Advances in Biological Chemistry
Vol. 2 No. 2 (2012) , Article ID: 18975 , 6 pages DOI:10.4236/abc.2012.22017
SSINCC: Simple separation of interacting nucleoprotein complex components
![]()
School of Pharmacy, University of Waterloo, Waterloo, Canada
Email: slavcev@uwaterloo.ca
Received 9 November 2011; revised 18 January 2012; accepted 21 March 2012
Keywords: DNA-protein-DNA interaction assay; Plasmid partition nucleoprotein complex; Specific nucleoprotein complex separation
ABSTRACT
Protein-DNA binding assays have been used in a variety of applications from fundamental studies regarding the binding process itself to serve as probes for the detection, quantification and separation of target analytes. Here we describe a novel method of analyzing and identifying intermolecular DNA interactions that allows for the simple separation of interacting nucleoprotein complex components (SSINCC), focusing specifically on DNA-DNA interactions using P1 plasmid active partition system nucleoprotein complexes as a model to demonstrate DNA sequence specificity and tolerance of composite factor complexity. Traditional and recent assays of proteinDNA interaction are summarized and compared with SSINC. Although SSINC is examined here employing P1 partition nucleoprotein complex as an example of DNA-DNA intermolecular association, universal applications of this methodology to nucleoprotein complex studies can be envisioned.
1. INTRODUCTION
The association between proteins and DNA is a fundamental requirement for most vital cellular functions including gene transcription, replication, recombination, repair, segregation, chromosomal stability, cell cycle progression, and epigenetic silencing. Protein-DNA binding assays have been used in a variety of fields, such as binding process to serve as probes for the detection, quantification and separation of target analytes [1]. These assays have also been used for the study of protein-DNA complex stoichiometry, the detection of DNA damage, and real-time separation of free and bound complexes by electrophoretic mobility. Plasmid biology, in particular plasmid partition, seeks to elucidate mechanisms of unit copy plasmid segregation systems and it is just one of many areas of scientific investigation that employs such biochemical assays. Plasmids serve as tractable model systems to study DNA segregation because they require only a DNA centromere, a centromere-binding protein and a force-generating ATPase. In this study we describe a novel DNA-DNA capture method, and use it to examine the ability of plasmid partition proteins to pair plasmids at their centromeres.
Segregation of the plasmid P1 is directed by the par locus, which is comprised of the parA-parB operon and the downstream cis-acting site parS [2]. The P1 parABS system is genetically and mechanistically very similar to sopABC of the E. coli F plasmid [3]. ParB is the protein that binds specifically to partition site parS. It forms dimers in solution [4] and recognizes two distinct asymmetric repeat sequences, Box A and Box B, within the parS site [5,6] that border an E. coli integration host factor (IHF) binding site [4,7]. Although it is strongly suspected, a central question in partition mechanism about whether newly replicated plasmids are paired during the reaction has yet to be elucidated. The oligomerization of ParB at parS sites is postulated to promote the pairing of P1 plasmids during the partition process prior to their separation and relocation to the 1/4 positions of the cell. P1 ParA is a weak ATPase with a Walker type ATPase site, the activity of which is stimulated 4 to 5 fold by ParB [8]. Although the exact molecular mechanism of ParA activity in P1 partition has yet to be discerned, ParA functions as a dimer and upon binding ATP stimulates DNA-DNA (parS-parS) association in a ParB+, parS+-dependent manner. It has been proposed that the hydrolysis of ATP by ParA is responsible for separating paired replicated plasmids.
Here, we describe a novel method of analyzing and identifying intermolecular DNA interactions that allows for the simple separation of interacting nucleoprotein complex components (SSINCC), focusing specifically on DNA-DNA interactions by using P1 plasmid active partition system nucleoprotein complexes as a model to demonstrate DNA sequence specificity and tolerance of composite factor complexity (Figure 1). While this sensitive system using biotin-streptavidin-DNA coated magnetic beads and radiolabeled DNA capture assay is particularly useful in studying plasmid pairing and segregation, the application of this method can be extended to any studies involving intermolecular DNA-DNA interactions that are formed via simple or composite nucleoprotein interactions; for example, various recombination and initiation of replication molecular systems.
2. MATERIALS AND METHODS
Reagents, media, and buffers. The suppliers for reagents were as follows: restriction enzymes and enzymes for DNA manipulations, Promega; antibiotics, Sigma and Fisher Scientific; radioisotopes, Amersham Biosciences; Streptavidin-magnetic bead (SAM) suspension, Dynalbiotech; Magnetic separation stand, Promega; Adenosine triphosphate, Boehringer Mannheim; Adenosine 5’-O-3- thiotriphosphate, Roche. Bacterial cells were grown in Luria Bertani (LB) medium. Antibiotics, when present, were used at the following concentrations: ampicillin, 100 µg/ml; chloramphenicol, 50 µg/ml; and kanamycin, 50 µg/ml. Buffers were made as follows: TN buffer, 0.1 M NaCl, 0.01 M Tris-HCl pH 7.6; DNA capture buffer (DCB), 0.05 M HEPES-KOH, 0.1 M NaCl, 1 mM
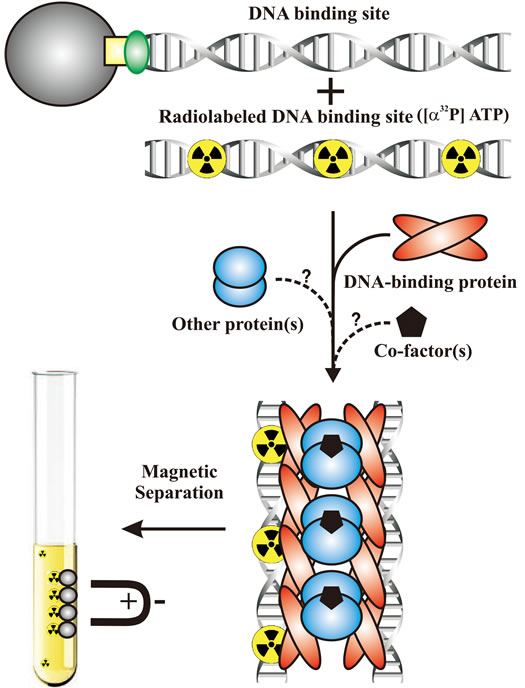
Figure 1. In vitro separation of interacting nucleoprotein complex components. A simple in vitro assay measuring the ability of a nucleoprotein components to associate SAM-conjugated DNA target site fragments with free radiolabeled DNA target site fragments, thereby capturing the complex. The capture of free radiolabeled DNA is then measured by a scintillation counter.
EDTA, 0.01 M MgCl2; Nucleotides (ATP, ATPγS or ADP), when present, were included at 2 mM.
Preparation of DNA-biotin-streptavidin-magnetic beads. DNA fragments were constructed as follows: A 1, 032- bp biotinylated parS fragment was PCR amplified from pLG44 using primers 5’-GCTTTGTGCTGTT-GGCGACG-3’ and Biotin-5’CTGAACAGGCAGGAT-CATCGCG-3’; a 932-bp biotiny-lated lacI fragment was amplified from pLG49 using primers 5’-CGCGA-CGTTATACGATGTCGCAGA-3’ and Biotin-5’-CGC-GCCCTTCACCGCCTGGCCCTG-3’. An α32P ATP-labeled 1, 012-bp parS fragment was amplified from pLG44 using primers 5’GTATCCGAAGCACTATTAA-GCATC-3’ and 5’-CACTTGCCCACGCTGTAGCG-3’. 11X DNA-SAM premixes were made in a total volume of 600 µl as follows: A 1 ml aliquot of SAM suspension was washed three times via lateral magnetic separation of beads with 1 ml of DCB, and resuspended to a total volume of 1 ml with DCB. Next, 500 µl of SAM was incubated with 50 µl (~2 pmol) of biotinylated DNA fragment and incubated with frequent mixing at 37˚C and a minimum of 120 minutes at 30˚C. A 50 µl aliquot was digested with EcoRV and found to bind approximately 50 fmol of biotinylated parS or lacI fragment. Beads were then washed twice with 1 ml of ParB buffer and resuspended to total volume of 520 µl in the same buffer. Final suspensions contained 0.02% BSA, 0.02% sonicated salmon sperm DNA and 10 mM MgCl2 and were incubated at 37˚C for 60 minutes. This procedure was used to prepare beads bound to biotinylated parS (premix A), biotinylated lacI (premix B), or no DNA (premix C). Premixes containing nucleotide possessed 0.002 M ATP, ATPγS, or ADP. Radiolabeled DNA capture assay.
Additions to 50 µl of premix: Nucleotide, if applicable, was added to each sample to 0.002 M; Next, 2 µl (~0.2 pmol) of α32P ATP-labeled parS fragment was added to each sample. Next, DCB was added to each sample as needed to ensure that each sample per assay possessed the same volume. Finally, proteins (ParB added last) were added as required to each sample. Samples were incubated at 37˚C with frequent gentle mixing for 15 minutes. Sample tubes were then placed into magnetic separation columns, supernatant was carefully removed and beads were washed once with 200 µl of DCB pre-warmed to 37˚C and +/−2 mM of appropriate nucleotide as required, and beads were resuspended in 100 µl of ice cold DCB without nucleotide. Samples were vortexed vigorously for 30 seconds and the entire volume was spotted onto glass fiber filters. Amounts of incorporated [α32P] ATP were measured with a scintillation counter. Maximal activity of total 32P-labelled parS DNA generally ranged between 50,000 to 200,000 cpm, allowing for accurate and reproducible measurement of parS capture.
3. RESULTS
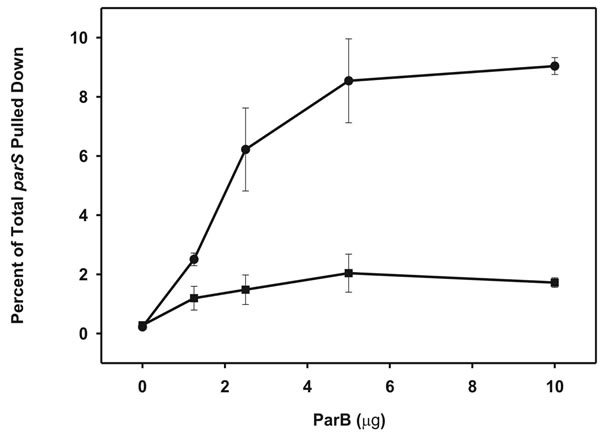
An oligomerizing DNA-binding protein associates DNA fragments in a sequence-specific manner. In this study we examined the hypothesis that an immobilized specific DNA sequence, parS, which serves as the target site for the DNA-binding proteins IHF and ParB could be complexed with free parS sites by ParB. Furthermore, we expected that the strength of this intermolecular DNA interaction could be modulated by the P1 partition ATPase, ParA, in a nucleoprotein-dependent manner. The pairing reaction in vitro was verified using a biotinylated DNA fragment carrying parS that could capture a radioactive but non-biotinylated fragment onto streptavidin-coated magnetic beads (SAM; Figure 1). As a negative control we used a biotinylated DNA fragment without parS from the lacI gene. We bound biotinylated parS, or lacI fragments to streptavidin-SAM beads based on the high affinity interaction between biotin and streptavidin (see Materials and Methods). After adding IHF and ParB to the mixture, we measured ParB binding of SAM-bound parS fragments (~50 fmol) to [α32P] ATP-radio-labeled soluble parS fragments based on the percentage of total radiolabeled parS fragment (~200 fmol) that could be pulled down (Figure 2(a)). We saw a relatively small but reproducible association of non-biotinylated parS in the presence of ParB. Optimal association of SAM-parS and free parS was achieved at 0.04 mM ParB, at which level almost 9% of free radiolabeled parS in the mixture was captured. In contrast, SAM-lacI beads, and beads without DNA were very poor at capturing radiolabeled parS (≤2% of total) at all ParB titration levels, indicating that parS capture is conferred via specific ParB-parS interactions. Next, as a negative control, we assayed the ability of the minimal DNA binding fragment of ParB, ParB (142 - 333) that lacks the N-terminal of ParB; this fragment retains parS site binding capability, but lacks the N-terminal domain that enables ParB-mediated oligomerization and hence intermolecular complexing of parS fragments (Figure 2(b)) [9]. Compared to full length ParB, ParB (142 – 333) poorly captured free parS (<1%). These findings indicate that the pairing of parS fragments by ParB is sequence-specific and cannot be conferred solely by the DNA-binding region of ParB; association also requires the N-terminal of ParB that confers oligomerization capability with adjacent ParB molecules upon binding DNA; a phenotype necessary for intermolecular association of parS fragments. DNA-DNA interactions are modulated by the ATPase partition protein in a nucleotide-dependent manner. While the addition of ParA-ATP to ParB and parS did not have much effect on base-line ParB-parS pairing activity (2.5% - 5%; Figure 3),
 (a)
(a) (b)
(b)
Figure 2. Association of target DNA fragments via a homo-oligomerizing centromere binding protein. (a) ParBmediated capture of [α32P] ATP-labeled parS fragment (~50 fmol) measured following incubation with IHF (0.2 µg) + Agarose-parS (~50 fmol) (●), and IHF + Agarose-lacI (~50 fmol) (■); (b) Capture of αP32-ATP-labeled parS fragment measured following incubation with ParB + IHF (0.2 µg) + Agarose-parS (~50 fmol) (●), and ParB (142 - 333) + IHF + Agarose-parS. Each reaction includes 0.02% BSA, 0.02% sonicated salmon sperm DNA was incubated for 15 min at 37˚C. Error bars represent variation from a minimum of three repeated trials.
in the presence of ATPγS, a nonhyrolyzable analogue of ATP, ParA-ATPγS was a powerful stimulant of DNA DNA complexation dramatically enhancing capture of radiolabled parS DNA to more than three times that of ParB alone. In contrast, as expected, ParA-ADP not only did not enhance binding, but actually reduced binding to below that seen by ParB alone. This finding may suggest that ParA-ADP sequesters or somehow inactivates ParB. Modulations in DNA-DNA intermolecular interaction by ParA-NTP were repeatedly discernable indicating the ability of this system to detect modulations in DNA-DNA intermolecular association conferred by a secondary

Figure 3. Influence of nucleoprotein complex factors that modulate association of the target DNA fragments. Capture of [α32P] ATP-labeled parS fragment (~50 fmol) measured following various incubations with nucleotide. ATP 2nm + SAM-parS (~50 fmol) (●); ATPγS (2 mM) + SAM-parS (~50 fmol) (■); ADP (2 mM) + SAM-parS (~50 fmol) (♦). Protein additions were at following levels: IHF (0.2 µg), ParB (2.5 µg), ParA (titrated). Open symbol (highly overlapped for the three nucleotide assays) in each assay denotes a 10 µg of ParA, 0.2 µg IHF in the absence of ParB control; semi-filled symbol represents all maximal protein reagents in the absence of parS. Each reaction included 0.02% BSA, 0.02% sonicated salmon sperm DNA was incubated for 15 min at 37˚C. Error bars represent variation from a minimum of three repeated trials.
component that is a modulator of the nucleoprotein complex and DNA-DNA interaction capabilities. We have used this assay to further characterize the pairing reaction in vitro, and showed it is consistent with in vivo assays that also demonstrate plasmid pairing inside bacterial cells [10].
4. DISCUSSION
We have developed a novel method to measure intermolecular association of DNA that is sequence-specific and can sensitively and reproducibly capture modulations of composite and dynamic nucleoprotein complexes. ParBmediated pairing of parS sites in vitro was based on the ability of ParB to link immobilized parS to free radiolabeled parS fragments and capture the complex. We noted that pairing could be achieved in the presence of adequate ParB and IHF. The complex was further modulated in a predictable fashion by the addition of the partition ATPase, ParA, in a nucleotide-dependent manner. We note a limitation of this approach is that it does not provide any information regarding the stoichiometry of the ParB-parS complexes being formed and whether these fragments are isolated as pairs or clusters comprised of several parS fragments. However, this system was sensitive to ParB-sequence specificity, ParB oligomerization capabilities and was able to detect even minor modulations of complex formation induced by ParA-nt, in a predictable manner and with reproducibility and adequate sensitivity. We also expect that the maximal signal generated by this assay is limited for the following reasons. First, ParB binding to parS in vivo is promoted by the superhelicity of the plasmid, a phenotype which is lost due to the use of short linear DNA fragments. Second, due to the short length of the parS substrate used in this assay, the potential for ParB spreading to promote parS-parS pairing could be limited, although about 400 bp of ParB spreading on a supercoiled plasmid was found sufficient to allow uncompromised partition [11]. More recent studies, in which plasmid stability was determined after spreading was restricted by the introduction of roadblocks on either side of parS, revealed that extensive spreading was, in fact, not required for plasmid partition and the exact role that spreading plays in DNA partition and transcription regulation has yet to be clarified [12]. Third, we cannot control which parS fragments (soluble or SAM-bound) are complexed by ParB, nor the stoichiometry of these associations, since immobilized parS fragments are just as likely to associate with themselves as with free parS fragments and vice versa; and while these interactions undoubtedly occur, they would not produce a measurable signal.
The described method using biotin-streptavidin-DNA coated magnetic beads and radiolabeled DNA capture assay can be applied to other biological systems where protein-DNA interactions are predominant. This new method can serve as additional methodology to study the interactions of regulatory proteins with the specific DNA sequences that they recognize and to which they bind in vivo. Traditional standard assays for protein-DNA interaction include Nitrocellulose filter-binding assay, Footprinting, Electrophoretic mobility shift assay (EMSA), and Southern and Western blotting [13,14], Electrophoretic mobility shift assay (EMSA), first described by Fried and Crothers [15], is a popular method used for detection of protein-DNA interactions. The technique is based on the observation that protein-DNA complexes migrate more slowly than free DNA molecules when subjected to non-denaturing polyacrylamide or agarose gel electrophoresis. Because the rate of DNA migration is shifted or retarded upon protein binding, the assay is also referred to as a gel shift or gel retardation assay. Adding a protein-specific antibody to the binding components creates an even larger complex (antibody-protein-DNA) which migrates even slower during electrophoresis, this is known as a supershift. Although EMSA allows simultaneous analysis of a large population of fragments, it can be time-consuming, difficult to quantitate and may be used to detect only high affinity protein binding sites. Shifted DNA fragments containing protein binding sites are identified by using additional procedures, i.e., gel elution, PCR amplification, cloning and sequencing [3]. Because of the “caging” effect within the gel matrix, the DNA-protein interactions can be stabilized and the corresponding shifted complexes can be detected as discrete bands; although in some cases, complexes may dissociate and do not produce detectable shifted bands. The SSINCC methodology described here to capture DNA circumvents such problems since there is no need for gel electrophoresis or other harsh treatments that may trigger dissociation of protein-DNA complex; hence it provides reasonably sensitive and specific data regarding the interaction. DNase I footprinting has also been used to determine the site of interaction of most sequence-specific DNA binding proteins and has been most extensively applied to the study of transcription factors. Because of the need of protein excess and the visualization of footprint by a partial DNA digestion ladder, the foot printing technique requires considerably more material than would a gel shift and cannot itself distinguish individual components of protein-DNA complexes. Moreover, bound proteins usually have other effects on the normal cleavage by DNase I. Unfortunately, DNase I does not cleave DNA indiscriminately, some sequences being very rapidly attacked while others remain unscathed even after extensive digestion [16]. This results in rather uneven “ladder” of digestion products after electrophoresis which in turn limits the resolution of the technique. Another widely used technique, chromatin immunoprecipitation [17] including nChIP, xChIP, and ChIP-on-chip, allows the detection of the in vivo interaction of specific proteins with specific genomic regions and are widely used in epigenetics [17- 19]. The basic steps in this technique are fixation, sonication, immunoprecipitation, and analysis of the immunoprecipitated DNA. Although ChIP is a highly versatile tool, the procedure requires the optimization of reaction conditions and can be quite laborious. While SSINCC is likely less sensitive than ChIP, separated complexes are separated in their free form without chemical modification and are highly specific with low background.
Recent advances have been made in protein-DNA binding assays using capillary electrophoresis, laserinduced fluorescence, fluorescence polarization, molecular beacons, and affinity chromatography. Fluorescence spectroscopy can be used as a sensitive non-destructive technique for the characterization of proteinDNA interactions [20]. In addition, fluorescence polarization is used to screen known consensus sequence and target protein. The DNA-protein interaction is detected by measuring the difference in rotational motion in solution when the consensus sequence labelled with a fluorophore such as fluorescein is excited by polarized light [21]. More recently, surface plasmon resonance, SPR, has been employed to monitor protein-DNA Interactions [22,23]. The SPR effect relies on changes in the refractive index of solutions adjacent to the immobilised surface. This optical technique has the advantage that it generates real-time data (both for kinetic and other analyses) but is quite expensive. Very similarly, SSINCC offers reasonable sensitivity, relatively inexpensive instrumentation and a non-destructive environment. ELISA microplate capture assays is another popular technique which uses immobilized DNA probes to capture specific protein-DNA interactions and confirm protein identities and relative amounts with target specific antibodies [24]. Although, this method is sensitive when performed with enzyme-labeled antibodies and a chemiluminescent substrate, detecting less than 0.2 pg of the target protein per well but it requires antibodies with affinity for DNAbound native proteins (i.e., supershift antibodies) and ELISA assay kits are available for only a few targets.
The proposed simple SSINCC capture assay employing biotin-streptavidin-DNA coated magnetic beads and radiolabeled bait can be applied to study protein-DNA interaction at large. The technique need not be limited to DNA-DNA interaction analysis and is likely extendable to the capture, separation and identification of protein components of a nucleoprotein complex in various biological systems such as transcription factor/promoter interactions. Proteins often function as components of larger complexes to perform a specific function, and formation of these complexes may be regulated. For example, intracellular signaling events often require transient and/or regulated protein-protein interactions for propagation, and protein binding to a specific DNA sequence, RNA molecule or metabolite is often regulated to modulate a particular cellular function. Thus, characterizing protein complexes can offer important insights into protein function. DNA-protein interactions are important in gene regulation, recombination, repair, transcription and translation, and understanding these interactions at the molecular level is of paramount importance, which in many cases is responsible for various abnormalities.
5. ACKNOWLEDGEMENTS
We thank Barbara Funnell and Lori Ing for help, reagents, and support during the initial development of this assay at the University of Toronto. This work was supported by grant 37997 from the Canadian Institutes of Health Research (to B.F.) and funding by the University of Waterloo (R.A.S.).
![]()
![]()
REFERENCES
- Pavski, V. and Le, X.C. (2003) Ultrasensitive proteinDNA binding assays. Current Opinion Biotechnology, 14, 65-73. doi:10.1016/S0958-1669(02)00016-2
- Abeles, A.L., Friedman, S.A. and Austin, S.J. (1985) Partition of unit-copy miniplasmids to daughter cells: III. The DNA sequence and functional organization of the P1 partition region. Journal of Molecular Biology, 185, 261- 272. doi:10.1016/0022-2836(85)90402-4
- Kaer, K., Matlik, K., Metsis, M. and Speek, M. (2008) Combination of native and denaturing PAGE for the detection of protein binding regions in long fragments of genomic DNA. BMC Genomics, 9, 272.
- Funnell, B.E. (1991) The P1 plasmid partition complex at parS. The influence of Escherichia coli integration host factor and of substrate topology. The Journal of Biological Chemistry, 266, 14328-14337.
- Davis, M.A. and Austin, S.J. (1988) Recognition of the P1 plasmid centromere analog involves binding of the ParB protein and is modified by a specific host factor. The EMBO Journal, 7, 1881-1888.
- Funnell, B.E. and Gagnier, L. (1993) The P1 plasmid partition complex at parS. II. Analysis of ParB protein binding activity and specificity. The Journal of Biological Chemistry, 268, 3616-3624.
- Funnell, B.E. (1988) Participation of Escherichia coli integration host factor in the P1 plasmid partition system. Proceedings of the National Academy of Sciences USA, 85, 6657-6661. doi:10.1073/pnas.85.18.6657
- Davis, M.A., Martin, K.A. and Austin, S.J. (1992) Biochemical activities of the ParA partition protein of the P1 plasmid. Molecular Microbiology, 6, 1141-1147. doi:10.1111/j.1365-2958.1992.tb01552.x
- Surtees, J.A. and Funnell, B.E. (1999) P1 ParB domain structure includes two independent multimerization domains. Journal of Bacteriology, 181, 5898-5908.
- Kaur, T., Al Abdallah, Q., Nafissi, N., Wettig, S., Funnell, B.E. and Slavcev, R.A. (2011) ParAB-mediated intermolecular association of plasmid P1 parS sites. Virology, 421, 192-201. doi:10.1016/j.virol.2011.09.027
- Rodionov, O., Lobocka, M. and Yarmolinsky, M. (1999) Silencing of genes flanking the P1 plasmid centromere. Science, 283, 546-549. doi:10.1126/science.283.5401.546
- Rodionov, O. and Yarmolinsky, M. (2004) Plasmid partitioning and the spreading of P1 partition protein ParB. Molecular Microbiology, 52, 1215-1223. doi:10.1111/j.1365-2958.2004.04055.x
- Helwa, R. and Hoheisel, J.D. (2010) Analysis of DNAprotein interactions: From nitrocellulose filter binding assays to microarray studies. Analytical and Bioanalytical Chemistry, 398, 2551-2561.
- Zhang, J.-B., Pan, Z.-X., Lin, F., Ma, X.-S. and Liu, H.-L. (2009) Biochemical methods for the analysis of DNAprotein interactions. Hereditas, 31, 325-336. doi:10.3724/SP.J.1005.2009.00325
- Fried, M. and Crothers, D.M. (1981) Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Research, 9, 6505-6525. doi:10.1093/nar/9.23.6505
- Drew, H.R. (1984) Structural specificities of five commonly used DNA nucleases. Journal of Molecular Biology, 176, 535-557. doi:10.1016/0022-2836(84)90176-1
- Reimer, J.J. and Turck, F. (2010) Genome-wide mapping of protein-DNA interaction by chromatin immunoprecipitation and DNA microarray hybridization (ChIP-chip). Part A: ChIP-chip molecular methods. Methods in Molecular Biology, 631, 139-160.
- Pascual-Ahuir, A. and Proft, M. (2012) Quantification of protein-DNA interactions by in vivo chromatin immunoprecipitation in yeast. Methods in Molecular Biology, 809, 149-156.
- Li, M.L., Wang, W. and Lu, Z.H. (2010) Genomic analysis of DNA-protein interaction by chromatin immunoprecipitation. Hereditas, 32, 219-228. doi:10.3724/SP.J.1005.2010.00219
- Favicchio, R., Dragan, A.I., Kneale, G.G. and Read, C.M. (2009) Fluorescence spectroscopy and anisotropy in the analysis of DNA-protein interactions. Methods in Molecular Biology, 543, 589-611. doi:10.1007/978-1-60327-015-1_35
- Lundblad, J.R., Laurance, M. and Goodman, R.H. (1996) Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Molecular Endocrinology, 10, 607-612. doi:10.1210/me.10.6.607
- McDonnell, J.M. (2001) Surface plasmon resonance: Towards an understanding of the mechanisms of biological molecular recognition. Current Opinion Chemical Biology, 5, 572-577. doi:10.1016/S1367-5931(00)00251-9
- Stockley, P.G. and Persson, B. (2009) Surface plasmon resonance assays of DNA-protein interactions. Methods in Molecular Biology, 543, 653-669.
- Brand, L.H., Kirchler, T., Hummel, S., Chaban, C. and Wanke, D. (2010) DPI-ELISA: A fast and versatile method to specify the binding of plant transcription factors to DNA in vitro. Plant Methods, 6, 25. doi:10.1186/1746-4811-6-25