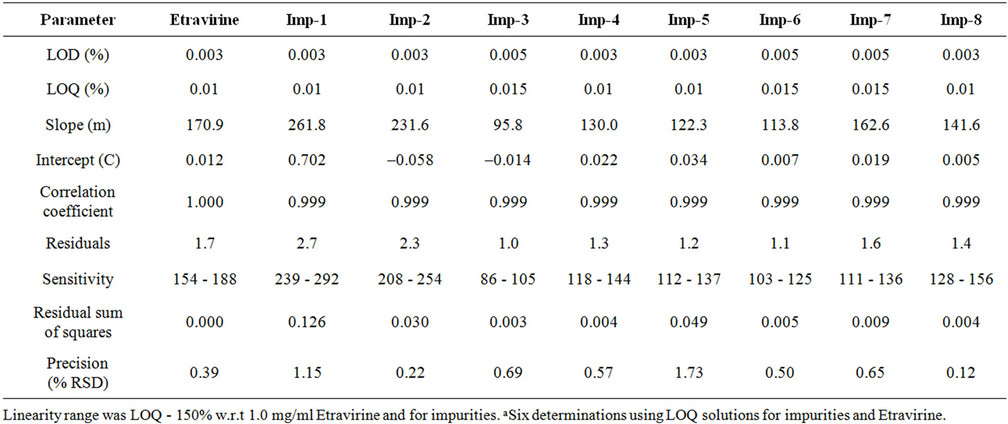
Table 3. Regression and precision data.
concentration for imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 is below 10.0%.
3.6. Linearity & Range
Linearity test solutions for the method are prepared from a stock solution at six concentration levels from LOQ to 150% of the Etravirine and impurities concentration (LOQ, 50 (0.05%), 80 (0.08%), 100 (0.10%), 120 (0.12%) and 150 (0.15%). The linear calibration plot for the method is obtained over the tested calibration range (50% - 150% level) and the obtained correlation coefficient is greater than 0.999. The results revealed an excellent correlation between the peak area and analyte concentration. The linear calibration plot for the related substance method is determined over the calibration range (LOQ to 0.15% w r to analyte concentration) for imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7, imp-8 and Etravirine, a correlation coefficient of greater than 0.999 is obtained. The linearity is checked for the related substance method over the same concentration range for three consecutive days. The % RSD values of the slope and y-intercept of the calibration curves are within 5.0%. These results showed an excellent correlation between the peak areas and concentrations of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7, imp-8 and Etravirine (Table 3). Residuals are within ±10% scattered with respect to 100% concentration response. Sensitivities are scattered within 10% with respect to 100% concentration sensitivity.
3.7. Accuracy
Accuracy of the method is evaluated in triplicate at four concentration levels LOQ level, 50 (0.05%), 100 (0.10%) and 150 (0.15%), and the percentage recoveries are also calculated.
Etravirine did show the presence of imp-1, imp-2, imp-4 and imp-5 at minor concentrations, and also contained 0.01% of unknown. Standard addition and recovery experiments are conducted to determine the accuracy of the related substance method for the quantification of all eight impurities (imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8) in the drug substance. The study is carried out in triplicate at LOQ level, 0.05%, 0.10% and 0.15% of the analyte concentration (1.0 mg/ml). The percentage of recoveries for imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 are calculated. The percentage recovery of Etravirine impurities in the drug substances i.e. imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 ranged from 97.10 to 104.11, 96.99 to 100.25, 99.25 to 100.20, 98.52 to 104.11, 95.86 to 101.10, 97.11 to 100.89, 100.11 to 104.13 and from 98.05 to 103.00 respectively.
The HPLC chromatograms of spiked samples at the 0.10% level of all eight impurities in the Etravirine drug substance sample are shown in Figure 2.
3.8. Robustness
To determine the robustness of the developed method, the experimental conditions are altered and the resolution between Etravirine and imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 is evaluated. The flow rate of the mobile phase is 0.6 mL/min. To study the effect of the flow rate on the resolution, the flow rate is changed by 0.1 units (to 0.5 and 0.7 mL·min−1).
The effect of the column temperature on the resolution is studied at 35˚C and 45˚C instead of 40˚C . In all these varied conditions, the components of the mobile phase remained constant. In all of the deliberately varied chromatographic conditions (flow rate and column temperature), the resolution between the closely eluting impurities, namely imp-5 and compound, is greater than 2.0, illustrating the robustness of the method. The variability of Etravirine and the impurities area response is within ±2% and within ±5%, respectively.
3.9. Solution Stability and Mobile Phase Stability
The solution stability of Etravirine in the impurities method was carried out by leaving both the sample and reference standard solutions in tightly capped volumetric flasks at room temperature for 48 hours. The same sample solutions are assaying for in 6-hours intervals over the study period. The mobile phase stability was also examined by assaying the freshly prepared sample solutions against freshly prepared reference standard solutions for 24 h intervals up to 5 days. The prepared mobile phase remained constant during the study period. The % RSD of the Etravirine assay is calculated for the mobile phase and solution stability experiments. The solution stability of Etravirine and its impurities in the related substance method is carried out by leaving a spiked sample solution in a tightly capped volumetric flask at room temperature for 48 h. The content of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 is determined at 6 h intervals up to the study period. The mobile phase stability is also investigated for 5days by injecting the sample solutions for every 24h interval. The content of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 is determined in the test solutions. The prepared mobile phase remained constant during the study period. The % RSD of assaying Etravirine during the solution stability and mobile phase stability experiments is within 2%. No significant changes are observed in the content of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 during the solution stability and mobile phase stability experiments when performed using the related substances method. The results of the solution and mobile phase stability experiments confirm that the sample solutions and mobile phase used during the related substance determinations are stable up to 48 h. Mobile phase is proved to be stable up to 5 days.
3.10. Results of Forced Degradation Studies
Degradation is not observed in Etravirine stressed samples subjected to light, humidity, heat, acid and oxidative stress conditions. Significant degradation of the drug substance and product is detected under base hydrolysis, leading to the formation of one major unknown degradation product at 0.94 RRT (Figure 2). Peak purity test results derived from the PDA detector confirmed that the Etravirine peak and the degraded peaks are homogeneous and pure in all of the analyzed stress samples. Degradation studies are carried out for the stress samples (at 500 µg/ml) against a qualified reference standard of Etravirine. The mass balance of the stressed samples is close to 99.0% (shows in Table 4). The assay of Etravirine is unaffected by the presence of imp-1, imp-2, imp-3, imp-4, imp-5, imp-6, imp-7 and imp-8 and its degradation products, confirming the stability-indicating power of the developed LC method.
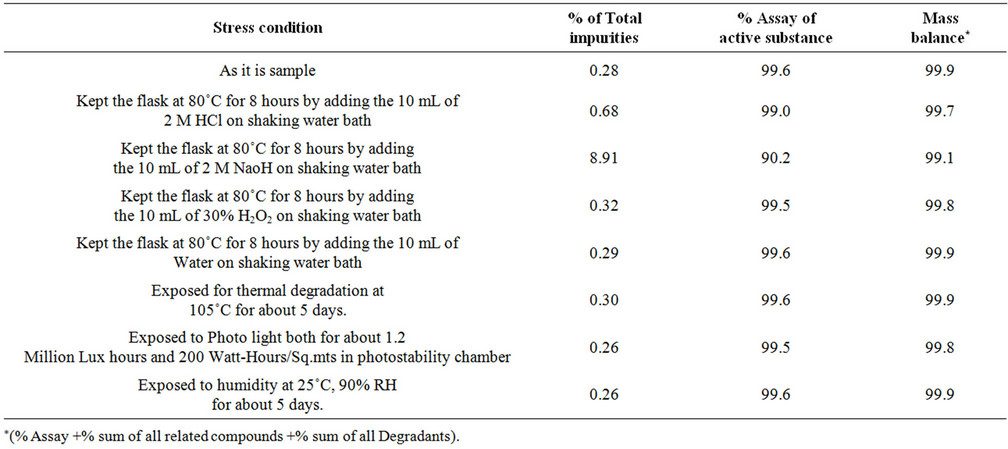
Table 4. Mass balance.
 (a)
(a)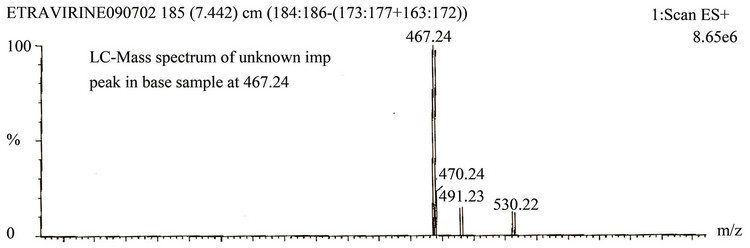 (b)
(b)
Figure 3. Typical LC Mass Spectrum.
3.11. Identification of Major Degradation Product (RRT at about 0.94) Formed in Base Hydrolysis (Stress Condition)
A LCMS study was carried to determine the m/z value of the major degradation product formed under base hydrolysis using an Agilent 2010 EV series liquid chromatography system coupled with triple quadrapole mass spectrometer. Method conditions are described in LC-Mass Spectrum conditions. The m/z value obtained for the degradation product resolving at 0.94 RRT in ESI positive mode was 467.24 (M + 1) (Figure 3).
4. Conclusion
The degradation conditions of Etravirine is established as per ICH recommendations. The gradient UP-LC method developed and used for stress studies also fit for quantitative, related substance and assay determination of Etravirine. The behavior of Etravirine under various stress conditions was studied and observed that all the degradation products and process impurities are well separated from the drug substance demonstrates the stability-indicating power of the method. The method is validated as per ICH recommendations. The developed UP-LC method is stability indicating which can be used to carry out the analysis of Etravirine in routine analysis of production samples and stability samples.
5. Acknowledgements
The authors wish to thank the management of Hetero labs limited, Hyderabad, Andhra Pradesh, INDIA for supporting this work. They also thank their colleagues in the analytical laboratory for their cooperation in carrying out this work.
REFERENCES
- M. Rowley, “The Discovery of Raltegravir, an Integrals Inhibitor for the Treatment of HIV Infection,” Progress in Medicinal Chemistry, Vol. 46, No. 5, 2008, pp. 1-28. doi:10.1016/S0079-6468(07)00001-X
- Y. Van Herrewege, G. Vanham, J. Michiels, K. Fransen, L. Kestens, K. Andries, P. Janssen and P. Lewi, “A series of Diaryltriazines and Diarylpyrimidines Are Highly Potent Nonnucleoside Reverse Transcriptase Inhibitors with Possible Applications as Microbicides,” Antimicrobial Agents and Chemotherapy, Vol. 48, No. 10, 2004, pp. 3684-3689. doi:10.1128/AAC.48.10.3684-3689.2004
- C. Mordant, B. Schmitt, E. Pasquier, C. Demestre, L. Queguiner, C. Masungi, A. Peeters, L. Smeulders, E. Bettens, K. Hertogs, J. Heeres, P. Lewi and J. Guillemont, “Synthesis of Novel Diarylpyrimidine Analogues of TMC278 and Their Antiviral Activity Against HIV-1 Wildtype and Mutant Strains,” European Journal of Medicinal Chemistry, Vol. 42, No. 5, 2007, pp. 567-579. doi:10.1016/j.ejmech.2006.11.014
- F. Goebel, A. Yakovlev, A. L. Pozniak, E. Vinogradova, G. Boogaerts, R. Hoetelmans, M. P. Béthune, M. Peeters and B. Woodfall, “Short-Term Antiviral Activity of TMC278—A Novel NNRTI—In Treatment-Naive HIV- 1-Infected Subjects”, AIDS, Vol. 20, No. 13, 2006, pp. 1721-1726. doi:10.1097/01.aids.0000242818.65215.bd
- C. Fang, J. D. Bauman, K. Das, A. Remorino, E. Arnold and R. M. Hochstrasser, “Two-Dimensional Infrared Spectra Reveal Relaxation of the Nonnucleoside Inhibitor TMC278 Complexed with HIV-1 Reverse Transcriptase,” Proceedings of the National Academy of Sciences, Vol. 105, No. 5, 2007, pp. 1472-1477. doi:10.1073/pnas.0709320104
- A. D’Avolio, M. Simiele, M. Siccardi, L. Baietto, M. Sciandra, V. Oddone, et al., “A HPLC-MS Method for the Simultaneous Quantification of Fourteen Antiretroviral Agents in Peripheral Blood Mononuclear Cell of HIV Infected Patients Optimized Using Medium Corpuscular Volume Evaluation,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 25, No. 4, 2011, pp. 779-788.
- A ahamed, G. Krishnamurthy, H. S. Bhojya naik and S. Ramesha, “Development and Validation of Stability Indicating Ultra Performance Liquid Chromatographic Method for Etravirine,” International Journal of Pharmacy and Pharmaceutical Sciences, Vol. 4, No. 1, 2011, pp. 255-261.
- L. Else, V. Watson, J. Tjia, A. Hughes, M. Siccardi, S. Khoo, et al., “Validation Of A Rapid And Sensitive HighPerformance Liquid Chromatography—Tandem Mass Spectrometry (HPLC-MS/MS) Assay For The Simultaneous Determination Of Existing And New Antiretroviral Compounds,” Journal of Chromatography B, Vol. 878, No. 19, 2010, pp. 1455-1465. doi:10.1016/j.jchromb.2010.03.036
- International Conference on Harmonisation, “Validation of Analytical Procedures: Text and Methodology,” Q2 (R1), 2005.
- International Conference on Harmonisation, “Stability Testing of New Drug Substances and Products Q1A (R2),” International Federation of Pharmaceutical Manufacturers Associations, Geneva, 2003.
- International Conference on Harmonisation, “Photo Stability Testing of New Drug Substances and Products,” Q1B, IFPMA, Geneva, 1996.
- S. Singh and M. Bakshi, “Guidance on Conduct of Stress Tests to Determine Inherent Stability of Drugs,” Pharmaceutical Technology Online, Vol. 24, 2000, pp. 1-14.
- International Conference on Harmonisation, “Guidelines on Validation of Analytical Procedures Definitions and Terminology,” Tripartite Guideline, EMEA, 1994.
- J. T. Carstensen, “Drug Stability Principles and Practices,” 2nd Edition, Marcel Decker, New York, 1995, pp. 3-4.
- M. Bakshi and S. Singh, “Development of Validated Stability Indicating Assay Methods-Critical Review,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 28, No. 6, 2002, pp. 1011-1040. doi:10.1016/S0731-7085(02)00047-X
- The United States Pharmacopeial Convention, “Validation of Compendial Methods,” The United States Pharmacopeia, 32nd Edition, USP32, 2008.
- J. T Carstensen and C. T. Rhodes, “Drug Stability Principles and Practices,” 3rd Edition, Marcel Dekker, New York, 2000.
NOTES
*Corresponding author.