Advances in Biological Chemistry
Vol.3 No.6(2013), Article ID:40930,15 pages DOI:10.4236/abc.2013.36065
Isolation and molecular characterization of a novel pseudomonas putida strain capable of degrading organophosphate and aromatic compounds
![]()
Department of Biology and Biochemistry, University of Houston, Houston, USA
Corresponding Author: *riyer@uh.edu
Copyright © 2013 *Rupa Iyer et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 8 October 2013; revised 12 November 2013; accepted 27 November 2013
Keywords: organophosphates; aromatic hydrocarbons; bioremediation; Pseudomonas putida
ABSTRACT
A bacterial strain designated in this study as POXN01 was found to be capable of degrading the synthetic organophosphorus pesticides paraoxon and methyl parathion. The strain was initially isolated through enrichment technique from rice field soil near Harlingen, Texas. Phylogenetic analysis based on 16S rRNA, gyrB and rpoD gene alignments identified the POXN01 isolate as a new strain of Pseudomonas putida, which is closely related to the recently discovered nicotine-degrading strain Pseudomonas putida S16. While being unable to metabolize nicotine, the POXN01 isolate was observed to actively proliferate using monocyclic aromatic hydrocarbons, in particular toluene, as nutrients. Search for the genetic determinants of paraoxon catabolism revealed the presence of organophosphorus-degrading gene, opd, identical to the one from Sphingobium fuliginis (former Flavobacterium sp. ATCC 27551). Assimilation of aromatic compounds likely relies on phc ARKLMNOPQ gene cluster for phenol, benzene and toluene catabolism, and on benRABCDKGEF cluster for benzoate catabolism. The observed versatility of POXN01 strain in degradation of xenobiotics makes it useful for the multi-purpose bioremediation of contaminated sites in both agricultural and industrial environmental settings.
1. INTRODUCTION
Quality of life achieved by modern society would be impossible without tremendous development of chemical industry within the past century. As a side effect of this growth, large amounts of man-made chemicals have been released into the environment either intentionally (as fertilizers, pesticides, or waste deposits) or by accident. Consequently, a substantial number of terrestrial and marine habitats today are contaminated by various xenobiotic compounds, many of which are harmful to living organisms.
Among other xenobiotics, organophosphorus (OP) compounds are of great concern considering their environmental impact and threat to human health. These synthetic chemicals are potent cholinesterase inhibitors once intended for military use, but at the present time, widely employed as insecticides. Occupational exposure to the OP pesticides in agricultural industries and self-poisoning with OP compounds causes significant health problems [1-6]. In addition, there are risks associated with major international efforts to destroy the stockpiled chemical warfare agents [7].
Despite their high toxicity, OP pesticides are extensively used in developing countries [8]. Exposure to organophosphate pesticides both purposely and accidentally results in millions of poisonings worldwide and 200,000 to 500,000 deaths annually throughout Asia and the Western Pacific [9-12]. While being removed from large-scale use in developed countries, organophosphates still may threaten metropolitan water supplies through unintentional contamination [13,14].
Removing environmentally hazardous OP compounds through detoxification by microbial enzymes has become the focus of biodegradation research over the last few decades. Previous reports have identified several organophosphate-degrading bacterial species, among them are Sphingobium fuliginis (former Flavobacterium sp. ATCC 27551) [15,16] and Brevundimonas diminuta GM (former Pseudomonas diminuta GM) [17,18], both expressing identical organophosphate hydrolase enzymes. Organophosphate hydrolase is a zinc-containing homodimeric membrane protein that has been found to hydrolyze paraoxon at a rate approaching the diffusion limit [19]. The enzyme is encoded by an opd (organophosphate degradation) gene and is capable of hydrolyzing a wide range of oxon and thion organophosphates [20,21]. The opd gene has been detected in soil microorganisms belonging to different taxonomic groups, which points to its horizontal spread using mobile genetic elements (transposons, plasmids, phages) as transfer vehicles [22- 28].
Currently, the isolation of a microorganism capable of degrading a selected xenobiotic compound is a routine task. However, modern DNA sequencing technologies allow us to quickly assess biotransformation potentials of the newly discovered strains well beyond the originally targeted activity. A strain that can simultaneously degrade multiple types of xenobiotics will be a preferable tool for decontamination of heavily polluted sites like landfills and sewage collectors. It can also be included in a variety of environmental clean-up scenarios as a multipurpose remedy for pollution.
We have isolated a bacterial strain capable of degradation of the OP pesticides paraoxon and methyl parathion as well as several commonly used industrial aromatic hydrocarbons. In the present paper, we describe characterization of this microorganism along with identification of molecular constituents responsible for its ability to detoxify xenobiotics.
2. MATERIALS AND METHODS
2.1. Ethics Statement
The soil sample collected for this study was taken from a rice field on private land by a student as part of a training program in biotechnology funded by the Texas Workforce Commission from 2011-2012. The student was given consent and access to the field by the landowner prior to collecting the sample. Sample collection included only a small portion of the topsoil and did not endanger any protected species.
2.2. Chemicals, Enzymes and Oligonucleotides
Paraoxon (O,O-diethyl O-p-nitrophenyl phosphate), methyl parathion (O,O-dimethyl O-p-nitrophenyl phosphorothioate), L-(-)nicotine, toluene, benzene, and phenol were obtained from Sigma-Aldrich (St. Louis, MO). Other chemicals were either from Sigma-Aldrich (St. Louis, MO) or from Fisher Scientific (Pittsburgh, PA). Egg white lysozyme and DNase-free ribonuclease A were obtained from Sigma-Aldrich (St. Louis, MO). Proteinase K was from Promega (Madison, WI). Synthetic deoxyoligonucleotides (Table 1) were purchased from Eurofins MWG (Huntsville, AL).
2.3. Media for Bacterial Growth
A carbon-deficient minimal medium (CSM) had the following composition: 0.2 g L−1 MgSO4 * 7H2O; 0.08 g L−1 Ca(NO3)2 * 4H2O; 0.005 g L−1 FeSO4 * 7H2O; 4.8 g L−1 K2HPO4; 1.2 g L−1 KH2PO4. Immediately prior to inoculation, it was supplemented with an appropriate carbon source. Luria-Bertani medium (LB) was prepared from the following components: 10 g L−1 BactoTryptone (Difco Laboratories, Detroit, MI); 5 g L−1 yeast extract (Difco Laboratories, Detroit, MI); 10 g L−1 NaCl. The pH of both mediums was adjusted to 7.5. For growth on a solid surface, the media were supplemented with 1.5 g L−1 BactoAgar (Difco Laboratories, Detroit, MI).
2.4. Isolation of Paraoxon-Metabolizing Microbial Strains from a Soil Sample
Soil samples were collected from a rice field near Harlingen (Cameron County, TX) at 26˚11′N, 97˚35′W. Air-dried soil (1 g) was suspended in 50 mL of the LB medium. The suspension was kept for 2 days on a rotary shaker at 30˚C and 200 rpm. Insoluble materials were allowed to settle out and an aliquot (100 μL) from the cleared supernatant was used to inoculate 3 mL of CSM supplemented with 0.1 mg mL−1 paraoxon. The culture was incubated for 1 week on a rotary shaker at 30˚C and 200 rpm. 100 μL of the bacterial suspension was transferred into 3 mL of fresh paraoxon-containing CSM, and the incubation step was repeated. After six consecutive subcultivations, the bacteria were plated on a CSM agar containing variable concentrations of paraoxon (0.1 - 2.0 mg mL−1). After overnight incubation at 30˚C, one distinct isolate that included large white or creamy-white circular colonies with irregular margins was found on the plates. This isolate, designated as POXN01, was determined to be a Gram-negative rod-shaped bacterium, which exhibited noticeable paraoxonand methyl parathion-degrading activity and was capable of growing on minimal medium with paraoxon as a carbon source.

Table 1. Primers used for PCR and sequencing. M represents an equimolar mixture of A and C.
2.5. Effect of OP and Aromatic Compounds on Bacterial Growth
To assess the effect of xenobiotics on the growth of POXN01 strain, the bacterial suspension was spread on CSM agar plates supplemented with the increasing concentrations of organophosphate or monocyclic aromatic compounds. Paraoxon and methyl parathion concentration in agar was 100, 200 and 400 μg mL−1, nicotine concentration was 8, 16 and 33 mg mL−1, and toluene and benzene concentration was 33, 50 and 66 mg mL−1. A 200 μl of 10,000-fold diluted (paraoxon, methyl parathion) or 5000-fold diluted (nicotine, toluene, benzene) stationary culture of POXN01 strain was evenly spread over the surface of an appropriate agar medium (plate diameter 10 cm, volume of solid medium 30 mL). The plates were incubated at 30˚C for 20 hours, and photographed for further analysis.
2.6. Degradation of OP and Aromatic Compounds by Resting Cells
Bacterial cultures were grown in LB medium (150 mL) on a rotary shaker at 37˚C and 200 rpm until cell density reached OD600 equal to 1.0. Cells were harvested by centrifugation for 10 min at 600 g and 4˚C, washed three times with 50 mM potassium phosphate (pH 7.0), and finally resuspended in 25 mL of the same buffer. The degradation experiment was initiated by mixing the cell suspension with an appropriate amount of a target compound. Concentration of paraoxon and methyl parathion in the degradation mixtures was 100 μg mL−1, while concentration of nicotine, toluene, benzene and phenol was 3 mg mL−1. The mixtures were incubated at 30˚C under constant shaking (120 rpm). Aliquots were taken from the mixtures at regular intervals, centrifuged to remove cells and insoluble materials, and analyzed either for the removal of target compound or for the accumulation of its degradation products in aqueous phase. Decomposition of OP pesticides was monitored by measuring absorbance of the released metabolic product p-nitrophenol at 405 nm. Degradation of nicotine, toluene, benzene and phenol was tracked by UV spectrometry within the range of 220 - 340 nm. TLC analysis of the selected samples was done on Silica Alu Foil plates (Sigma-Aldrich, St. Louis, MO) and Whatman KC18F plates (Fisher Scientific, Pittsburgh, PA) in solvent systems chloroform : ethanol : methanol: 0.5 M NaOH 30 : 15 : 2 : 1.5 (v/v) and methanol: water 2:1 (v/v), respectively.
2.7. Isolation of Cellular DNA
Total cellular DNA was extracted using the CTAB method [29] with the following modifications: cells were resuspended in TE buffer containing lysozyme (50,000 units mL−1) and ribonuclease A (300 Kunitz units mL−1), and incubated for 1 hour at 37˚C. Immediately prior to lysis, proteinase K (>30,000 units g−1) was added to a final concentration 0.25 mg mL−1. Cells were lysed with 0.5% (w/v) SDS for one hour at 37˚C. The subsequent removal of polysaccharides and residual proteins was performed as described in the original protocol.
2.8. 16S rDNA Sequencing
Fragments of the 16S rRNA gene were amplified by PCR from cellular DNA using 16S rDNA-specific primers (Table 1) [30-32] and PCR Master Mix (Promega, Madison, WI). The PCR conditions were as follows: initial denaturation at 94˚C for 4.5 min, 32 cycles consisting of denaturation at 94˚C for 0.5 min, annealing at 52˚C for 0.5 min, and extension at 72˚C for 1 min, and final elongation at 72˚C for 4 min. PCR products were column-purified [33] and sequenced bi-directionally by the dye-terminator method with the same primers used for amplification. Sequencing was performed by SeqWright, Inc. (Houston, TX).
2.9. Random Sequencing of Genomic DNA
Purified cellular DNA (5 μg) of the POXN01 isolate was converted into a shotgun DNA library using a Paired End DNA Sample Prep Kit (Illumina, Inc., San Diego, CA) according to the manufacturer's instructions. Sequencing was performed on an Illumina Genome Analyzer II with a paired-end module at the Solexa Sequencing Core Facility of M. D. Anderson Cancer Center (University of Texas, Houston, TX). After removal of duplicates and low quality sequences from the raw dataset with the FASTX toolkit [34], a total of 3.46 million usable pairs of 36 nucleotide-long reads was obtained for a total of 249.14 million nucleotides. For a 6 Mb bacterial genome, this would correspond to 41× coverage at the even distribution of reads. The reads were assembled into contigs using Velvet 1.2.07 [35] or Edena 3 dev120626 [36] de novo assemblers after several preliminary runs were undertaken for parameter optimization. Eventually the Velvet assembly was performed with a hash length of 19, coverage cut-off of 2 and an expected coverage of 12.5×. Other parameters were found to lesser affect length and number of the assembled contigs. The Edena assembly was performed with a minimum overlap size of 18 and a coverage depth limit of 3. Both sets of contigs were evaluated batchwise against an NCBI-maintained nucleotide sequence database, nt (as of July 27, 2012), using NCBI-BLAST-2.2.25 + software [37,38]. Sequences of the genes of interest from the POXN01 isolate were reconstructed from the contigs exhibiting high similarity to the homologues from closely related bacterial species. The reconstruction from contigs was supplemented with alignments of the short reads to the homologous genes in order to validate the reconstructed sequences. The alignments were performed using either Mosaik 1.0.1388 [39] or BWA 0.5.9 [40] software packages.
2.10. Phylogenetic Analysis of Gene Sequences
Multiple DNA sequence alignments were performed using ClustalX 2.0.12 and corrected manually when necessary [41]. Phylogenetic affiliations were evaluated using a Phylip 3.69 software package [42]. Evolutionary distances for the neighbor-joining method of sequence clustering were calculated using the F84 model of nucleotide substitutions [43]. Maximum likelihood modeling of DNA sequence evolution was done with transitiontransversion ratio set at 2. The topology of generated dendrograms was validated by bootstrapping with 1000 replicates for neighbor-joining trees and 200 replicates for maximum likelihood trees. Trees were visualized using TreeGraph 2.0.45 [44].
2.11. Detection and Identification of Organophosphorus Hydrolase gene
The presence of an organophosphorus hydrolase gene in the bacterial genome was assessed by PCR with forward and reverse primers derived from the sequence of the parathion hydrolase gene of Sphingobium fuliginis (former Flavobacterium sp. strain ATCC 27551), accession number M29593 (Table 1) [30-32]. PCR was performed under the following conditions: initial denaturation at 94˚C for 4.5 min, 32 cycles consisting of denaturation at 94˚C for 0.5 min, annealing at 55˚C for 0.5 min, and extension at 72˚C for 1 min, and final chain elongation at 72˚C for 4 min. PCR products were analyzed by electrophoresis in a 2% agarose gel with 1 × TBE as the running buffer. When necessary, the obtained DNA fragments were excised from the gel, purified as described in [33], and sequenced by the dideoxy-terminator method at SeqWright, Inc. (Houston, TX).
2.12. Nucleotide Sequence Accession Numbers
POXN01 nucleotide sequences were deposited in GenBank under the following accession numbers: KC189953 (benABCDKGEF operon), KC189954 (benR gene), KC 189955 (catBCA operon), KC189956 (gyrB gene), KC 189957 (opd gene fragment), KC189958 (pcaIJFTBDC gene cluster), KC189959 (pcaRK gene cluster), KC152 907 (phcARKLMNOPQ gene cluster), KC189960 (rpoD gene), KC189961 (16S rRNA gene fragment), KC189 962 (todX gene), KC189963 (ttg2ABCDEFG gene cluster), KC189964 (ttg8 gene), KC189965 (ttgRABC gene cluster).
3. RESULTS AND DISCUSSION
The bacterial strain designated as POXN01 was isolated from rice field soil by enriching for bacteria capable of using synthetic OP pesticide paraoxon as the sole source of carbon. When grown in pure culture in paraoxonsupplemented CSM medium, the isolate promoted a color change of the culture medium to yellow, which pointed to its ability to hydrolyze paraoxon to p-nitrophenol and diethylphosphate. The isolate was observed to form colonies on CSM agar supplemented with 100 - 400 μg mL−1 paraoxon or methyl parathion as the only available carbon sources (Figure 1). Thus, the strain is capable of both degrading the OP pesticides and utilizing products of their degradation as nutrients. Paraoxon is apparently a better substrate for the POXN01 isolate than methyl parathion since paraoxon decomposition by POXN01 resting cells proceeds noticeably faster (Figure 2).
Initial attempt to identify the isolate relied on sequence analysis of 16S rRNA genes probed by PCR with standard 16S rDNA-specific primers. A fragment of 16S rDNA from strain POXN01 was obtained by PCR with primer pair 27F-U1510R. BLAST analysis revealed the amplicon to be identical to a number of sequences from GenBank database, and all of them belonged to environmental isolates related to the genus Pseudomonas. However, these isolates did not apparently form a coherent phylogenetic group, and the majority of them were not characterized beyond 16S rDNA sequencing. Thus, the exact taxonomic position of the POXN01 strain remained obscure.
Further insight into the phylogenetic affiliation of the POXN01 isolate was obtained from random sequencing of its genomic DNA using the Illumina short read sequencing system. The collected short reads were as
 (a)
(a)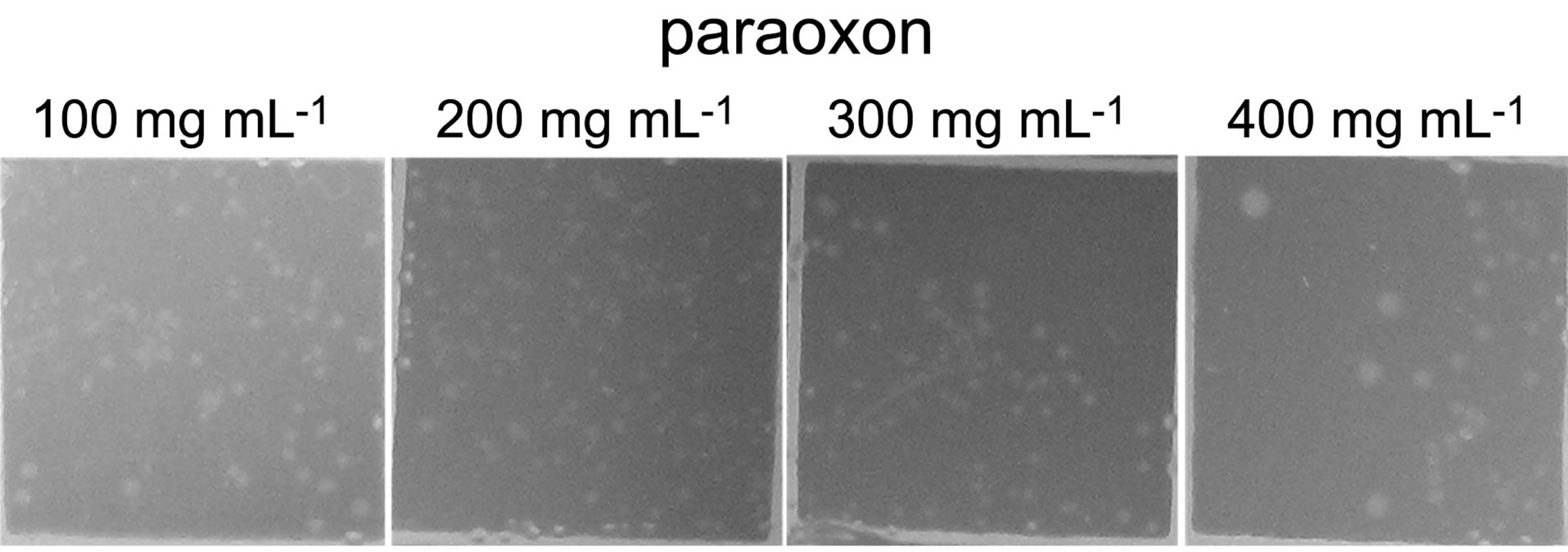 (b)
(b)
Figure 1. Growth of POXN01 isolate on CSM agar plates supplemented with methyl parathion (a) or paraoxon (b). A culture dilution factor of 10 - 4 was used for plate inoculation. Images depict colony growth within representative 1 cm2 frames.
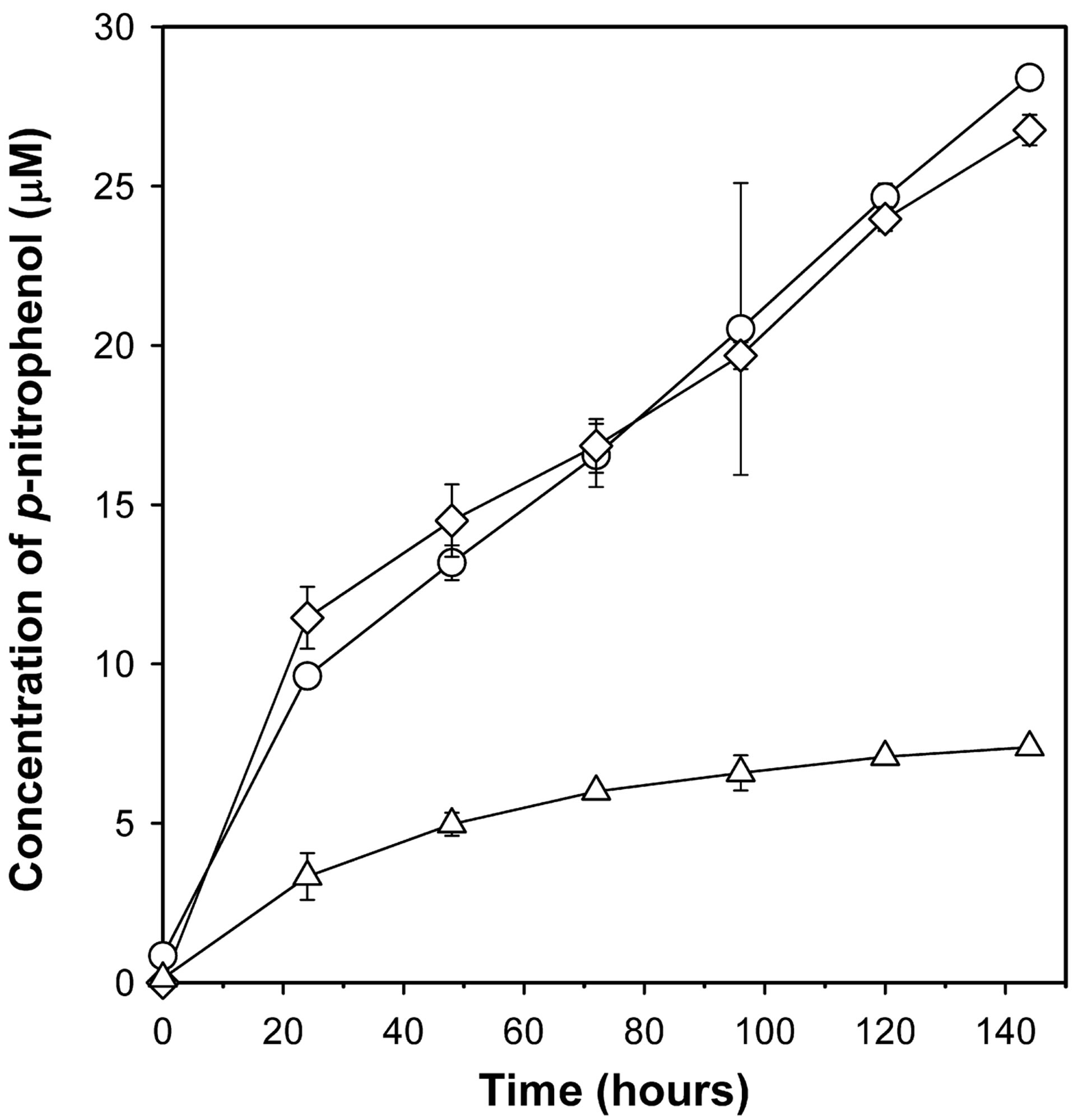
Figure 2. Degradation of paraoxon and methyl parathion by POXN01 isolate. Starting concentration of paraoxon and methyl parathion for degradative analysis was 100 μg mL-1. Concentration of pnitrophenol, the metabolic byproduct of paraoxon and methyl parathion, in the media was derived from the absorbance at 405 nm. P. putida strain CBF10-2 harboring the opd gene (67) was used as a positive control. All values have been corrected for the spontaneous organophosphate hydrolysis by subtracting the amount of p-nitrophenol formed in the presence of E. coli JM109/pUC19 cells (non-OP-degrading strain). Shown are paraoxon degradation by POXN01 isolate (diamonds), methyl parathion degradation by POXN01 isolate (triangles), and paraoxon degradation by P. putida CBF10-2 (circles).
sembled using Velvet or Edena assemblers into two respecttive sets of contigs. Each set was used to perform a BLAST similarity search through the NCBI's nt nucleotide sequence database. In both cases the vast majority of top scoring hits were against Pseudomonas putida nucleotide sequences. Among bacteria with sequenced complete genomes, a nicotine-degrading strain P. putida S16 exhibited the highest degree of similarity to the POXN01 isolate (Table 2).
It is known that 16S rDNA similarity analysis alone does not provide satisfactory intrageneric resolutions for pseudomonads, and therefore, it is often supplemented by sequence comparison of other conserved genes [46-50]. Two housekeeping genes, gyrB and rpoD, are frequently utilized as molecular markers to elaborate phylogenetic relationships within the genus Pseudomonas [48,51-53]. Sequences of the POXN01 gyrB and rpoD genes were recovered from the obtained contigs of POXN01 genomic DNA and individually aligned with the homologous genes from type strains of 105 species belonging to genus Pseudomonas. This was combined with analogous


Table 2. Summary of POXN01 genome sequence assembly. Only contigs longer than 100 bp were considered. BLAST search was performed with E-value cut-off 10−6. For each mentioned P. putida strain, the whole genome sequence is available.
alignment of 16S rDNA genes. An unrooted neighborjoining tree was computed from concatenated individual alignments joined in the following order: 16S rDNA - gyrB - rpoD. The POXN01 isolate was found grouped with P. monteilii within P. putida group of species (Figure S1). The refined analysis of POXN01 taxonomic affiliation was performed as above and included 20 strains of Pseudomonas putida, 8 other species belonging to the P. putida intrageneric group (P. fulva, P. parafulva, P. entomophila, P. monteilii, P. mosselii, P. plecoglossicida, P. cremoricolorata, and P. oryzihabitans), and P. aeruginosa taken as an outgroup. In both maximum likelihood and neighbor-joining trees, the POXN01 isolate was clustered together with P. putida S16 known for its ability to efficiently metabolize nicotine [54,55], P. putida BH, which can use phenol as a nutrient [56,57], and P. putida A10L, which can feed on 2-hydroxy-2- phenylacetate [58] (Figure 3). This whole branch is located somewhat closer to P. monteilii than to P. putida type strain, ATCC12633, which explains the observed association of POXN01 isolates with P. monteilii rather than with P. putida on the genus-wide tree.
Since the decomposition of paraoxon by the POXN01 strain apparently proceeds via the hydrolysis of a phosphoester bond yielding diethylphosphate and p-nitrophenol, the obtained fragments of its genomic DNA sequence were searched for the presence of an organophosphorus hydrolase gene. However, the sequenced part of the POXN01 genome did not contain any segment with significant similarity to the genes of known OPdegrading enzymes. Therefore, we attempted to identify organophosphorus hydrolase gene by PCR. Among OPhydrolyzing enzymes, the most studied so far is a product of opd gene, which was found in Sphingobium fuliginis [27,59], Brevundimonas diminuta GM [17,27], and in several representatives of genera Pseudomonas and Agrobacterium (in the latter case the gene is designnated opdA) [24-26,60]. Detection of the gene in the POXN01 strain was performed according to an established protocol that relies on analytical PCR with opdspecific primers [32]. PCR on the whole POXN01 genomic DNA yielded a single amplicon of the expected size for each of the primer pairs F196-R757, F196-R840, F450-R757, and F450-R840 (Figure 4). The largest amplicon obtained with primer pair F196-R977 was sequenced, and its sequence was found to be identical to that of the opd gene from S. fuliginis (Table 3).
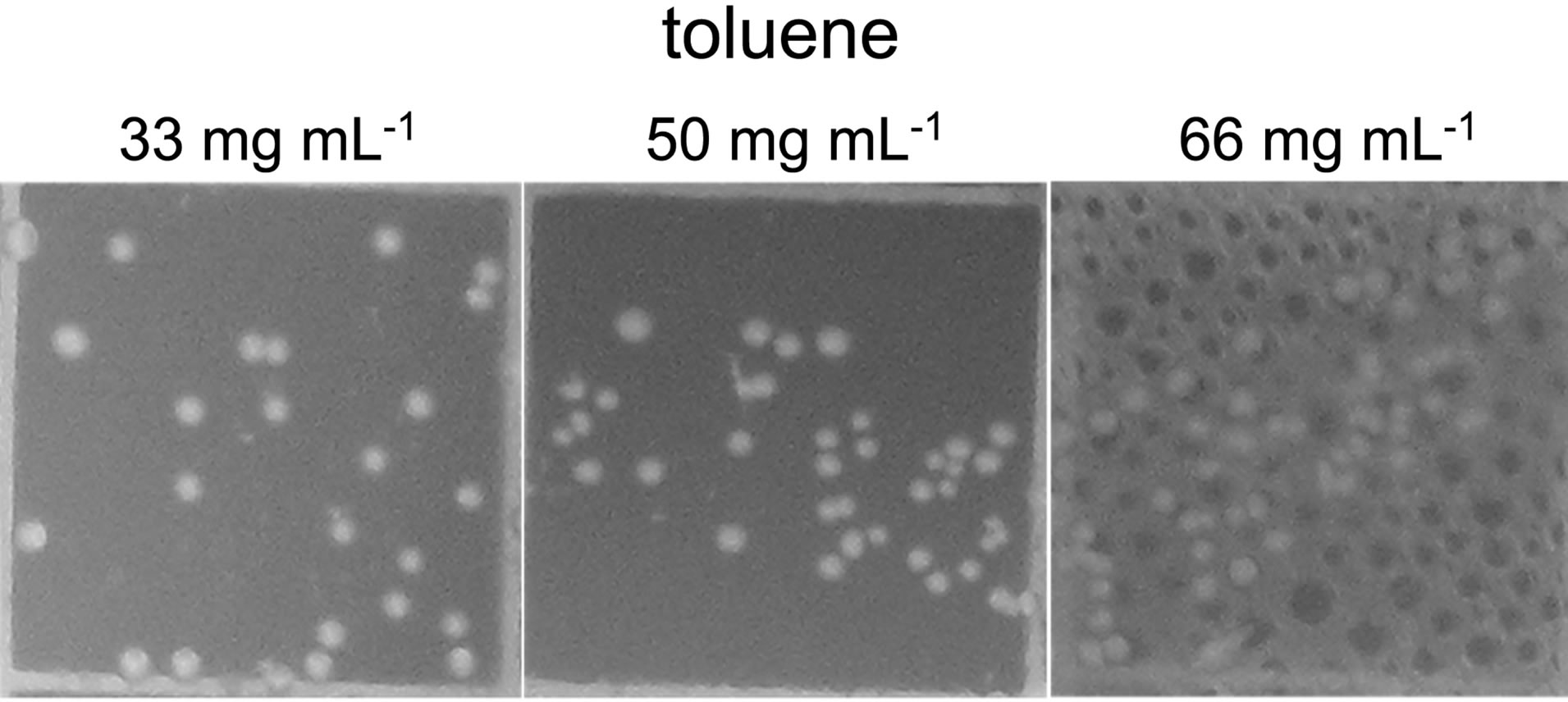
Availability of sequenced fragments of POXN01 genome made it possible to search for the genes involved in degradation of other xenobiotics. In spite of high overall similarity to P. putida S16, no nicotine catabolism genes were identified in the POXN01 contigs. Furthermore, the POXN01 strain was unable to grow on nicotine-supplemented CSM and its resting cells did not degrade nicotine (data not shown). At the same time, POXN01 genome was found to contain several gene clusters related to the catabolism of monocyclic aromatic hydrocarbons (Table 3). In agreement with this finding, the POXN01 strain formed colonies on benzeneand toluene-supplemented CSM agar (Figure 5). Incubation of POXN01 resting cells with benzene, phenol, or toluene in phosphate buffer resulted in significant alteration of the UV absorption spectra of the solutions, which implies formation of water-soluble products of decomposition of these aromatic compounds (Figure 6). This was accompanied by accumulation of fluorescent metabolite(s) detected by TLC analysis (Figure S2). Overall, these observations revealed the ability of POXN01 strain to tolerate presence of various aromatic hydrocarbons, and to degrade
 (a)
(a)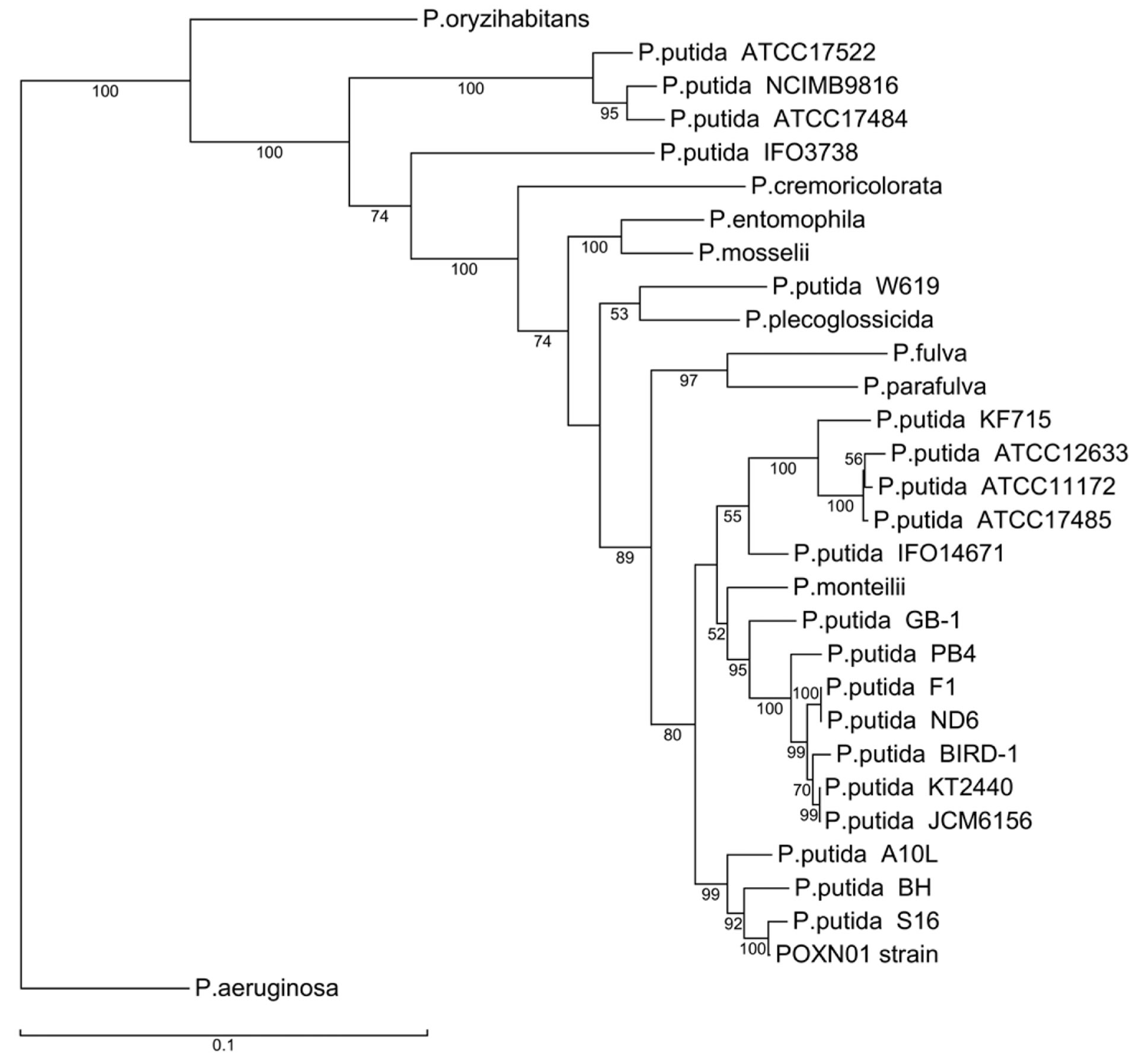 (b)
(b)
Figure 3. Phylogenetic trees derived from concatenated alignments of 16S rRNA - gyrB - rpoD genes of the POXN01 isolate and species belonging to the Pseudomonas putida intrageneric group. The trees were rooted using Pseudomonas aeruginosa as an outgroup. Bootstrap values expressed as percentages of the total number of replicates are shown next to each node (values below 50% are not shown). Scale of the total number of replicates are shown next to each node (values below 50% are not shown). Scale.

Figure 4. Detection of the opd gene in the genomic DNA of the POXN01 isolate. Electrophoretic analysis of PCR products obtained by amplification of the genomic DNA of the POXN01 strain with opd-specific primer pairs F450-R757 (lane 1), F450-R840 (lane 2), F196-R757 (lane 3) and F196-R840 (lane 4). The expected length of PCR fragments is 304 bp, 390 bp, 584 bp and 670 bp, respectively.
 (a)
(a) (b)
(b)
Figure 5. Growth of POXN01 isolate on CSM agar plates supplemented with toluene (a) and benzene (b). A culture dilution factor of 2 × 10 - 4 was used for plate inoculation. Images depict colony growth within representative 1 cm2 frames.
them in the course of assimilation.
The initial steps of aromatic hydrocarbon degradation by the POXN01 strain likely involve phc and ben gene clusters. phc (phenol catabolism) cluster includes genes for catechol 1,2-dioxygenase (phcA), sigma54-dependent transcriptional activator (phcR), six subunits of multicomponent phenol/benzene hydroxylase (phcKLMNOP), and conserved exported protein of unknown function (designated here as phcQ). Phenol/benzene hydroxylase can oxidize both phenol and benzene to catechol, which is then converted to cis,cis-muconate through intradiol cleavage catalyzed by catechol 1,2-dioxygenase [61]. ben cluster consists of nine genes involved in benzoate uptake and degradation: transcriptional regulator (benR), three subunits of benzoate 1,2-dioxygenase (benABC), 1,6-dihydroxycyclohexa-2,4-diene-1-carboxylate dehydrogenase (benD), two putative benzoate transporters (benK, benE), catechol 1,2-dioxygenase (designated here as benG), and benzoate-specific outer membrane porin (benF). The enzymes encoded in ben cluster convert benzoate to cis,cis-muconate via 1,2-cis-dihydroxybenzoate and catechol (62). Thus, phenol, benzene, and benzoate upper degradation pathways converge at cis,cismuconate, which is further processed by the enzymes of catBCA operon and pca regulon to tricarboxylic acid cycle intermediate, succinyl-CoA (Figure 7). At the same time, no close homologs of the known genes for the specialized toluene degradation enzymes have been found. While this may certainly result from incompleteness of the available POXN01 genome sequence, there is a possibility that the toluene ring fission is initiated by phenol/benzene hydroxylase. Indeed, this enzyme exhibits quite a relaxed specificity towards their aromatic hydrocarbon substrates, and can oxidize toluene albeit at a lower rate than phenol and benzene [63]. Similarly, it might hydroxylate p-nitrophenol released during paraoxon hydrolysis, thus making it susceptible to dioxigenase-mediated ring cleavage.
An ability of POXN01 strain to proliferate in the environment containing toxic substances while using them as nutrients implies tight control over their uptake and intracellular concentration. The strain maintains homeostasis using TtgABC efflux pump to expel aromatic hydrocarbons and some antibiotics from the cytoplasm [64,65]. It is supplemented by another putative transporter complex, Ttg2, which has been shown to signifycantly increase host tolerance of toluene [66]. Hydrocarbon uptake is mediated by a TodX-like outer membrane channel protein with a hatch domain regulating a passage of hydrophobic compounds to the periplasm [67]. In addition, the strain carries ttg8 gene encoding a conserved protein of unknown function, which plays an important role in resistance to toluene [66]. The protein exhibits a pronounced similarity to O-antigen polymerases, and as such might control permeability of cell envelope to xenobiotics.
The discovered biotransformation capabilities of POXN01 strain may find application in a variety of tasks. The strain can be used to speed up mineralization of OP pesticides in agricultural systems and sewage water, to degrade chemical warfare agents, and to contain spills of petroleum hydrocarbons and related industrial chemicals. Further studies on POXN01 metabolism might reveal other useful properties of this microorganism.
5. ACKNOWLEDGEMENTS
Partial funding for this research was supplied by the Texas Workforce
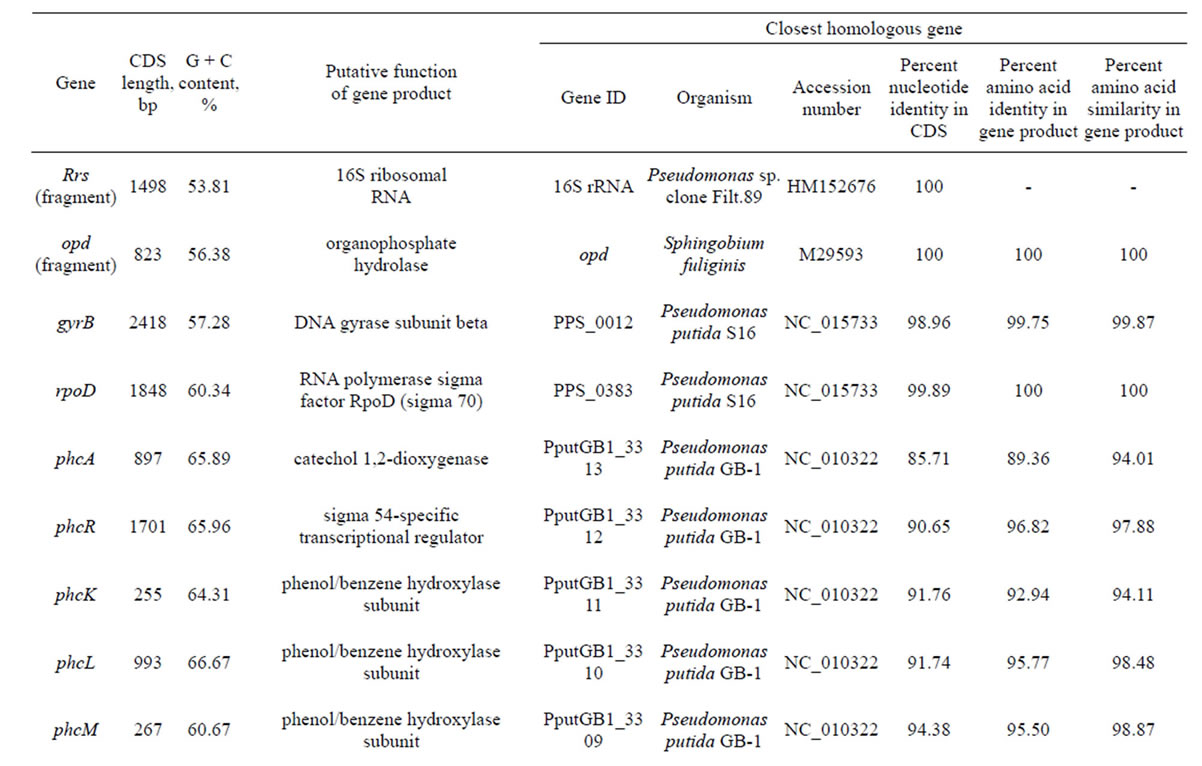

Table 3. Genes identified in POXN01 strain.


continued
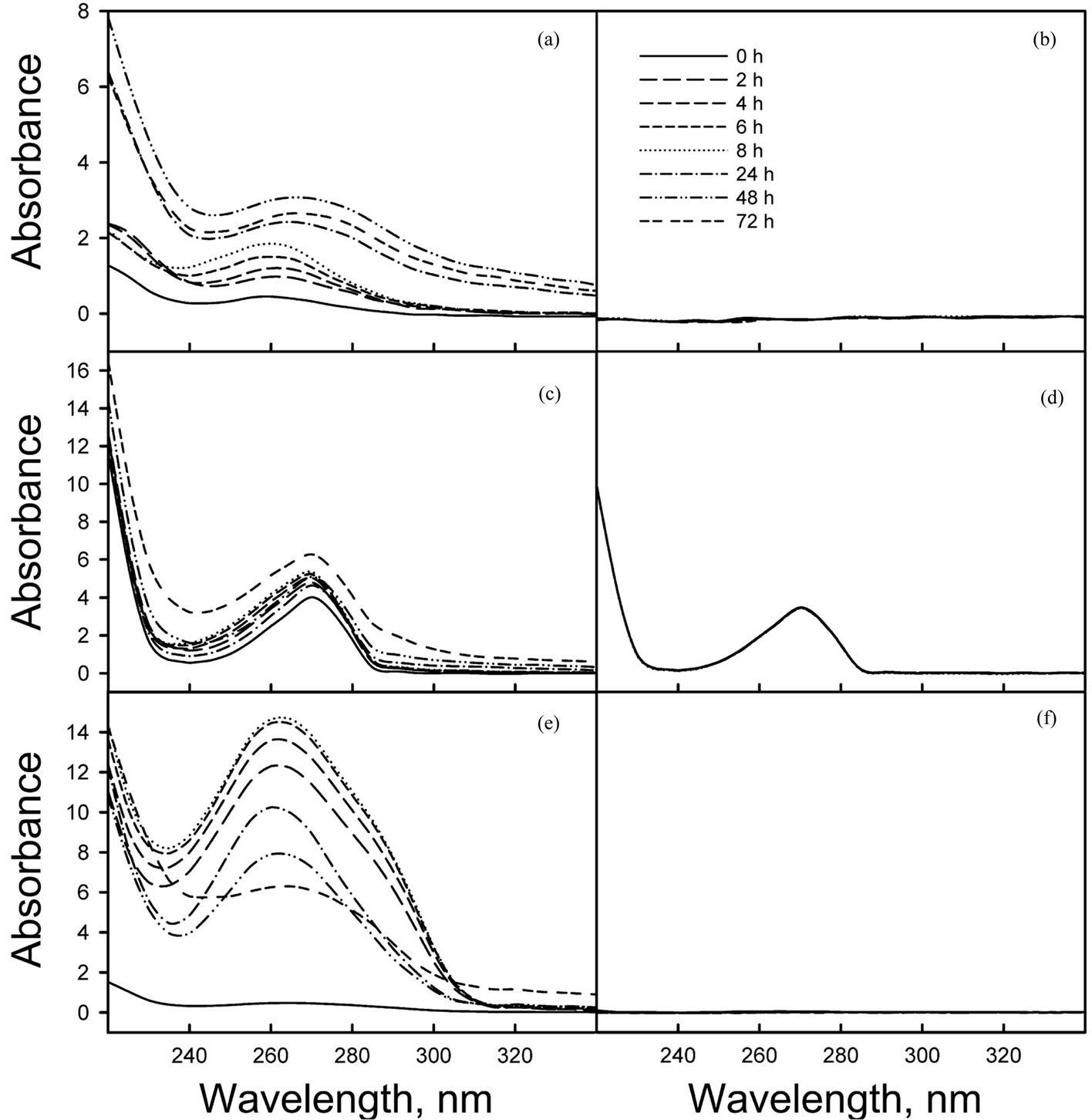
Figure 6. Spectral analysis of degradation of aromatic compounds by POXN01 resting cells. Benzene (a), (b), phenol (c), (d) or toluene (e), (f) were resuspended in 0.05 M potassium phosphate (pH 7.0) and incubated either with (a), (c), (e) or without (b), (d), (f) POXN01 cells as described in Materials in Methods. At indicated times, samples were withdrawn and centrifuged to remove cells and to separate aqueous and organic phases. UV spectra of clarified aqueous phases were measured against 0.05 M potassium phosphate (pH 7.0).
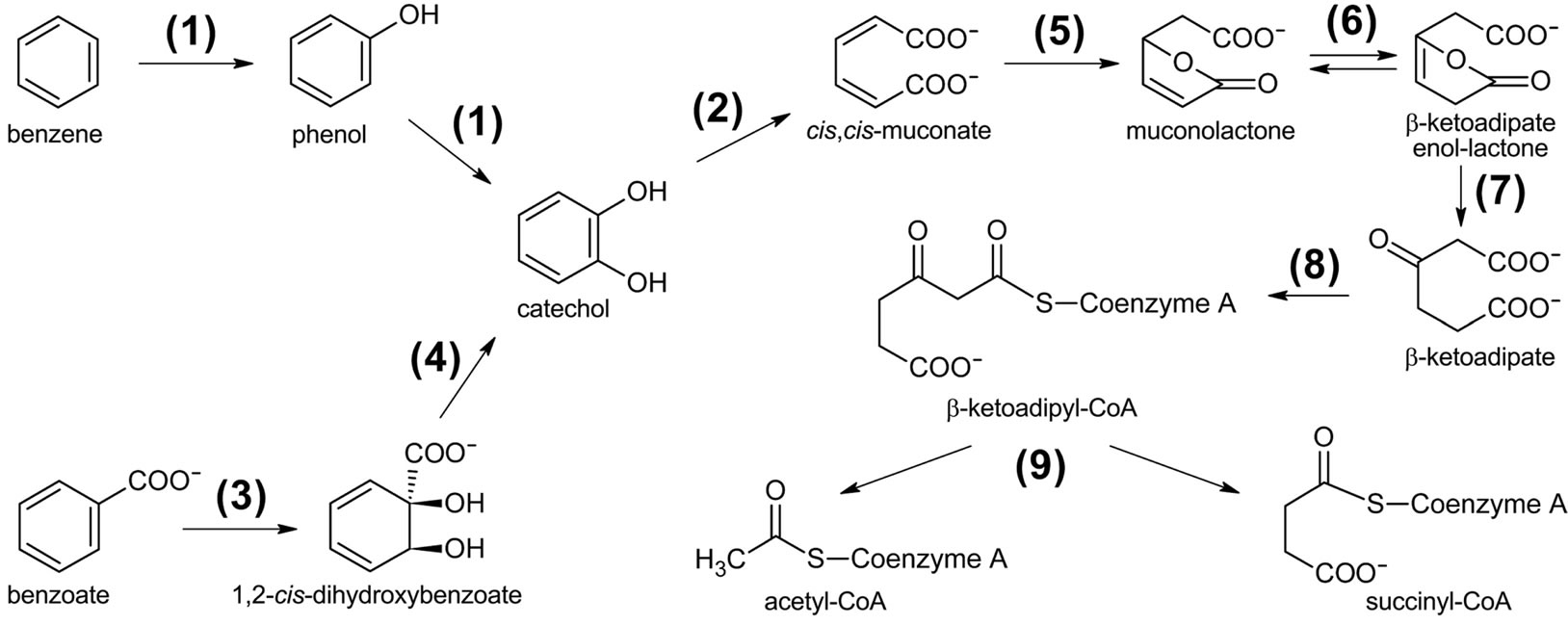
Figure 7. Putative pathway for the degradation of aromatic compounds by the POXN01 isolate. Gene products involved are as follows: (1) phenol/benzene hydroxylase (phcKLMNOP), (2) catechol 1,2-dioxygenase (phcA, benG, catA), (3) benzoate 1,2-dioxigenase (benABC), (4) 1,6-dihydroxycyclohexa-2,4-diene-1-carboxylate dehydrogenase (benD), (5) muconate cycloisomerase (catB), (6) muconolactone delta-isomerase (catC), (7) beta-ketoadipate enol-lactone hydrolase (pcaD), (8) beta-ketoadipate:succinyl-CoA transferase (pcaIJ), (9) beta-ketoadipyl-CoA thiolase (pcaF).
Commission (TWC 2811wpb001 Title: Building a 21st Century Biotechnology Workforce). The authors would like to thank Roland Tsai, biotechnology undergraduate major, Kevin Smith, biotechnology lab manager, and Ashish Damania, bioinformatics undergraduate major, for assistance with sample collection, preparation and data analysis respectively.
REFERENCES
- Grillo, G., Karalliedde, L. and Senanayake, N. (1989) Organophosphorus insecticide poisoning. British Journal of Anaesthesia, 63, 736-750. http://dx.doi.org/10.1093/bja/63.6.736
- van der Hoek, W. and Konradsen, F. (2006) Analysis of 8000 hospital admissions for acute poisoning in a rural area of Sri Lanka. Clinical Toxicology (Philadelphia), 44, 225-231. http://dx.doi.org/10.1080/15563650600584246
- van der Hoek, W., Konradsen, F., Athukorala, K. and Wanigadewa, T. (1998) Pesticide poisoning: A major health problem in Sri Lanka. Social Science & Medicine, 46, 495-504. http://dx.doi.org/10.1016/S0277-9536(97)00193-7
- Eddleston, M., Eyer, P., Worek, F., Mohamed, F., Senarathna, L., von Meyer, L., Juszczak, E., Hittarage, A., Azhar, S., Dissanayake, W., Sheriff, M.H., Szinicz, L., Dawson, A.H. and Buckley, N.A. (2005) Differences between organophosphorus insecticides in human self-poisoning: A prospective cohort study. Lancet, 366, 1452-1459. http://dx.doi.org/10.1016/S0140-6736(05)67598-8
- Jeyaratnam, J., Lun, K.C. and Phoon, W.O. (1987) Survey of acute pesticide poisoning among agricultural workers in four Asian countries. Bulletin of the World Health Organization, 65, 521-527. http://dx.doi.org/10.1002/ajim.20623
- Calvert, G.M., Karnik, J., Mehler, L., Beckman, J., Morrissey, B., Sievert, J., Barrett, R., Lackovic, M., Mabee, L., Schwartz, A., Mitchell, Y. and Moraga-McHaley, S. (2008) Acute pesticide poisoning among agricultural workers in the United States, 1998-2005. American Journal of Industrial Medicine, 51, 883-898.
- US Congress, Office of Technology Assessment (1992) Disposal of chemical weapons: Alternative technologiesBackground Paper. US Government Printing Office. http://www.fas.org/ota/reports/9210.pdf.
- Eddleston, M., Karalliedde, L., Buckley, N., Fernando, R., Hutchinson, G., Isbister, G., Konradsen, F., Murray, D., Piola, J.C., Senanayake, N., Sheriff, R., Singh, S., Siwach, S.B. and Smit, L. (2002) Pesticide poisoning in the developing world: A minimum pesticides list. Lancet, 360, 1163-1167. http://dx.doi.org/10.1016/S0140-6736(02)11204-9
- Buckley, N.A., Karalliedde, L., Dawson, A., Senanayake, N. and Eddleston, M. (2004) Where is the evidence for treatments used in pesticide poisoning? Is clinical toxicology fiddling while the developing world burns? Journal of Toxicology-Clinical Toxicology, 42, 113-116. http://dx.doi.org/10.1081/CLT-120028756
- Jeyaratnam, J. (1990) Acute pesticide poisoning: A major global health problem. World Health Statistics Quarterly, 43, 139-144.
- World Health Organization (1990) The public health impact of pesticides use in agriculture. WHO, Geneva, 128. http://whqlibdoc.who.int/publications/1990/9241561394.pdf. Accessed 1 July 2012
- Kesavachandran, C.N., Fareed, M., Pathak, M.K., Bihari, V., Mathur, N. and Srivastava, A.K. (2009) Adverse health effects of pesticides in agrarian populations of developing countries. In: Whitacre, D.M., Ed., Reviews of Environmental Contamination and Toxicology, 200, Springer Science + Business Media, New York, 33-52.
- Coupe, R.H., Manning, M.A., Foreman, W.T., Goolsby, D.A. and Majewski, M.S. (2000) Occurrence of pesticides in rain and air in urban and agricultural areas of Mississippi, April-September 1995. Science of the Total Environment, 248, 227-240. http://dx.doi.org/10.1016/S0048-9697(99)00545-8
- Schipper, P.N., Vissers, M.J. and van der Linden, A.M. (2008) Pesticides in groundwater and drinking water wells: Overview of the situation in the Netherlands. Water Science and Technology, 57, 1277-1286. http://dx.doi.org/10.2166/wst.2008.255
- Sethunathan, N. and Yoshida, T. (1973) A Flavobacterium sp. that degrades diazinon and parathion. Canadian Journal of Microbiology, 19, 873-875. http://dx.doi.org/10.1139/m73-138
- Kawahara, K., Tanaka, A., Yoon, J. and Yokota, A. (2010) Reclassification of a parathione-degrading Flavobacterium sp. ATCC 27551 as Sphingobium fuliginis. Journal of General and Applied Microbiology, 56, 249-255. http://dx.doi.org/10.2323/jgam.56.249
- Serdar, C.M., Gibson, D.T., Munnecke, D.M. and Lancaster, J.H. (1982) Plasmid involvement in parathion hydrolysis by Pseudomonas diminuta. Applied and Environmental Microbiology, 44, 246-249.
- Segers, P., Vancanneyt, M., Pot, B., Torck, U., Hoste, B., Dewettinck, D., Falsen, E., Kersters, K. and De Vos, P. (1994) Classification of Pseudomonas diminuta Leifson and Hugh 1954 and Pseudomonas vesicularis Busing, Doll, and Freytag 1953 in Brevundimonas gen. nov. as Brevundimonas diminuta comb. nov. and Brevundimonas vesicularis comb. nov., respectively. International Journal of Systematic Bacteriology, 44, 499-510. http://dx.doi.org/10.1099/00207713-44-3-499
- Caldwell, S.R., Newcomb, J.R., Schlecht, K.A. and Raushel, F.M. (1991) Limits of diffusion in the hydrolysis of substrates by the phosphotriesterase from Pseudomonas diminuta. Biochemistry, 30, 7438-7444. http://dx.doi.org/10.1021/bi00244a010
- Dumas, D.P., Caldwell, S.R., Wild, J.R. and Raushel, F.M. (1989) Purification and properties of the phosphotriesterase from Pseudomonas diminuta. Journal of Biological Chemistry, 264, 19659-19665.
- Dumas, D.P., Wild, J.R. and Raushel, F.M. (1989) Diisopropylfluorophosphate hydrolysis by a phosphotriesterase from Pseudomonas diminuta. Biotechnology and Applied Biochemistry, 11, 235-243.
- Siddavattam, D., Khajamohiddin, S., Manavathi, B., Pakala, S. and Merrick, M. (2003) Transposon-like organization of the plasmid-borne organophosphate degradation (opd) gene cluster found in Flavobacterium sp. Applied and Environmental Microbiology, 69, 2533-2539. http://dx.doi.org/10.1128/AEM.69.5.2533-2539.2003
- Somara, S., Manavathi, B., Tebbe, C. and Siddavattam, D. (2002) Localisation of identical organophosphorus pesticide degrading (opd) genes on genetically dissimilar indigenous plasmids of soil bacteria: PCR amplification, cloning and sequencing of the opd gene from Flavobacterium balustinum. Indian Journal of Experimental Biology, 40, 774-779.
- Horne, I., Qiu, X., Russell, R.J. and Oakeshott, J.G. (2003) The phosphotriesterase gene opdA in Agrobacterium radiobacter P230 is transposable. FEMS Microbiology Letters, 222, 1-8. http://dx.doi.org/10.1016/S0378-1097(03)00211-8
- Horne, I., Sutherland, T.D., Harcourt, R.L., Russell, R.J. and Oakeshott, J.G. (2002) Identification of an opd (organophosphate degradation) gene in an Agrobacterium isolate. Applied and Environmental Microbiology, 68, 3371-3376. http://dx.doi.org/10.1128/AEM.68.7.3371-3376.2002
- Chaudhry, G.R., Ali, A.N. and Wheeler, W.B. (1988) Isolation of a methyl parathion-degrading Pseudomonas sp. that possesses DNA homologous to the opd gene from a Flavobacterium sp. Applied and Environmental Microbiology, 54, 288-293.
- Harper, L.L., McDaniel, C.S., Miller, C.E. and Wild, J.R. (1988) Dissimilar plasmids isolated from Pseudomonas diminuta MG and a Flavobacterium sp. (ATCC 27551) contain identical opd genes. Applied and Environmental Microbiology, 54, 2586-2589.
- Pandeeti, E.V., Chakka, D., Pandey, J.P. and Siddavattam, D. (2011) Indigenous organophosphate-degrading (opd) plasmid pCMS1 of Brevundimonas diminuta is selftransmissible and plays a key role in horizontal mobility of the opd gene. Plasmid, 65, 226-231. http://dx.doi.org/10.1016/j.plasmid.2011.02.003
- Wilson, K. (1990) Preparation of genomic DNA from bacteria. In: Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A. and Struhl, K. Eds., Current Protocols in Molecular Biology, John Wiley & Sons, Inc., New York.
- Frank, J.A., Reich, C.I., Sharma, S., Weisbaum, J.S., Wilson, B.A. and Olsen, G.J. (2008) Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Applied and Environmental Microbiology, 74, 2461-2470. http://dx.doi.org/10.1128/AEM.02272-07
- Baker, G.C., Smith, J.J. and Cowan, D.A. (2003) Review and re-analysis of domain-specific 16S primers. Journal of Microbiological Methods, 55, 541-555. http://dx.doi.org/10.1016/j.mimet.2003.08.009
- Sprenger, W.W., Dijkstra, A., Zwart, G.J., Agterveld, M.P., Noort, P.C. and Parsons, J.R. (2003) Competition of a parathion-hydrolyzing Flavobacterium with bacteria from ditch water in carbon-, nitrateand phosphate-limited continuous cultures. FEMS Microbiology Ecology, 43, 45-53. http://dx.doi.org/10.1111/j.1574-6941.2003.tb01044.x
- Borodina, T.A., Lehrach, H. and Soldatov, A.V. (2003) DNA purification on homemade silica spin-columns. Analytical Biochemistry, 321, 135-137. http://dx.doi.org/10.1016/S0003-2697(03)00403-2
- Hannon, G.J. (2010) FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit.
- Zerbino, D.R. and Birney, E. (2008) Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research, 18, 821-829. http://dx.doi.org/10.1101/gr.074492.107
- Hernandez, D., François, P., Farinelli, L., Osteras, M. and Schrenzel, J. (2008) De novo bacterial genome sequencing: Millions of very short reads assembled on a desktop computer. Genome Research, 18, 802-809. http://dx.doi.org/10.1101/gr.072033.107
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403-410.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K. and Madden, T.L. (2009) BLAST+: Architecture and applications. BMC Bioinformatics, 10, 421. http://dx.doi.org/10.1186/1471-2105-10-421
- Smith, D.R., Quinlan, A.R., Peckham, H.E., Makowsky, K., Tao, W., Woolf, B., Shen, L., Donahue, W.F., Tusneem, N., Stromberg, M.P., Stewart, D.A., Zhang, L., Ranade, S.S., Warner, J.B., Lee, C.C., Coleman, B.E., Zhang, Z., McLaughlin, S.F., Malek, J.A., Sorenson, J.M., Blanchard, A.P., Chapman, J., Hillman, D., Chen, F., Rokhsar, D.S., McKernan, K.J., Jeffries, T.W., Marth, G.T. and Richardson, P.M. (2008) Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Research, 18, 1638-1642. http://dx.doi.org/10.1101/gr.077776.108
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25, 1754-1760. http://dx.doi.org/10.1093/bioinformatics/btp324
- Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J. and Higgins, D.G. (2007) Clustal W and Clustal X version 2. Bioinformatics, 23, 2947-2948. http://dx.doi.org/10.1093/bioinformatics/btm404
- Felsenstein, J. (1989) PHYLIP-Phylogeny inference package. Cladistics, 5, 164-166.
- Felsenstein, J. and Churchill, G.A. (1996) A Hidden Markov Model approach to variation among sites in rate of evolution. Molecular Biology and Evolution, 13, 93-104. http://dx.doi.org/10.1093/oxfordjournals.molbev.a025575
- Stöver, B.C. and Müller, K.F. (2010) TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics, 11, 7. http://dx.doi.org/10.1186/1471-2105-11-7
- Iyer, R., Iken, B. and Tamez, T. (2012) Isolation, molecular and biochemical identification of Paraoxon-metabolizing Pseudomonas species. Journal of Bioremediation and Biodegradation, 2, 2011, 5. http://www.omicsonline.org/2155-6199/2155-6199-2-132.pdf.
- Anzai, Y., Kim, H., Park, J.Y., Wakabayashi, H. and Oyaizu, H. (2000) Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. International Journal of Systematic and Evolutionary Microbiology, 50, 1563-1568. http://dx.doi.org/10.1099/00207713-50-4-1563
- Moore, E.R.B., Mau, M., Arnscheidt, A., Bottger, E.C., Hutson, R.A., Collins, M.D., VandePeer, Y., DeWachter, Y. and Timmis, K.N. (1996) The determination and comparison of the 16S rRNA gene sequences of species of the genus Pseudomonas (sensu stricto) and estimation of the natural intrageneric relationships. Systematic and Applied Microbiology, 19, 478-492. http://dx.doi.org/10.1016/S0723-2020(96)80021-X
- Mulet, M., Lalucat, J. and García-Valdés, E. (2010) DNA sequence-based analysis of the Pseudomonas species. Environmental Microbiology, 12, 1513-1530.
- Ait Tayeb, L., Ageron, E., Grimont, F. and Grimont, P.A.D. (2005) Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Research in Microbiology, 156, 763- 773. http://dx.doi.org/10.1016/j.resmic.2005.02.009
- Hilario, E., Buckley, T.R. and Young, J.M. (2004) Improved resolution on the phylogenetic relationships among Pseudomonas by the combined analysis of atpD, carA, recA and 16S rDNA. Antonie van Leeuwenhoek, 86, 51- 64. http://dx.doi.org/10.1023/B:ANTO.0000024910.57117.16
- Peix, A., Ramírez-Bahena, M.H. and Velázquez, E. (2009) Historical evolution and current status of the taxonomy of genus Pseudomonas. Infection, Genetics and Evolution, 9, 1132-1147. http://dx.doi.org/10.1016/j.meegid.2009.08.001
- Yamamoto, S. and Harayama, S. (1998) Phylogenetic relationships of Pseudomonas putida strains deduced from the nucleotide sequences of gyrB, rpoD and 16S rRNA genes. International Journal of Systematic and Evolutionary Microbiology, 48, 813-819. http://dx.doi.org/10.1099/00207713-48-3-813
- Yamamoto, S., Kasai, H., Arnold, D.L., Jackson, R.W., Vivian, A. and Harayama, S. (2000) Phylogeny of the genus Pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology, 146, 2385-2394.
- Wang, S., Liu, Z., Tang, H., Meng, J. and Xu, P. (2007) Characterization of environmentally friendly nicotine degradation by Pseudomonas putida biotype A strain S16. Microbiology, 153, 1556-1565. http://dx.doi.org/10.1099/mic.0.2006/005223-0
- Tang, H., Yao, Y., Wang, L., Yu, H., Ren, Y., Wu, G. and Xu, P. (2012) Genomic analysis of Pseudomonas putida: Genes in a genome island are crucial for nicotine degradation. Scientific Reports, 2, 377. http://dx.doi.org/10.1038/srep00377
- Fujita, M., Ike, M. and Kamiya, T. (1993) Accelerated phenol removal by amplifying the gene expression with a recombinant plasmid encoding catechol-2,3-oxygenase. Water Research, 27, 9-13. http://dx.doi.org/10.1016/0043-1354(93)90189-O
- Takeo, M., Maeda, Y., Okada, H., Miyama, K., Mori, K., Ike, M. and Fujita, M. (1995) Molecular cloning and sequencing of the phenol hydroxylase gene from Pseudomonas putida BH. Journal of Fermentation and Bioengineering, 79, 485-488. http://dx.doi.org/10.1016/0922-338X(95)91266-8
- Shimao, M., Nakamura, T., Okuda, A. and Harayama, S. (1996) Characteristics of transposon insertion mutants of mandelic acid-utilizing Pseudomonas putida strain A10L. Bioscience, Biotechnology and Biochemistry, 60, 1051- 1055. http://dx.doi.org/10.1271/bbb.60.1051
- Mulbry, W.W. and Karns, J.S. (1989) Parathion hydrolase specified by the Flavobacterium opd gene: Relationship between the gene and protein. Journal of Bacteriology, 171, 6740-6746.
- [61] Rani, N.L. and Lalithakumari, D. (1994) Degradation of methyl parathion by Pseudomonas putida. Canadian Journal of Microbiology, 40, 1000-1006. http://dx.doi.org/10.1139/m94-160
- [62] Santos, P.M. and Sa-Correia, I. (2007) Characterization of the unique organization and co-regulation of a gene cluster required for phenol and benzene catabolism in Pseudomonas sp. M1. Journal of Biotechnology, 131, 371-378. http://dx.doi.org/10.1016/j.jbiotec.2007.07.941
- [63] Cowles, C.E., Nichols, N.N. and Harwood, C.S. (2000) BenR, a XylS homologue, regulates three different pathways of aromatic acid degradation in Pseudomonas putida. Journal of Bacteriology, 182, 6339-6346. http://dx.doi.org/10.1128/JB.182.22.6339-6346.2000
- [64] Cafaro, V., Notomista, E., Capasso, P. and Di Donato, A. (2005) Regiospecificity of two multicomponent monooxygenases from Pseudomonas stutzeri OX1: Molecular basis for catabolic adaptation of this microorganism to methylated aromatic compounds. Applied and Environmental Microbiology, 71, 4736-4743. http://dx.doi.org/10.1128/AEM.71.8.4736-4743.2005
- [65] Ramos, J.L., Duque, E., Godoy, P. and Segura, A. (1998) Efflux pumps involved in toluene tolerance in Pseudomonas putida DOT-T1E. Journal of Bacteriology, 180, 3323-3329.
- [66] Ramos, J.L., Duque, E., Gallegos, M.T., Godoy, P., RamosGonzalez, M.I., Rojas, A., Teran, W. and Segura, A. (2002) Mechanisms of solvent tolerance in gram-negative bacteria. Annual Review of Microbiology, 56, 743-768. http://dx.doi.org/10.1146/annurev.micro.56.012302.161038
- [67] Kim, K., Lee, S., Lee, K. and Lim, D. (1998) Isolation and characterization of toluene-sensitive mutants from the toluene-resistant bacterium Pseudomonas putida GM73. Journal of Bacteriology, 180, 3692-3696.
- [68] Hearn, E.M., Patel, D.R. and van den Berg, B. (2008) Outer-membrane transport of aromatic hydrocarbons as a first step in biodegradation. Proceedings of the National Academy of Sciences of the United States of America, 105, 8601-8606. http://dx.doi.org/10.1073/pnas.0801264105
Abbreviations
OP: Organophosphorus;
CSM: Carbon-Deficient media;
TLC: Thin Layer Chromatography;
UV: Ultraviolet)PCR: Polymerase Chain Reaction.
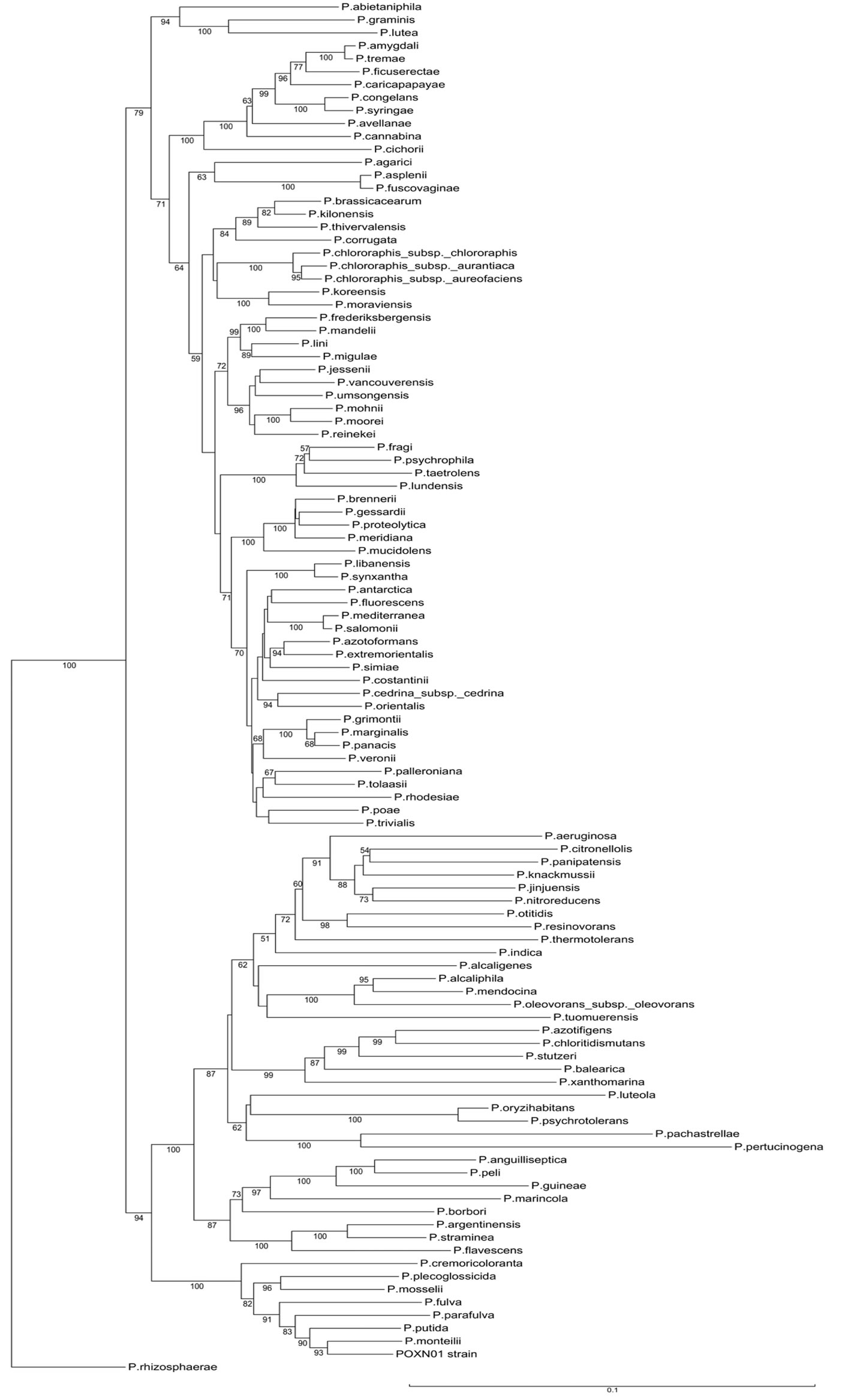
Figure S1. Neighbor-joining tree derived from concatenated alignments of 16S rRNA - gyrB - rpoD genes of the POXN01 isolate and type species of the genus Pseudomonas. Bootstrap values expressed as percentages of the total number of replicates are shown next to each node (values below 50% are not shown). Scale bar represents 10% sequence difference.

(a) (b)
Figure S2. TLC analysis of products formed in the course of phenol degradation by POXN01 resting cells. Phenol degradation experiment was performed as described in Materials and Methods. Samples were collected at indicated times, cleared by centrifugation and kept frozen at (−80) ˚C prior analysis. Ascending TLC was carried out on Whatman KC18F plates using methanol: water 2:1 (v/v) as an eluent. Spots were visualized either under UVC light (200 - 280 nm) (a) or UVA light (340 - 400 nm) (b).
NOTES
*Corresponding author.