 Pharmacology & Pharmacy, 2011, 2, 256-265 doi:10.4236/pp.2011.24033 Published Online October 2011 (http://www.SciRP.org/journal/pp) Copyright © 2011 SciRes. PP Modulation of c-Jun NH2-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome Enri Borda1,2, Daniela Passafaro1, Silvia Reina1,2, Leonor Sterin-Borda1,2 1Pharmacology Unit, School of Dentistry, Buenos Aires University, Buenos Aires, Argentina; 2National Research Council of Argen- tina (CONICET), Buenos Aires, Argentina. Email: enri@farmaco.odon.uba.ar Received June 27th, 2011; revised July 20th, 2011; accepted September 14th, 2011. ABSTRACT Background: We wanted to determine (via an immunopharmacological approach) whether the c-Jun NH2 terminal kinase (JNK) cascade is phosphorylated in the submandibular gland by carbachol and cholinergic autoantibodies (IgG) present in the sera of patients with primary Sjögren’s syndrome (pSS) by interaction and activation of salivary gland muscarinic acetylcholine receptors (mAChRs). Methods: The JNK, PGE2 and NOS assays were measured in rat sub- mandibular gland with pSS IgG and carbachol alone or in the presence of different blocker agents. Results: pSS IgG- activated M3 mAChRs stimulated JNK phosphorylation whereas the activation of M1 mAChRs by carbachol stimulated JNK phosphorylation involving calcium-activated mechanism. The intracellular pathway leading to pSS IgG-induced biological effects on JNK activity involved activation of protein kinase C (PKC), inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) enzymes. Also, activation of COX-2 and COX-1 by pSS IgG and carbachol-induced PGE2 generation were involved. Conclusion: These results may contribute to better understanding the modulatory role of JNK enzymes by cholinergic autoantibodies from pSS patients acting on mAChR in rat submandibular gland. Keywords: JNK, pSS IgG, Autoantibodies, PGE2, NOS, Carbachol, Cholinergic Antagonists, Enzymatic Inhibitors 1. Introduction Primary Sjögren’s syndrome (pSS) is a systemic auto- immune disease of unknown etiology. Many autoanti- bodies have been reported to be present in pSS [1]. In particular; antibodies to the antigens SS-A/Ro and SS-B/ La are commonly used in the diagnosis of pSS [2]. The presence of subtypes M1 [3], M3 [4,5] and M4 [6] specific autoantibodies in 83% - 90% persons with pSS [7] is an important advance towards understanding the pathogene- sis of pSS not only in terms of impaired glandular func- tion, but also because of peripheral parasympathetic dysfunction associated with the disease [8,9]. IgG molecules from serum samples from patients with pSS have been reported to bind to glandular cholinore- ceptors and act as partial agonists. IgG molecules have been reported to not only activate the receptor, but also to impair the response to the authentic cholinergic agonist [10], suggesting a defect in post-receptor signaling [11]. The most direct mechanism for preventing target organs from carrying out their function is that of early agonist- mediated activation of cholinoreceptors initiated by autoantibodies [12] which bind to, and persistently acti- vate, cholinoreceptors [13]. Subsequently, the agonistic activity displayed by these autoantibodies may induce the desensitization [14], internalization and/or intracellular degradation of cholinoreceptors, leading to a progressive decrease in the expression and activity of these receptors [12]. Xerostomia and keratoconjunctivitis sicca result from lymphocyte infiltration of the salivary gland [15] and lachrymal gland [16]. The infiltrating cells interfere with glandular function by cell-mediated glandular destruction and production of autoantibodies that interfere with cho- linoreceptors [17]. Dental caries resulting from the loss of salivary flow may be associated with periodontal dis- ease [18]. Prostaglandins (PGs) are among the most relevant lo- cal mediators that participate in the modulation of acini cell functions under basal conditions [19]. PGs are re-  Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome257 2 leased in large amounts during inflammation or in early stages of autoimmune diseases. In particular, overpro- duction of PGs has been shown to occur in neuroinflam- matory diseases [20]. Also, nitric oxide (NO) plays a key part in the pathophysiology of systemic and chronic in- flammatory disease and in the neurodegenerative process [21]. Recent evidence has indicated that there is constant crosstalk between NO and PGs biosynthesis pathways involved in the pathological mechanisms underlying cer- tain inflammatory disorders [21,22]. In general, muscarinic acetylcholine receptor (mAChR) subtypes are grouped according to their functional cou- pling. This can be via mobilization of intracellular cal- cium (M1, M3, M5) through the activation of phospholi- pase C (PLC), which results in the release of the second messenger inositol 1,4,5-triphosphate (IP3) or by inhibit- tion of adenylate cyclase (M2, M4), which results in re- duction of the intracellular levels of cyclic adenosine monophosphate [23]. The same receptor may generate more than one set of intracellular second messengers and considerable crosstalk exists between signaling cascades [24]. The ability of these receptors to stimulate or inhibit cell growth has been attributed to differences in cell models, but the mechanisms involved in these cell type- dependent differences in growth response are unknown [25]. Mitogen-activated protein kinases (MAPKs) are acti- vated by a diverse array of extracellular stimuli and regu- late various cellular responses [26,27]. MAPK family members include c-Jun NH2-terminal kinase (JNK) [28]. JNK is activated by cellular stress (ultraviolet and gamma radiation), osmotic and heat shock, inhibitors of protein synthesis, and inflammatory cytokines (tumor necrosis factor (TNF)-α, interleukin (IL)-1), but also weakly by growth factors (epidermal growth factor, EGF) [28,29]. JNK activation has been implicated in the im- mune response, oncogenic transformation, apoptosis [28, 30] and activation of two major transcription factors: activator protein 1 (AP-1) and nuclear factor-kappa B (NF-kB) [31,32]. In turn, AP-1 and NF-kB induce the transcription of several genes involved in acute and chronic inflammation as well as diseases of connective tissue [32]. The gene for the inducible isoform of nitric oxide synthase (iNOS) is involved in inflammation [32]. MAPK regulation by G protein-coupled receptors (GPCRs) appears to be a widespread phenomenon. It is also likely to mediate the proliferative and hypertrophic responses of cells to various hormones, neurotransmitters and local mediators that act at this class of receptor [33]. JNK activation has also been demonstrated for several GPCRs, including M1 and M2 mAChR [34-36], angio- tensin [37], α1-adrenergic [38], thrombin [39] and endo- thelin-1 [40]. Some studies support the notion of mobile- zation of intracellular calcium and activation of protein kinase C (PKC) [41] in cholinergic receptor-mediated JNK activation [35,37]. 2. Aim The aim of the present work was to examine the effect of cholinergic autoantibodies present in the sera of pSS pa- tients and the authentic cholinergic agonist carbachol on JNK phosphorylation. We found that M1 and M3 mAChRs were coupled to JNK in the submandibular glands of rats. However, carbachol preferentially stimulated M1 mAChRs whereas pSS IgG stimulated M3 mAChRs. Both, the ac- tivation of M3 and M1 mAChRs by pSS IgG and car- bachol stimulated JNK phosphorylation. The pSS IgG stimulation effect appeared to be mediated by activation of PKC, iNOS and cyclo-oxygenase-2 (COX-2) whereas the carbachol stimulatory effect on JNK was mainly as- sociated with intracellular calcium-activated endothelial nitric oxide synthase (eNOS) and neuronal nitric oxide synthase (nNOS) with COX-1 stimulation. Also, the re- sults indicated that in the JNK activation phenomenon, by pSS IgG and carbachol on salivary gland participated cholinergic M3 and M1 receptor, respectively. Our results suggested that activation of JNK by pSS IgG indicate that the enzyme may be involved in the pathological process of chronic sialodenitis present in the course of pSS. 3. Methodology 3.1. Drugs Carbachol, pirenzepine, J104291, verapamil, calphostin C and thapsigargin were obtained from Sigma-Aldrich; FR-122047, DuP 697, methylisothiourea sulphate, L- NIO and NZ were from Tocris Cookson (Ellisville, MO, USA). Stock solutions were freshly prepared in the ap- propriate buffers. The drugs were diluted in a water bath to achieve the final concentrations stated in the text. 3.2. Animals Male Wistar rats weighing 250 - 300 g from the Pharma- cologic Bioterium (School of Dentistry, University of Buenos Aires) were used throughout. The animals housed in standard environmental conditions were fed with a commercial pellet diet and water ad libitum. For surgical removal of submandibular glands, the animals were sac- rificed using ether. The experimental protocol followed the Guide to The Care and Use of Experimental Animals (DHEW Publication, NIH 80-23). 3.3. Subjects and Serological Tests Females (age, 35 - 55 years) who had been diagnosed 7 - 15 y previously and who had been free of treatment for 8 Copyright © 2011 SciRes. PP  Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome 258 2 months were selected from the metropolitan area of Buenos Aires. The study population was 25 women with pSS who presented with dry mouth, and 18 healthy women (mean age, 45 ± 10 years) without systemic disease (control group). The diagnosis of SS was based on four or more of the criteria published elsewhere [42]. Biopsy results, degree of xerostomia and keratocon- junctivitis sicca, and the results of serological tests in the different groups were the same as previously reported [10]. 3.4. Peptides A 25-mer peptide (K-R-T-V-P-D-N-Q-C-F-I-Q-F-L-S- N-P-A-V-T-F-G-T-A-I) and a 24-mer peptide (E-R-T- M-L-A-G-Q-C-Y-I-Q-F-L-S-Q-P-I-T-F-G-T-A-M) corre- sponding to the amino-acid sequence of the second ex- tracellular loop of the human M3 mAChRs and M1 mAChRs, respectively, were synthesized from F-moc- amino acids activated using the 1-hydroxy benzo tria- zole/dicyclo hexyl carbodimide (HOBt/DCC) strategy and an automatic peptide synthesizer (Model 431A, Ap- plied Biosystems, Foster City, CA, USA). The peptides were desalted, purified by high-performance liquid chromatography (HPLC), and subjected to amino-ter- minal sequence analysis using automatic Edman degra- dation and an Applied Biosystems 470A Sequencer. An unrelated 25-mer peptide (S-G-S-G-S-G-S-G-S-G-S-G- S-G-S-G-S-G-S-G-S-G-S-G-S) was synthesized as a ne- gative control. 3.5. Purification of Human IgG The serum IgG fraction from patients with pSS and from normal individuals (control) was isolated using protein G affinity chromatography as described elsewhere [9]. Briefly, sera were loaded onto the protein G affinity column (Sigma-Aldrich, St Louis, MO, USA) equili- brated with 1 M Tris-HCl (pH 8.0) and the columns were washed with 10 volumes of the same buffer. The IgG fraction was eluted with 100 mM glycine-HCl, pH 3.0, and immediately neutralized. The concentration and pu- rification of IgG were determined using a radial immu- nodiffusion assay. 3.6. JNK Assay Slices of submandibular glands of rats (20 mg) were in- cubated for 30 min in 500 μL of Krebs-Ringer bicarbon- ate (KRB) buffer and gassed with 5% CO2 in O2 at 37˚C. pSS IgG or carbachol were added 15 min before the end of the incubation period, and blockers added 15 min be- fore the addition of different concentrations of pSS IgG or carbachol. The submandibular gland was then ho- mogenized in 1.0 mL of cell lysis buffer (product 9803). We then followed the manufacturer instructions for the PathScan Total and Phospho-SAPK/JNK kit (Sandwich ELISA Kit; Cell Signalling Technology Incorporated, Beverly, MA, USA). JNK results were expressed as the optical density at 450 nm (OD 450 nm). 3.7. PGE2 Assay Rat submandibular gland slices (20 mg) were incubated for 60 min in 500 μL of KRB and gassed with 5% CO2 in O2 at 37˚C. pSS IgG or carbachol were added 30 min before the end of the incubation period and blockers added 30 min before the addition of different concentra- tions of pSS IgG or carbachol. The submandibular gland was then homogenized in a 1.5-mL polypropylene mi- crocentrifuge tube. We then followed the manufacturer instructions for the PGE2 Biotrak Enzyme Immune Assay System (ELISA; Amersham Biosciences, Piscataway, NJ, USA). PGE2 results were expressed as picograms per milligram of tissue wet weight (pg/mg tissue wet wt). 3.8. Nitric Oxide Synthase (NOS) Assay NOS activity was measured in rat submandibular gland tissue by production of [U-14C]-citrulline from [U-14C]- arginine according to the procedure described for brain slices [43]. Briefly, after 20-min preincubation in KRB solution, tissues were transferred to 500 mL of pre- warmed KRB equilibrated with 5% CO2 in O2 in the presence of [U-14C]-arginine (0.5 mCi). Drugs were added and the mixture incubated for 20 min under 5% CO2 in O2 at 37˚C. Tissues were then homogenized with an Ultraturrax homogenizer in 1 mL of medium contain- ing 20 mM HEPES (pH 7.4), 0.5 mM ethyleneglycol tetra-acetic acid (EGTA), 0.5 mM ethylenediamine tetra- acetic acid (EDTA), 1 mM dithiothreitol, 1 mM leu- peptin and 0.2 mM phenylmethylsulphonyl fluoride (PMSF) at 4˚C. After centrifugation at 20,000 × g for 10 min at 4˚C, supernatants were applied to 2-mL columns of Dowex AG 50 WX-8 (sodium form). [14C]-citrulline was eluted with 3 mL of water and quantified by liquid scintillation counting. The results were expressed as pi- comol per gram tissue wet weight (pmol/g/tissue wet wt). 3.9. Statistical Analices The unpaired Student’s t-test was used to determine sta- tistical significance. Analysis of variance (ANOVA) and a post-hoc test (Dunnett’s method or the Student-New- man-Keuls test) were employed if a pairwise multiple comparison procedure was necessary. p < 0.05 was con- sidered significant. 3.10. Ethical Approval of the Study Protocol The study protocol was approved by the Ethics Commit- tee of the School of Dentistry at Buenos Aires University (Buenos Aires, Argentina). The studies were conducted Copyright © 2011 SciRes. PP 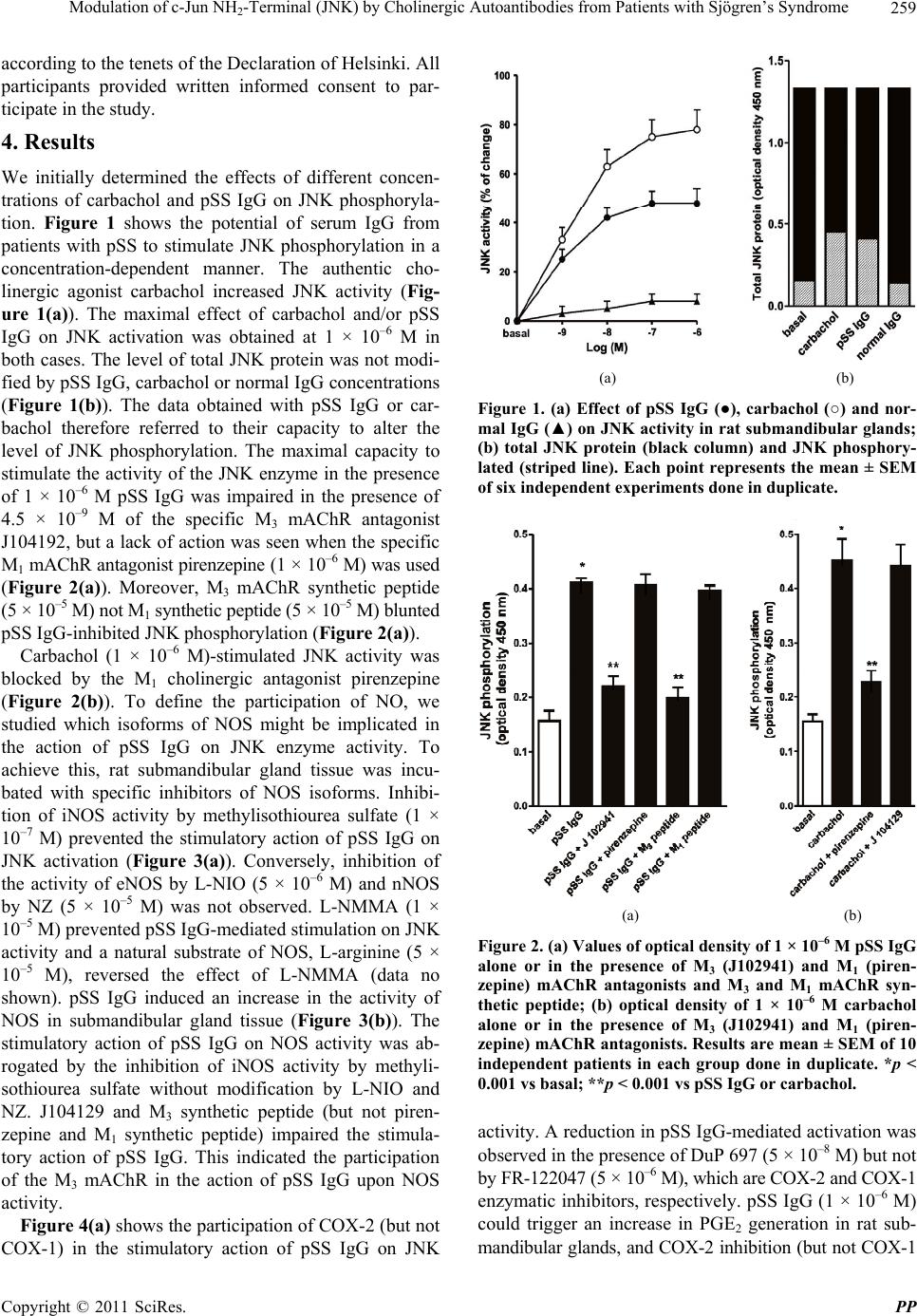 Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome259 2 according to the tenets of the Declaration of Helsinki. All participants provided written informed consent to par- ticipate in the study. 4. Results We initially determined the effects of different concen- trations of carbachol and pSS IgG on JNK phosphoryla- tion. Figure 1 shows the potential of serum IgG from patients with pSS to stimulate JNK phosphorylation in a concentration-dependent manner. The authentic cho- linergic agonist carbachol increased JNK activity (Fig- ure 1(a)). The maximal effect of carbachol and/or pSS IgG on JNK activation was obtained at 1 × 10–6 M in both cases. The level of total JNK protein was not modi- fied by pSS IgG, carbachol or normal IgG concentrations (Figure 1(b)). The data obtained with pSS IgG or car- bachol therefore referred to their capacity to alter the level of JNK phosphorylation. The maximal capacity to stimulate the activity of the JNK enzyme in the presence of 1 × 10–6 M pSS IgG was impaired in the presence of 4.5 × 10–9 M of the specific M3 mAChR antagonist J104192, but a lack of action was seen when the specific M1 mAChR antagonist pirenzepine (1 × 10–6 M) was used (Figure 2(a)). Moreover, M3 mAChR synthetic peptide (5 × 10–5 M) not M1 synthetic peptide (5 × 10–5 M) blunted pSS IgG-inhibited JNK phosphorylation (Figure 2(a)). Carbachol (1 × 10–6 M)-stimulated JNK activity was blocked by the M1 cholinergic antagonist pirenzepine (Figure 2(b)). To define the participation of NO, we studied which isoforms of NOS might be implicated in the action of pSS IgG on JNK enzyme activity. To achieve this, rat submandibular gland tissue was incu- bated with specific inhibitors of NOS isoforms. Inhibi- tion of iNOS activity by methylisothiourea sulfate (1 × 10–7 M) prevented the stimulatory action of pSS IgG on JNK activation (Figure 3(a)). Conversely, inhibition of the activity of eNOS by L-NIO (5 × 10–6 M) and nNOS by NZ (5 × 10–5 M) was not observed. L-NMMA (1 × 10–5 M) prevented pSS IgG-mediated stimulation on JNK activity and a natural substrate of NOS, L-arginine (5 × 10–5 M), reversed the effect of L-NMMA (data no shown). pSS IgG induced an increase in the activity of NOS in submandibular gland tissue (Figure 3(b)). The stimulatory action of pSS IgG on NOS activity was ab- rogated by the inhibition of iNOS activity by methyli- sothiourea sulfate without modification by L-NIO and NZ. J104129 and M3 synthetic peptide (but not piren- zepine and M1 synthetic peptide) impaired the stimula- tory action of pSS IgG. This indicated the participation of the M3 mAChR in the action of pSS IgG upon NOS activity. Figure 4(a) shows the participation of COX-2 (but not COX-1) in the stimulatory action of pSS IgG on JNK (a) (b) Figure 1. (a) Effect of pSS IgG (●), carbachol (○) and nor- mal IgG (▲) on JNK activity in rat submandibular glands; (b) total JNK protein (black column) and JNK phosphory- lated (striped line). Each point represents the mean ± SEM of six independent experiments done in duplicate. (a) (b) Figure 2. (a) Values of optical density of 1 × 10–6 M pSS IgG alone or in the presence of M3 (J102941) and M1 (piren- zepine) mAChR antagonists and M3 and M1 mAChR syn- thetic peptide; (b) optical density of 1 × 10–6 M carbachol alone or in the presence of M3 (J102941) and M1 (piren- zepine) mAChR antagonists. Results are mean ± SEM of 10 independent patients in each group done in duplicate. *p < 0.001 vs basal; **p < 0.001 vs pSS IgG or carbachol. activity. A reduction in pSS IgG-mediated activation was observed in the presence of DuP 697 (5 × 10–8 M) but not by FR-122047 (5 × 10–6 M), which are COX-2 and COX-1 enzymatic inhibitors, respectively. pSS IgG (1 × 10–6 M) could trigger an increase in PGE2 generation in rat sub- mandibular glands, and COX 2 inhibition (but not COX-1 - Copyright © 2011 SciRes. PP 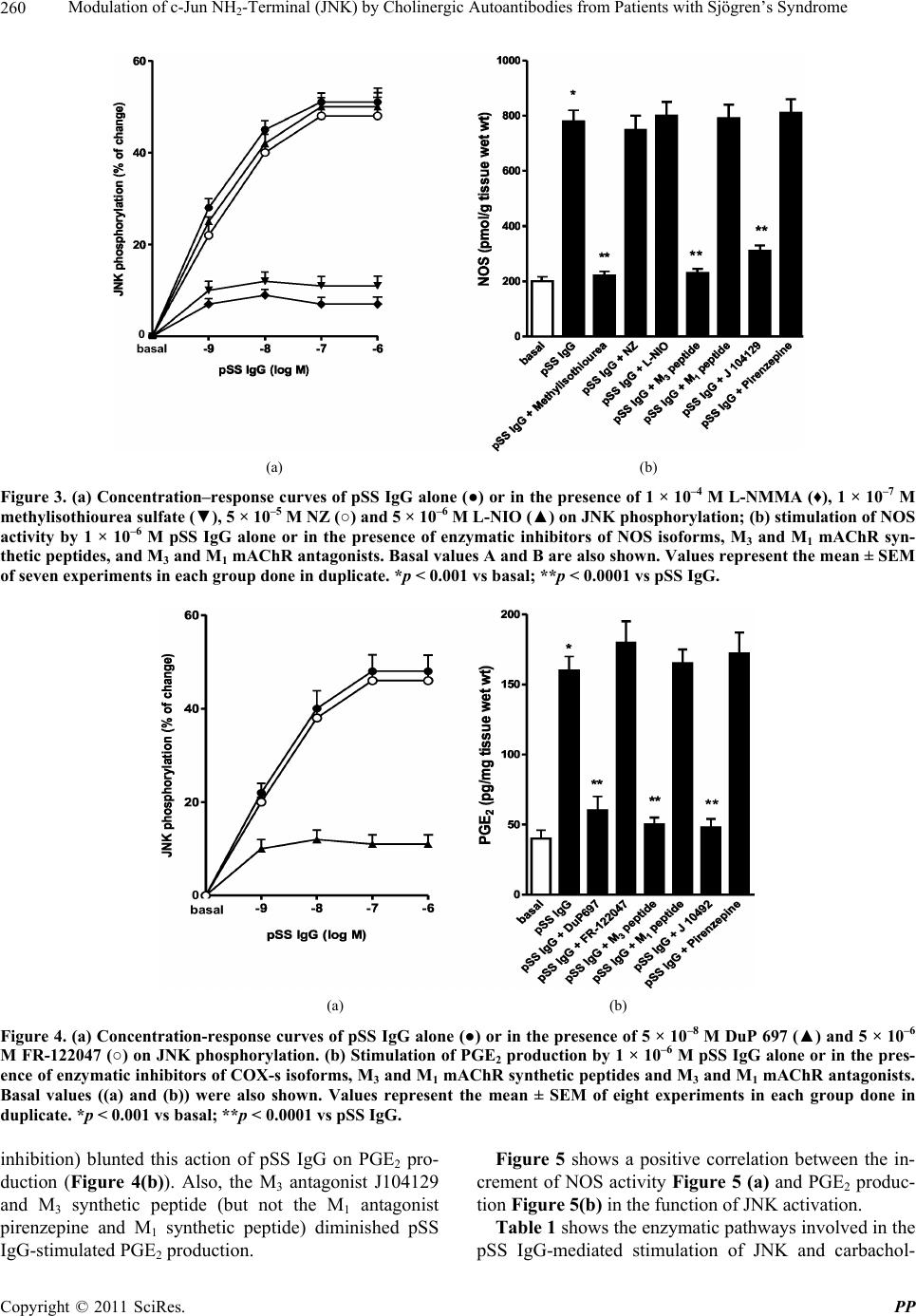 Modulation of c-Jun NH2-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome Copyright © 2011 SciRes. PP 260 0 (a) (b) Figure 3. (a) Concentration–response curves of pSS IgG alone (●) or in the presence of 1 × 10–4 M L-NMMA (♦), 1 × 10–7 M methylisothiourea sulfate (▼), 5 × 10–5 M NZ (○) and 5 × 10–6 M L-NIO (▲) on JNK phosphorylation; (b) stimulation of NOS activity by 1 × 10–6 M pSS IgG alone or in the presence of enzymatic inhibitors of NOS isoforms, M3 and M1 mAChR syn- thetic peptides, and M3 and M1 mAChR antagonists. Basal values A and B are also shown. Values represent the mean ± SEM of seven experiments in each group done in duplicate. *p < 0.001 vs basal; **p < 0.0001 vs pSS IgG. 0 (a) (b) Figure 4. (a) Concentration-response curves of pSS IgG alone (●) or in the presence of 5 × 10–8 M DuP 697 (▲) and 5 × 10–6 M FR-122047 (○) on JNK phosphorylation. (b) Stimulation of PGE2 production by 1 × 10–6 M pSS IgG alone or in the pres- ence of enzymatic inhibitors of COX-s isoforms, M3 and M1 mAChR synthetic peptides and M3 and M1 mAChR antagonists. Basal values ((a) and (b)) were also shown. Values represent the mean ± SEM of eight experiments in each group done in duplicate. *p < 0.001 vs basal; **p < 0.0001 vs pSS IgG. Figure 5 shows a positive correlation between the in- crement of NOS activity Figure 5 (a) and PGE2 produc- tion Figure 5(b) in the function of JNK activation. inhibition) blunted this action of pSS IgG on PGE2 pro- duction (Figure 4(b)). Also, the M3 antagonist J104129 and M3 synthetic peptide (but not the M1 antagonist pirenzepine and M1 synthetic peptide) diminished pSS IgG-stimulated PGE2 production. Table 1 shows the enzymatic pathways involved in the pSS IgG-mediated stimulation of JNK and carbachol- 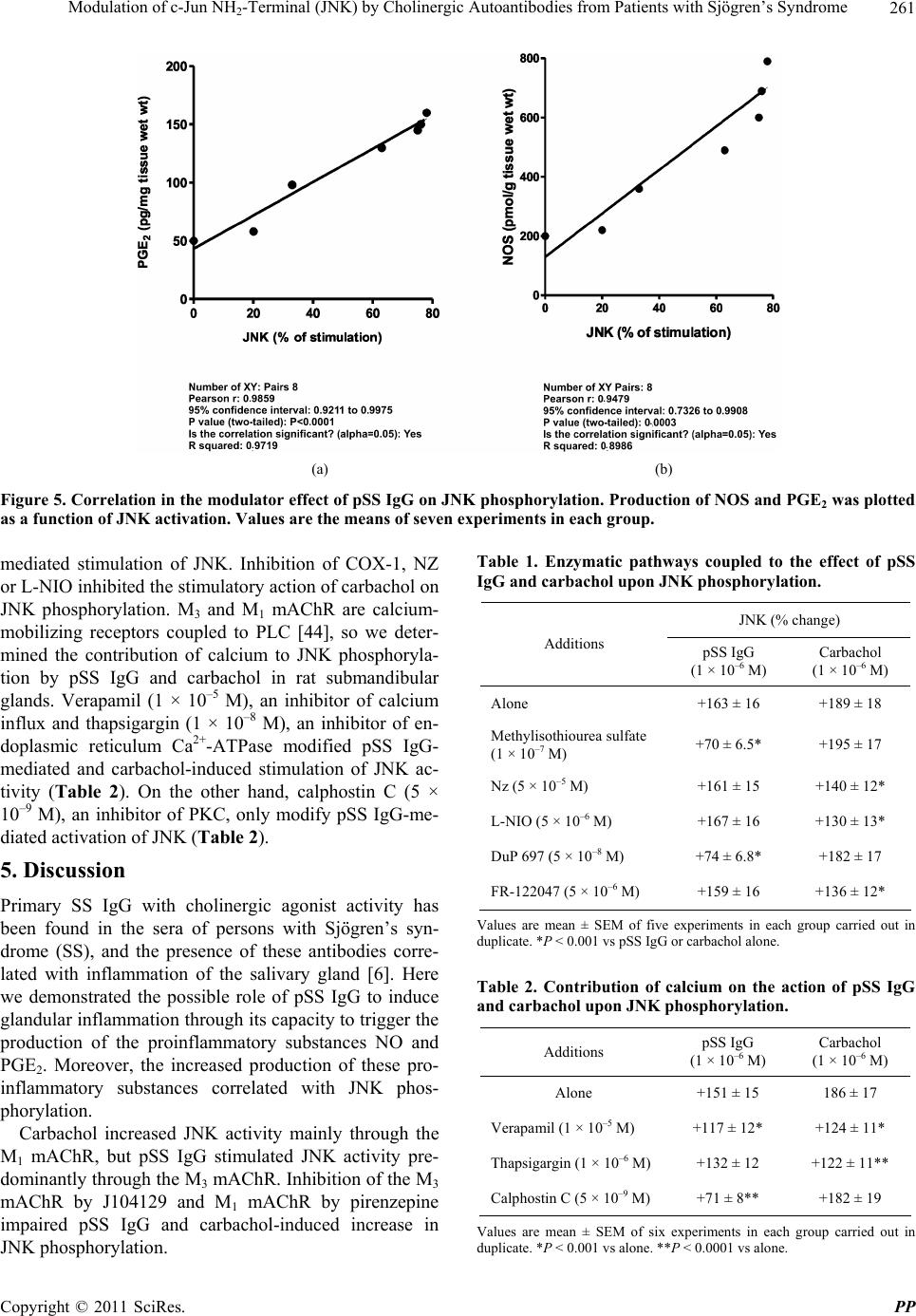 Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome261 2 . . . . . (a) (b) Figure 5. Correlation in the modulator effect of pSS IgG on JNK phosphorylation. Production of NOS and PGE2 was plotted as a function of JNK activation. Values are the means of seven experiments in each group. mediated stimulation of JNK. Inhibition of COX-1, NZ or L-NIO inhibited the stimulatory action of carbachol on JNK phosphorylation. M3 and M1 mAChR are calcium- mobilizing receptors coupled to PLC [44], so we deter- mined the contribution of calcium to JNK phosphoryla- tion by pSS IgG and carbachol in rat submandibular glands. Verapamil (1 × 10–5 M), an inhibitor of calcium influx and thapsigargin (1 × 10–8 M), an inhibitor of en- doplasmic reticulum Ca2+-ATPase modified pSS IgG- mediated and carbachol-induced stimulation of JNK ac- tivity (Table 2). On the other hand, calphostin C (5 × 10–9 M), an inhibitor of PKC, only modify pSS IgG-me- diated activation of JNK (Table 2). 5. Discussion Primary SS IgG with cholinergic agonist activity has been found in the sera of persons with Sjögren’s syn- drome (SS), and the presence of these antibodies corre- lated with inflammation of the salivary gland [6]. Here we demonstrated the possible role of pSS IgG to induce glandular inflammation through its capacity to trigger the production of the proinflammatory substances NO and PGE2. Moreover, the increased production of these pro- inflammatory substances correlated with JNK phos- phorylation. Carbachol increased JNK activity mainly through the M1 mAChR, but pSS IgG stimulated JNK activity pre- dominantly through the M3 mAChR. Inhibition of the M3 mAChR by J104129 and M1 mAChR by pirenzepine impaired pSS IgG and carbachol-induced increase in JNK phosphorylation. Table 1. Enzymatic pathways coupled to the effect of pSS IgG and carbachol upon JNK phosphorylation. JNK (% change) Additions pSS IgG (1 × 10–6 M) Carbachol (1 × 10–6 M) Alone +163 ± 16 +189 ± 18 Methylisothiourea sulfate (1 × 10–7 M) +70 ± 6.5* +195 ± 17 Nz (5 × 10–5 M) +161 ± 15 +140 ± 12* L-NIO (5 × 10–6 M) +167 ± 16 +130 ± 13* DuP 697 (5 × 10–8 M) +74 ± 6.8* +182 ± 17 FR-122047 (5 × 10–6 M) +159 ± 16 +136 ± 12* Values are mean ± SEM of five experiments in each group carried out in duplicate. *P < 0.001 vs pSS IgG or carbachol alone. Table 2. Contribution of calcium on the action of pSS IgG and carbachol upon JNK phosphorylation. Additions pSS IgG (1 × 10–6 M) Carbachol (1 × 10–6 M) Alone +151 ± 15 186 ± 17 Verapamil (1 × 10–5 M) +117 ± 12* +124 ± 11* Thapsigargin (1 × 10–6 M) +132 ± 12 +122 ± 11** Calphostin C (5 × 10–9 M) +71 ± 8** +182 ± 19 Values are mean ± SEM of six experiments in each group carried out in duplicate. *P < 0.001 vs alone. **P < 0.0001 vs alone. Copyright © 2011 SciRes. PP 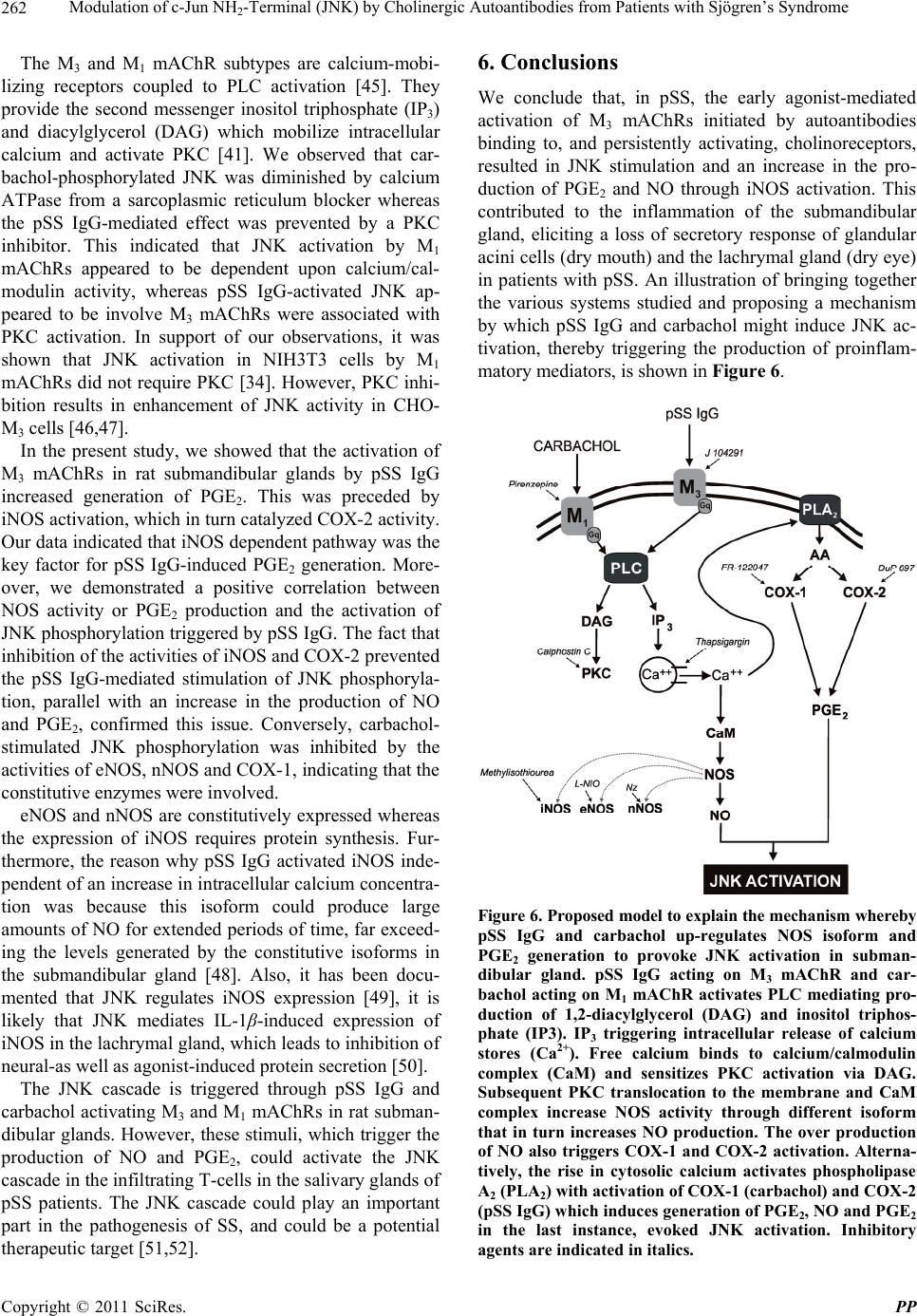 Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome 262 2 The M3 and M1 mAChR subtypes are calcium-mobi- lizing receptors coupled to PLC activation [45]. They provide the second messenger inositol triphosphate (IP3) and diacylglycerol (DAG) which mobilize intracellular calcium and activate PKC [41]. We observed that car- bachol-phosphorylated JNK was diminished by calcium ATPase from a sarcoplasmic reticulum blocker whereas the pSS IgG-mediated effect was prevented by a PKC inhibitor. This indicated that JNK activation by M1 mAChRs appeared to be dependent upon calcium/cal- modulin activity, whereas pSS IgG-activated JNK ap- peared to be involve M3 mAChRs were associated with PKC activation. In support of our observations, it was shown that JNK activation in NIH3T3 cells by M1 mAChRs did not require PKC [34]. However, PKC inhi- bition results in enhancement of JNK activity in CHO- M3 cells [46,47]. In the present study, we showed that the activation of M3 mAChRs in rat submandibular glands by pSS IgG increased generation of PGE2. This was preceded by iNOS activation, which in turn catalyzed COX-2 activity. Our data indicated that iNOS dependent pathway was the key factor for pSS IgG-induced PGE2 generation. More- over, we demonstrated a positive correlation between NOS activity or PGE2 production and the activation of JNK phosphorylation triggered by pSS IgG. The fact that inhibition of the activities of iNOS and COX-2 prevented the pSS IgG-mediated stimulation of JNK phosphoryla- tion, parallel with an increase in the production of NO and PGE2, confirmed this issue. Conversely, carbachol- stimulated JNK phosphorylation was inhibited by the activities of eNOS, nNOS and COX-1, indicating that the constitutive enzymes were involved. eNOS and nNOS are constitutively expressed whereas the expression of iNOS requires protein synthesis. Fur- thermore, the reason why pSS IgG activated iNOS inde- pendent of an increase in intracellular calcium concentra- tion was because this isoform could produce large amounts of NO for extended periods of time, far exceed- ing the levels generated by the constitutive isoforms in the submandibular gland [48]. Also, it has been docu- mented that JNK regulates iNOS expression [49], it is likely that JNK mediates IL-1β-induced expression of iNOS in the lachrymal gland, which leads to inhibition of neural-as well as agonist-induced protein secretion [50]. The JNK cascade is triggered through pSS IgG and carbachol activating M3 and M1 mAChRs in rat subman- dibular glands. However, these stimuli, which trigger the production of NO and PGE2, could activate the JNK cascade in the infiltrating T-cells in the salivary glands of pSS patients. The JNK cascade could play an important part in the pathogenesis of SS, and could be a potential therapeutic target [51,52]. 6. Conclusions We conclude that, in pSS, the early agonist-mediated activation of M3 mAChRs initiated by autoantibodies binding to, and persistently activating, cholinoreceptors, resulted in JNK stimulation and an increase in the pro- duction of PGE2 and NO through iNOS activation. This contributed to the inflammation of the submandibular gland, eliciting a loss of secretory response of glandular acini cells (dry mouth) and the lachrymal gland (dry eye) in patients with pSS. An illustration of bringing together the various systems studied and proposing a mechanism by which pSS IgG and carbachol might induce JNK ac- tivation, thereby triggering the production of proinflam- matory mediators, is shown in Figure 6. Figure 6. Proposed model to explain the mechanism whereby pSS IgG and carbachol up-regulates NOS isoform and PGE2 generation to provoke JNK activation in subman- dibular gland. pSS IgG acting on M3 mAChR and car- bachol acting on M1 mAChR activates PLC mediating pro- duction of 1,2-diacylglycerol (DAG) and inositol triphos- phate (IP3). IP3 triggering intracellular release of calcium stores (Ca2+). Free calcium binds to calcium/calmodulin complex (CaM) and sensitizes PKC activation via DAG. Subsequent PKC translocation to the membrane and CaM complex increase NOS activity through different isoform that in turn increases NO production. The over production of NO also triggers COX-1 and COX-2 activation. Alterna- tively, the rise in cytosolic calcium activates phospholipase A2 (PLA2) with activation of COX-1 (carbachol) and COX-2 (pSS IgG) which induces generation of PGE2, NO and PGE2 in the last instance, evoked JNK activation. Inhibitory agents are indicated in italics. Copyright © 2011 SciRes. PP  Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome263 2 7. Acknowledgements This work was supported by grants from Buenos Aires University (grant number UBACyT O 003) and the Na- tional Research & Technology Agency (PICT’s 02120 and 01647). The authors thank Mrs. Elvita Vannucchi and Mr. Alejandro Thorton for their expert technical as- sistance. There is no conflict of interest. REFERENCES [1] I. Marczinovits, L. Kovacs, A. Gyorgy, G. K. Toth, L. Dorgai and J. A. Molnar, “A Peptide of Human Mus- carinic Acetylcholine Receptor 3 Is Antigenic in Primary Sjögren’s Syndrome,” Journal of Autoimmunity, Vol. 24, No. 1, 2005, pp. 47-54. doi:10.1016/j.jaut.2004.11.005 [2] H. M. Moutsopoulos and N. Talal, “Immunologic Abnor- malities in Sjögren Syndrome,” In: N. Talal and H. M. Moutsopoulos, Eds., Sjögren’s Syndrome: Clinical and im- munopathological Aspects, Springer-Verlag, Berlin, 1987, pp. 258-265. [3] C. P. Leiros, L. Sterin-Borda, E. S. Borda, J. C. Goin and M. M. Hosey, “Desensitization and Sequestration of Hu- man M2 Muscarinic Acetylcholine Receptors by Autoan- tibodies from Patients with Chagas’ Disease,” Journal of Biology and Chemistry, Vol. 272, No. 20, 1999, pp. 12989-12993. [4] S. Bacman, L. Sterin-Borda, J. J. Camusso, R. Arana, O. Hubscher and E. Borda, “Circulating Antibodies against Rat Parotid Gland M3 Muscarinic Receptors in Sjögren Syndrome,” Clinical & Experimental Immunology, Vol. 104, No. 3, 1996, pp. 454-459. doi:10.1046/j.1365-2249.1996.42748.x [5] S. Bacman, A. Berra, L. Sterin-Borda and E. Borda, “Mu- scarinic Acetylcholine Receptor Antibodies as a New Marker of Dry Eye Sjögren Syndrome,” Investigative Ophthalmology & Vision Science, Vol. 42, No. 2, 2001, pp. 321-327. [6] S. Reina, L. Sterin-Borda, B. Orman and E. Borda, “Auto- antibodies against Submandibular Gland Musdcarinic Cho- linoceptor Subtypes in Primary Sjögren,” European Jour- nal of Inflammation, Vol. 3, No. 3, 2005, pp. 135-141. [7] L. Kovacs, I. Marczinovits, A. Gryorgy, G. Toth, L. Dor- gai and J. Pal, “Clinical Associations of Autoantibodies to Human Muscarinic Acetylcholine Receptor M3 in Pri- mary Sjögren’s Syndrome,” Rheumatology, Vol. 44, No. 8, 2005, pp. 1021-1025. doi:10.1093/rheumatology/keh672 [8] S. Reina, L. Sterin-Borda, B. Orman and E. Borda, “Hu- man mAChR Antibodfies from Sjögren Syndrome Sera Increase Cerebral Nitric Oxide Synthase Activity and Ni- tric Oxide Synthase mRNA Level,” Clinical Immunology, Vol. 113, No. 2, 2004, pp. 193-202. doi:10.1016/j.clim.2004.08.005 [9] S. Reina, B. Orman, L. Anaya, L. Sterin-Borda and E. Borda, “Cholinergic Autoantibodies in Sjögren Syndro- me,” Journal of Dental Research, Vol. 86, No. 9, 2007, pp. 832-836. doi:10.1177/154405910708600905 [10] A. Berra, L. Sterin-Borda, S. Bacman and E. Borda, “Role of Salivary IgA in the Pathogenesis of Sjögren Syndrome,” Clinical Immunology, Vol. 104, No. 1, 2002, pp. 49-57. doi:10.1006/clim.2002.5228 [11] L. J. Dawson, J. Stanbury, N. Venn, B. Hasdimir, S. N. Rogers and P. M. Smith, “Antimuscarinic Antibodies in Primary Sjögren’s Syndrome Reversibly Inhibit the Me- chanism of Fluid Secretion by Human Submandibular Salivary Acinar Cells,” Arthritis & Rheumatology, Vol. 54, No. 4, 2006, pp. 1165-1173. doi:10.1002/art.21764 [12] J. Li, Y. M. Ha, N. Y. Ku, S. Y. Choi, S. J. Lee, S. B. Oh, et al. “Inhibitory Effects of Autoantibodies on the Mus- carinic Receptors in Sjögren’s Syndrome,” Laboratory Investigation, Vol. 84, No. 11, 2004, pp. 1430-1438. doi:10.1038/labinvest.3700173 [13] S. A. Waterman, T. P. Gordon and M. Rischmueller, “In- hibitory Effects of Muscarinic Receptor Autoantibodies on Parasympathetic Neurotransmission in Sjögren’s Syn- drome,” Arthritis & Rheumatology, Vol. 43, No. 7, 2000, pp. 1647-1654. doi:10.1002/1529-0131(200007)43:7<1647::AID-ANR31 >3.0.CO;2-P [14] S. Cha, E. Singson, J. Cornelius, J. P. Yagna, H. J. Knot and A. B. Peck, “Muscarinic Acetylcholine Type-3 Recep- tor Desensitization Due to Chronic Exposure to Sjögren Syndrome-Associated Autoantibodies,” The Journal of Rheumatology, Vol. 33, No. 2, 2006, pp. 296-306. [15] D. B. Ferguson, “The Flow Rate and Composition of Hu- man Labial Gland Saliva,” Archives of Oral Biology, Vol. 44, Supplement 1, 1999, pp. S11-S14. doi:10.1016/S0003-9969(99)90004-3 [16] K. Tsubota, M. Kaido, Y. Yagi and T. Fujihara, “Diseases Associated with Ocular Surface Abnormalities: The Im- portance of Reflex Tearing,” British Journal of Ophthal- mology, Vol. 83, No. 1, 1999, pp. 89-91. doi:10.1136/bjo.83.1.89 [17] R. I. Fox, “Sjögren’s Syndrome,” Lancet, Vol. 366, No. 9482, 2005, pp. 321-331. doi:10.1016/S0140-6736(05)66990-5 [18] N. Ravald and T. List, “Caries and Periodontal Condi- tions in Patients with Primary Sjögren’s Syndrome,” Sweden Dental Journal, Vol. 22, No. 3, 1998, pp. 97-103. [19] J. Yuan, A. S. Bowman, M. Aljamali, M. R. Payne, J. S. Tucker, J. W. Dillwith, et al. “Prostaglandin E(2)-Stimu- lated Secretion of Protein in Salivary Glands of the Lone Star Tick via a Phosphoinositide Signaling Pathway,” In- sect Biochemical and Molecular Biology, Vol. 30, No. 11, 2000, pp. 1099-1106. [20] G. M. Pasinetti, “Cyclooxygenase and Alzheimer’s Dis- ease: Implications for Preventive Initiatives to Slow the Progression of Clinical Dementia,” Archives of Geron- tology and Geriatric, Vol. 33, No. 1, 2001, pp. 13-28. doi:10.1016/S0167-4943(01)00091-7 [21] V. Mollage, C. E. Muscoli, E. Masini, S. Cuzzocrea and D. Salvemini, “Modulation of Prostaglandin Biosynthesis by Nitric Oxide and Nitric Oxide Donors,” Pharmacol- ogical Review, Vol. 57, No. 2, 2005, pp. 217-252. Copyright © 2011 SciRes. PP  Modulation of c-Jun NH-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome 264 2 doi:10.1124/pr.57.2.1 [22] S. Ganzinelli, E. Borda and L. Sterin-Borda, “Autoanti- bodies from Schizophrenia Patients Induce Cerebral Cox-1/iNOS mRNA Expression with NO/PGE2/MMP-3 Production,” The International Journal of Neuropsycho- pharmacology, Vol. 13, No. 3, 2010, pp. 293-303. doi:10.1017/S1461145709990770 [23] E. C. Hulme, N. J. Birdsall and N. J. Buckley, “Mus- carinic Receptor Subtypes,” Annual Review of Pharma- cology and Toxicology, Vol. 30, No. 1, 1990, pp. 633-673. doi:10.1146/annurev.pa.30.040190.003221 [24] B. Nicke, K. Detjen and C. D. Logsdon, “Muscarinic Cho- linergic Receptors Activate Both Inhibitory and Stimula- tory Growth Mechanisms in NIH3T3 Cells,” The Journal of Biology and Chemistry, Vol. 274, No. 31, 1999, pp. 21701- 21706. doi:10.1074/jbc.274.31.21701 [25] J. S. Gutkind and K. C. Robbins, “Activation of Trans- forming G Protein-Coupled Receptors Induces Rapid Tyrosine Phosphorylation of Cellular Proteins, Including p125FAK and the p130 v-src Substrate,” Biochemistry and Biophysical Research Communication, Vol. 188, No. 1, 1992, pp.155-161. doi:10.1016/0006-291X(92)92363-3 [26] R. J. Davis, “The Mitogen-Activated Protein Kinase Sig- nal Transduction Pathway,” The Journal of Biology and Chemistry, Vol. 268, No. 20, 1993, pp. 14553-14556. [27] A. Minden and M. Karin, “Regulation and Function of the JNK Subgroup of MAP Kinases,” Biochemica et Bio- physica Acta, Vol. 1333, No. 2, 1993, pp. F85-F104. [28] S. Gupta, T. Barrett, A. J. Whitmarsh, J. Cavanagh, H. K. Sluss, B. Dérijard and R. J. Davis, “Selective Interaction of JNK Protein Kinase Isoforms with Transcription Fac- tors,” The EMBO Journal, Vol. 15, No. 11, 1996, pp. 2760-2770. [29] J. M. Kyriakis, P. Banerjee, E. Nikolakaki, T. Dai, E. A. Rubie, M. F. Ahmad, J. Avruch and J. R. Woodgett. “The Stress-Activated Protein Kinase Subfamily of c-Jun Kina- ses,” Nature, Vol. 369, No. 6476, 1994, pp. 156-160. doi:10.1038/369156a0 [30] J. M: Kyriakis and J. Avruch, “Sounding the Alarm: Pro- tein Kinase Cascades Activated by Stress and Inflamma- tion,” Journal of Biology and Chemistry, Vol. 271, No. 40, 1996, pp. 24313-24316. doi:10.1074/jbc.271.40.24313 [31] A. Paul, S. Wilson, C. M. Belham, C. J. Robinson, P. H. Scott, G. W. Gould and R. Plevin, “Stress-Activated Pro- tein Kinases: Activation, Regulation and Function,” Cel- lular Signalling, Vol. 9, No. 6, 1997, pp. 403-410. doi:10.1016/S0898-6568(97)00042-9 [32] C. A. Dinarello, “Proinflammatory Cytokines,” Chest, Vol. 118, No. 2, 2000, pp. 503-508. doi:10.1378/chest.118.2.503 [33] J. S. Gutkind, “The Pathways Connecting G Protein-Cou- pled Receptors to the Nucleus through Divergent Mito- gen-Activated Protein Kinase Cascades,” The Journal of Biology and Chemistry, Vol. 273, No. 4, 1998, pp. 1839- 1842. doi:10.1074/jbc.273.4.1839 [34] O. A. Coso, M. Chiariello, G. Kalinec, J. M. Kyriakis, J. Woodgett and J. S. Gutkind. “Transforming G Protein- Coupled Receptors Potently Activate JNK (SAPK). Evi- dence for a Divergence from the Tyrosine Kinase Signal- ing Pathway,” The Journal of Biology and Chemistry, Vol. 270, No. 10, 1995, pp. 5620-5624. [35] F. M. Mitchell, M. Russell and G. L. Johnson, “Differen- tial Calcium Dependence in the Activation of c-Jun Kinase and Mitogen-Activated Protein Kinase by Mus- carinic Acetylcholine Receptors in Rat 1A Cells,” Bio- chemistry Journal, Vol. 309, Part 2, 1995, pp. 381-384. [36] O. A. Coso, H. Teramoto, W. F. Simonds and J. S. Gut- kind, “Signaling from G Protein-Coupled Receptors to c-Jun Kinase Involves Beta Gamma Subunits of Hetero- trimeric G Proteins Acting on a Ras and Rac1-Dependent Pathway,” The Journal of Biology and Chemistry, Vol. 271, No. 8, 1996, pp. 3963-3696. doi:10.1074/jbc.271.8.3963 [37] I. E. Zohn, H. Yu, X. Li, A. D. Cox and H. S. Earp, “An- giotensin II Stimulates Calcium-Dependent Activation of c-Jun N-Terminal Kinase,” Molecular and Cellular Biol- ogy, Vol. 15, No. 11, 1995, pp. 6160-6168. [38] M. T. Ramirez, V. P. Sah, X. L. Zhao, J. J. Hunter, K. R. Chien and J. H. Brown, “The MEKK-JNK Pathway Is Stimulated by Alpha1-Adrenergic Receptor and Ras Ac- tivation and Is Associated with in Vitro and in Vivo Car- diac Hypertrophy,” The Journal of Biology and Chemis- try, Vol. 272, No. 22, 1997, pp. 14057-14061. doi:10.1074/jbc.272.22.14057 [39] P. S. Shapiro, J. N. Evans, R. J. Davis and J. A. Posada, “The Seven-Transmembrane-Spanning Receptors for En- dothelin and Thrombin Cause Proliferation of Airway Smooth Muscle Cells and Activation of the Extracellular Regulated Kinase and c-Jun NH2-Terminal Kinase Groups of Mitogen-Activated Protein Kinases,” The Journal of Bi- ology and Chemistry, Vol. 271, No. 10, 1996, pp. 5750- 5754. doi:10.1074/jbc.271.10.5750 [40] M. A. Bogoyevitch, A. J. Ketterman and P. H. Sugden, “Cellular Stresses Differentially Activate c-Jun N-Terminal Protein Kinases and Extracellular Signal-Regulated Protein Kinases in Cultured Ventricular Myocytes,” The Journal of Biology and Chemistry, Vol. 270, No. 50, 1995, pp. 29710- 29717. doi:10.1074/jbc.270.50.29710 [41] A. C. Newton, “Protein Kinase C: Structure, Function, and Regulation,” The Journal of Biology and Chemistry, Vol. 270, No. 48, 1995, pp. 28495-28498. [42] C. Vitali, S. Bombardieri, H. M. Moutsopoulos, G. Balestrieri, W. Bencivelli, R. M. Bernstein, et al. “Pre- liminary Criteria for the Classification of Sjögren’s Syn- drome. Results from a Prospective Concerted Action Sup- ported by the European Community,” Arthritis and Rheu- matology, Vol. 36, No. 3, 1993, pp. 340-347. doi:10.1002/art.1780360309 [43] T. Borda, A. Genaro, L. Sterin-Borda and G. Cremaschi, “Involvement of Endogenous Nitric Oxide Signalling System in Brain Muscarinic Acetylcholine Receptor Ac- tivation,” Journal of Neural Transmission, Vol. 105, No. 2-3, 1995, pp. 193-204. doi:10.1007/s007020050048 [44] M. P. Caulfield, “Muscarinic Receptor Characterization, Copyright © 2011 SciRes. PP  Modulation of c-Jun NH2-Terminal (JNK) by Cholinergic Autoantibodies from Patients with Sjögren’s Syndrome Copyright © 2011 SciRes. PP 265 Coupling and Function,” Pharmacology & Therapeutic, Vol. 58, No. 3, 1993, pp. 319-379. doi:10.1016/0163-7258(93)90027-B [45] M. J. Berridge, “Inositol Trisphosphate and Calcium Sig- nalling,” Nature, Vol. 361, No. 6410, 1993, pp. 315-325. doi:10.1038/361315a0 [46] P. G. Wylie, R. A. Challiss and J. L. Blank, “Regulation of Extracellular-Signal Regulated Kinase and c-Jun N-Termi- nal Kinase by G-Protein-Linked Muscarinic Acetylcholine Receptors,” Biochemistry Journal, Vol. 338, Part 3, 1999, pp. 619-628. doi:10.1042/0264-6021:3380619 [47] D. C. Hornigold, R. Mistry, P. D. Raymond, J. L. Blank and R. A. Challiss, “Evidence for Cross-Talk between M2 and M3 Muscarinic Acetylcholine Receptors in the Regu- lation of Second Messenger and Extracellular Signal- Regulated Kinase Signalling Pathways in Chinese Ham- ster Ovary Cells,” British Journal of Pharmacology, Vol. 138, No. 7, 2003, pp. 1340-1350. doi:10.1038/sj.bjp.0705178 [48] P. Kubes and D. M. McCafferty, “Nitric Oxide and Intes- tinal Inflammation,” The American Journal of Medicine, Vol. 109, No. 2, 2000, pp. 150-118. doi:10.1016/S0002-9343(00)00480-0 [49] M. T. Heneka and D. L. Feinstein, “Expression and Func- tion of Inducible Nitric Oxide Synthase in Neurons,” Journal of Neuroimmunology, Vol. 114, No. 1, 2001, pp. 8-18. doi:10.1016/S0165-5728(01)00246-6 [50] D. Zoukhri, E. Macari, S. H. Choi and C. L. Kublin, “c-Jun NH2-Terminal Kinase Mediates Interleukin-1Beta-Induced Inhibition of Lacrimal Gland Secretion,” Journal of Neu- rochemistry, Vol. 96, No. 1, 2006, pp. 126-135. doi:10.1111/j.1471-4159.2005.03529.x [51] K. Soejima, H. Nakamura, M. Tamai, A. Kawakami and K. Eguchi, “Activation of MKK4 (SEK1), JNK, and c- Jun in Labial Salivary Infiltrating T Cells in Patients with Sjögren’s Syndrome,” Rheumatology International, Vol. 27, No. 4, 2006, pp. 329-333. [52] S. Gonzalo, L. Grasa, M. P. Arrueba, M. A. Plaza and M. D. Murillo, “Lipopolysaccharide-Induced Digestive and Liver Disease,” Digestive and Liver Disease, Vol. 43, No. 4, 2011, pp. 277-285. doi:10.1016/j.dld.2010.10.009
|