 American Journal of Anal yt ical Chemistry, 2011, 2, 675-682 doi:10.4236/ajac.2011.26077 Published Online October 2011 (http://www.SciRP.org/journal/ajac) Copyright © 2011 SciRes. AJAC Simultaneous Separation and Quantification of Iron and Transition Species Using LC-ICP-MS Qinhong Hu Department of Earth and Environmental Sciences, The University of Texast, Arlington, USA E-mail: maxhu@uta.edu Received June 27, 2011; revised July 28, 2011; accepted August 5, 2011 Abstract Using liquid chromatography-inductively coupled plasma-mass spectrometry (LC-ICP-MS), this work inves- tigates the simultaneous separation and quantification of seven transition metal species (Fe, Mn, Co, Ni, Cu, Zn, and Cd), based on a separation scheme published by Dionex company that used the spectrophotometric method for quantification. The LC-ICP-MS method overcomes the shortcomings of conventional ferrozine approaches of measuring Fe(II) and total Fe by two separate runs and calculating Fe(III) by the difference of two runs. The advantage is particularly evident in that organo-iron species are found to be the predominant iron species in many natural waters, and the difference method cannot measure the concentration of Fe(III) because ferrozine will not complex with organo-iron species. In the work reported here, the LC-ICP-MS method is successfully applied to the separation of dissolved iron species, as well as six other divalent transi- tion metals in tap water, deionized water, river water, hot springs, and groundwater samples. Keywords: LC-ICP-MS, Fe(II), Fe(III), Organo-Fe, Transition Metals 1. Introduction Concentration determination of soluble reactive species is key to understanding biogeochemical processes in aquatic and terrestrial environments. Iron is one of the most reactive elements in aquatic and geological envi- ronments, and is involved in the cycling of many major chemicals, as well as trace elements [1]. For example, hydrous ferric oxides (e.g., ferrihydrite) are the most reactive soil components with respect to arsenic sorption and can take up hundreds of mg/kg As, either as As(III) or As(V) [2]. The reduction of Fe oxyhydroxides and release of arsenic has been invoked as a probable mechanism of elevated As concentration in groundwater used for drinking and responsible for the poisoning of millions of people [3,4] . Iron is present in th e hydrosphere under two oxidation states, Fe(II) and Fe(III), which are thermodynamically stable under anoxic and oxic conditions, respectively [5 ]. Measurements of both dissolved Fe(II) and Fe(III) con- centration are important in assessing iron’s contribution in mediating numerous biogeochemical processes that involves many elements [6]. Most analytical approaches require a separate analysis of dissolved Fe(II) and total dissolved Fe, and the calculation of Fe(III) by the differ- ence [4]. First proposed by Stookey [7], the ferrozine (monosodium salt hydrate of 3-(2-pyridyl)-5, 6-diphenyl- 1,2,4-triazine-p, p’-disulfonic acid) method is the most widely-used way of determining Fe(II) and Fe(III). Ferrozine reacts with Fe(II) to form a stable magenta complex species, with a maximum absorbance at 562 nm, which is measured spectrophotometrically. When Fe(III) is also present in the aqueous samples, either as a true dissolved complex or in colloids, a separate reduction step with hydroxylamine (NH4OH·HCl) is performed to measure the total iron, with the difference ascribed to Fe(III) [5,6]. The approach lacks sufficien t sensitivity for determining iron con centrations in natural waters at µg/L levels, and therefore a pre-concentration is usually re- quired. Alternative approaches to the ferrozine method have been proposed [6,8,9]. Yan [6] reported a method of on-line coupling of flow injection separation and pre- concentration with ICP-MS, with a sample volume of 2.5 mL and detection limit of 0.08 µg/L. However, the con- centration of Fe(II) was obtained as the difference be- tween the combin ed Fe(III) and Fe(II), and Fe(II I) alone. This was done by controlling the sample acidity range, and detecting the Fe(III) by the Fe(III)-pyrrolidinecar- bodithioate (PDC) complex. In summary, relatively little  Q. H. HU 676 has been published on the simultaneous measurement of both Fe(II) and Fe(I II) in a single run. Metal ions can exist in several different forms, which are determined by the extent of complexation and the oxidation state. In many aqueous samples, metal ions are present in their hydrated forms. Hydrated metal ions can also be complexed by weak ligands such as organic acids or amino acids. These ligands are generally displaced by the complexing agents used in liquid chromatography eluents, with the total of both hydrated and weakly com- plexed metal ion s determined [10]. Hydrated and weakly complexed transition metals can be separated as cations on a cation exchange column. By adding a carboxylic acid chelating agent to the eluent, the net charge on the metal is reduced, because the carboxylic acids are ani- onic in solutions above their pKas. The selectivity of the separation is related to the different degrees of associa- tion between the metals and the chelating agen ts produc- ing different net charges on the metal complexes. If strong enough chelating agents are used in high enough concentration, the net ch arge of the metal complexes can be negative. The resultant anionic metal complexes can be separated by an anion exchange process. The Dionex IonPac® CS-5A column has both cation and anion ex- change capacities, allowing metals to be separated as cations or anions on a single column. The 9 µm poly- meric pellicular packing of the CS-5A column has an ethylene-vinylbenzene divinylbenzene resin core, 55% cross-linking, consisting of two layers of latex particles, functionalized with both anion exchange alkyl quarter- nary amine (internal layer) and cation exchange sulfonic acid groups (outer layer), with capacities of 40 and 20 µequivalents, respectively. With pyridine-2, 6-dicarboxy- lic acid (PDCA) used as the chelating agent in the eluent, the transition metals are separated as anionic complexes in the analytical column of the IonPac CS-5A [10]. The following seven transition metals can be separated and quantified: iron, both ferric (Fe3+) and ferrous (Fe2+), divalent cations of copper, nickel, zinc, cobalt, cadmium, and manganese [10]. The present study was primarily focused on the separation of iron species, as well as the detection and prevalence of organo-Fe species in natural waters. However, the separation of the other six transi- tion metals will also be described in this paper, and some examples of applications in natural waters will be given. 2. Materials and Methods Except for Fe2+ and Fe3+, transition metal standards of 1000 mg/L were obtained from CPI International (Santa Rosa, CA). These stock solutions were purchased in dilute acid (1% - 2% nitric acid) solutions and diluted to different concentrations in deionzied (DI) water for LC- ICP-MS analyses. Ferrous ammonium sulphate (Mohr’s salt, [NH4]2[Fe][SO4]2·6H2O) was purchased from Alfa Aesar (Ward Hill, MA) and ferric chloride hexahydrate (iron III) from Mallinckrodt Baker (Phillipsburg NJ). These chemicals were used to prepare the standard solutions for Fe (I I) and Fe(III), res pective ly . Each ind ivi- dual transition metal was prepared to obtain the retention time from LC separation for peak identification. All solutions were prepared using purified deionzied (DI) water (Milli-Q Ultrapure Water Purification System, Millipore, Billerica, MA). As reported in Dionex (2011), the eluent for separating the transition metals includes the following mixture: 7.0 mM (PDCA), 66 mM potassium hydroxide, 74 mM for- mic acid, 5.6 mM potassium sulfate. Dionex carries the MetPac™ PDCA eluent concentrate, which is to be di- luted 5 times with 200 mL of the co ncentrate added with 800 mL DI water to make up 1-L eluent. To adjust the complexing ability of PDCA to optimize the separation time, we also purchased the individual chemicals in mak- ing th e eluent: PDCA from Alfa Aesar (Ward Hill, MA), potassium hydroxide from EMD Chemicals (Gibbstown, NJ), formic acid from EMD Chemicals, and potassium sulfate from Mallinckrodt Baker (Phillip sburg NJ). Both ferrous and ferric ions can be separated under the above separation conditions, using PDCA as the chelat- ing agent to form anionic complexes. Because the ferrous ion is easily oxidized to ferr ic iron, oxygen was removed from the eluent by degassing the eluent solution bottle with helium for half an hour. To remove oxygen from the analytical and guard columns, a solution of 0.1 M so- dium sulfite (12.6 g/L Na2SO3) was pumped through the columns for 2 hours befor e the sample analysis [10]. We used an advanced analytical method of LC-ICP- MS for separating and quantifying iron, and the other 6 transition metal species, by modifying the quantification method of Dionex [10], to enable sensitive and simulta- neous analyses in aqueous samples. Briefly, the LC sys- tem consisted of a PerkinElmer Series 200 Quaternary Pump and a Series 200 Autosampler (PerkinElmer/ SCIEX, Sheldon, CT) with an IonPac CS5A analytical and CG5A guard columns from Dionex (Sunnyvale, CA). Separation conditions included the following: mobile phase of MetPac PDCA eluent mixture at pH of 4.2 (ad- justed with ammoniu m hydroxid e, with 20% - 22% NH 3), flow rate 1.2 mL/min, and sample injection volume 50 µl. In the Technical Note of Dionex [10], the metal com- plexing agent 4-(2-pyrid ylazo) resorcinol (PAR) is added postcolumn to form a light-absorbing complex with the hydrated and weakly complexed metals. These transition metals are detected by measuring the absorbance at 530 nm of the complex. We used the ICP-MS for sensitive Copyright © 2011 SciRes. AJAC 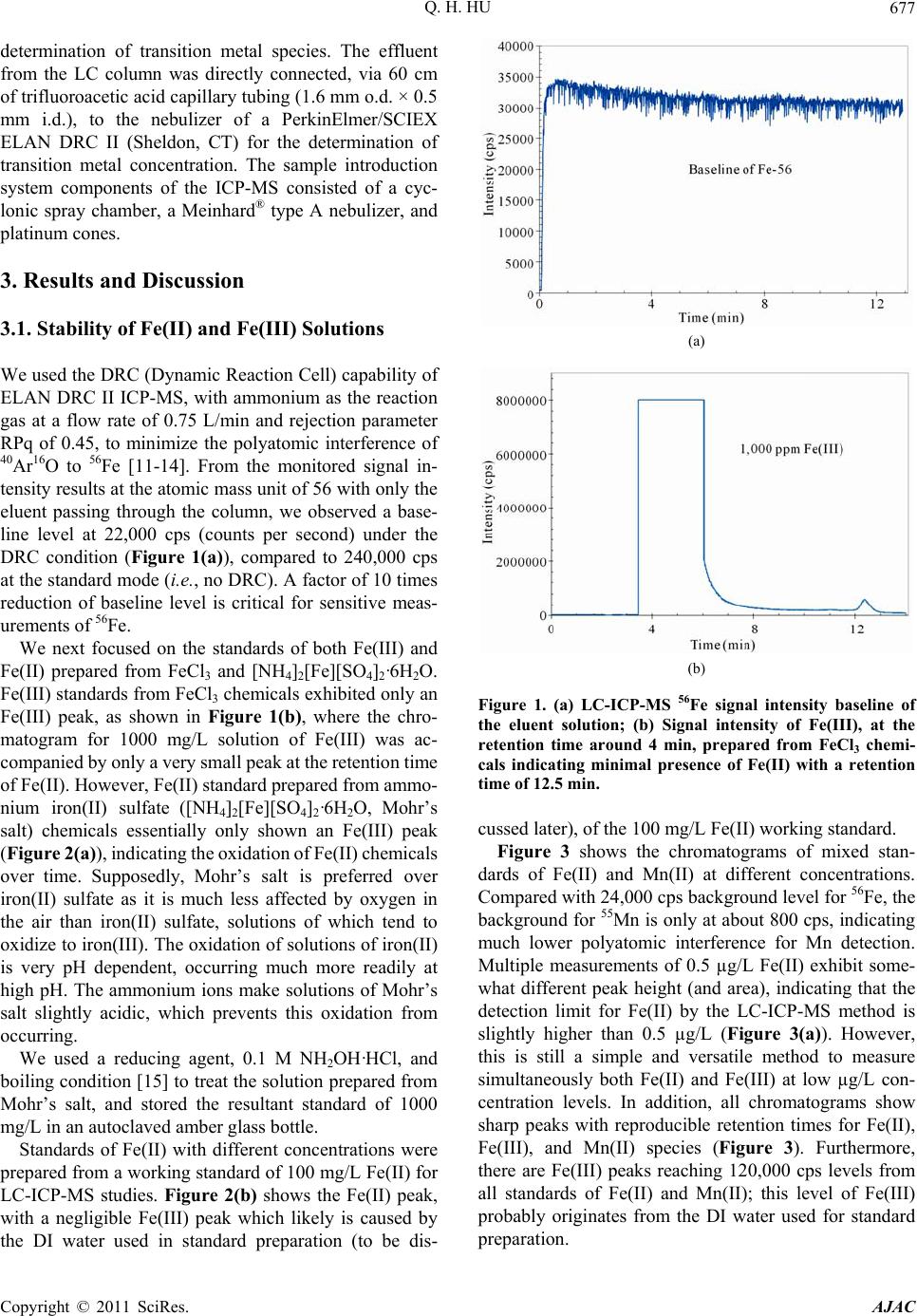 Q. H. HU677 determination of transition metal species. The effluent from the LC column was directly connected, via 60 cm of trifluoroacetic acid capillary tubing (1.6 mm o.d. × 0.5 mm i.d.), to the nebulizer of a PerkinElmer/SCIEX ELAN DRC II (Sheldon, CT) for the determination of transition metal concentration. The sample introduction system components of the ICP-MS consisted of a cyc- lonic spray chamber, a Meinhard® type A nebulizer, and platinum cones. 3. Results and Discussion 3.1. Stability of Fe(II) and Fe(III) Solutions We used the DRC (Dynamic Reaction Cell) capability of ELAN DRC II ICP-MS, with ammonium as the reaction gas at a flow rate of 0.75 L/min and rejection parameter RPq of 0.45, to minimize the polyatomic interference of 40Ar16O to 56Fe [11-14]. From the monitored signal in- tensity results at the atomic mass unit of 56 with only the eluent passing through the column, we observed a base- line level at 22,000 cps (counts per second) under the DRC condition (Figure 1(a)), compared to 240,000 cps at the standard mode (i.e., no DRC). A factor of 10 times reduction of baseline level is critical for sensitive meas- urements of 56Fe. We next focused on the standards of both Fe(III) and Fe(II) prepared from FeCl3 and [NH4]2[Fe][SO4]2·6H2O. Fe(III) standards from FeCl3 chemicals exhibited only an Fe(III) peak, as shown in Figure 1(b), where the chro- matogram for 1000 mg/L solution of Fe(III) was ac- companied by only a very small peak at the retention time of Fe(II). H ow e ver , Fe(II) standa rd prepared fr om amm o- nium iron(II) sulfate ([NH4]2[Fe][SO4]2·6H2O, Mohr’s salt) chemicals essentially only shown an Fe(III) peak (Figure 2(a)), indicating the oxidation of Fe(II) chemicals over time. Supposedly, Mohr’s salt is preferred over iron(II) sulfate as it is much less affected by oxygen in the air than iron(II) sulfate, solutions of which tend to oxidize to iron(III). The oxidation of solutions of iron(II) is very pH dependent, occurring much more readily at high pH. The ammonium ions make solutions of Mohr’s salt slightly acidic, which prevents this oxidation from occurring. We used a reducing agent, 0.1 M NH2OH·HCl, and boiling condition [15] to treat the solu tion prepared from Mohr’s salt, and stored the resultant standard of 1000 mg/L in an autoclaved amber glass bottle. Standards of Fe(II) with different concentrations were prepared from a working standard of 100 mg/L Fe(II) for LC-ICP-MS studies. Figure 2(b) shows the Fe(II) peak, with a negligible Fe(III) peak which likely is caused by the DI water used in standard preparation (to be dis- (a) (b) Figure 1. (a) LC-ICP-MS 56Fe signal intensity baseline of the eluent solution; (b) Signal intensity of Fe(III), at the retention time around 4 min, prepared from FeCl3 chemi- cals indicating minimal presence of Fe(II) with a retention time of 12.5 min. cussed later), of the 100 mg/L Fe(II) working standard. Figure 3 shows the chromatograms of mixed stan- dards of Fe(II) and Mn(II) at different concentrations. Compared with 24,000 cp s back ground level f or 56Fe, the background for 55Mn is only at about 800 cps, indicating much lower polyatomic interference for Mn detection. Multiple measurements of 0.5 µg/L Fe(II) exhibit some- what different peak height (and area), indicating that the detection limit for Fe(II) by the LC-ICP-MS method is slightly higher than 0.5 µg/L (Figure 3(a)). However, this is still a simple and versatile method to measure simultaneously both Fe(II) and Fe(III) at low µg/L con- centration levels. In addition, all chromatograms show sharp peaks with reproducible retention times for Fe(II), Fe(III), and Mn(II) species (Figure 3). Furthermore, there are Fe(III) peaks reaching 120,000 cps levels from all standards of Fe(II) and Mn(II); this level of Fe(III) probably originates from the DI water used for standard preparation. Copyright © 2011 SciRes. AJAC 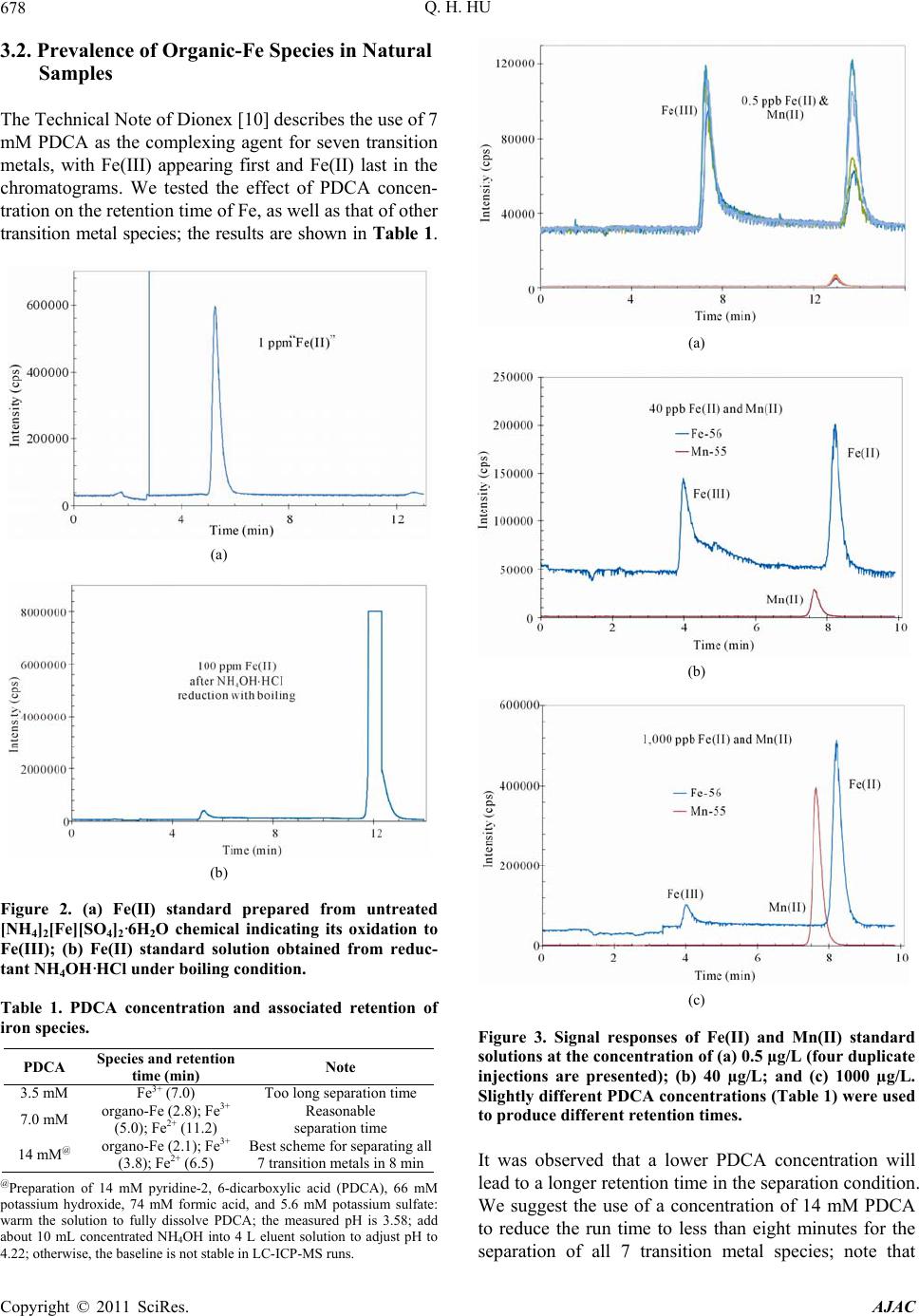 Q. H. HU 678 3.2. Prevalence of Organic-Fe Species in Natural Samples The Technical Note of Dionex [10] describes the use of 7 mM PDCA as the complexing agent for seven transition metals, with Fe(III) appearing first and Fe(II) last in the chromatograms. We tested the effect of PDCA concen- tration on the retention time of Fe, as well as that of other transition metal species; the results are shown in Table 1. (a) (b) Figure 2. (a) Fe(II) standard prepared from untreated [NH4]2[Fe][SO4]2·6H2O chemical indicating its oxidation to Fe(III); (b) Fe(II) standard solution obtained from reduc- tant NH4OH·HCl under boiling condition. Table 1. PDCA concentration and associated retention of iron species. PDCA Species and retention time (min) Note 3.5 mM Fe3+ (7.0) Too long separation time 7.0 mM organo-Fe (2.8); Fe3+ (5.0); Fe2+ (11.2) Reasonable separation time 14 mM@ organo-Fe (2.1); Fe3+ (3.8); Fe2+ (6.5) Best scheme for separating all 7 transition metals in 8 min @Preparation of 14 mM pyridine-2, 6-dicarboxylic acid (PDCA), 66 mM potassium hydroxide, 74 mM formic acid, and 5.6 mM potassium sulfate: warm the solution to fully dissolve PDCA; the measured pH is 3.58; add about 10 mL concentrated NH4OH into 4 L eluent solution to adjust pH to 4.22; otherwise, the baseline is not stable in LC-ICP-MS runs. (a) (b) (c) Figure 3. Signal responses of Fe(II) and Mn(II) standard solutions at the concentration of (a) 0.5 µg/L (four duplicate injections are presented); (b) 40 µg/L; and (c) 1000 µg/L. Slightly different PDCA concentrations (Table 1) were used to produce different retention time s. It was observed that a lower PDCA concentration will lead to a longer retention time in the sep aration cond ition. We suggest the use of a concentration of 14 mM PDCA to reduce the run time to less than eight minutes for the separation of all 7 transition metal species; note that Copyright © 2011 SciRes. AJAC 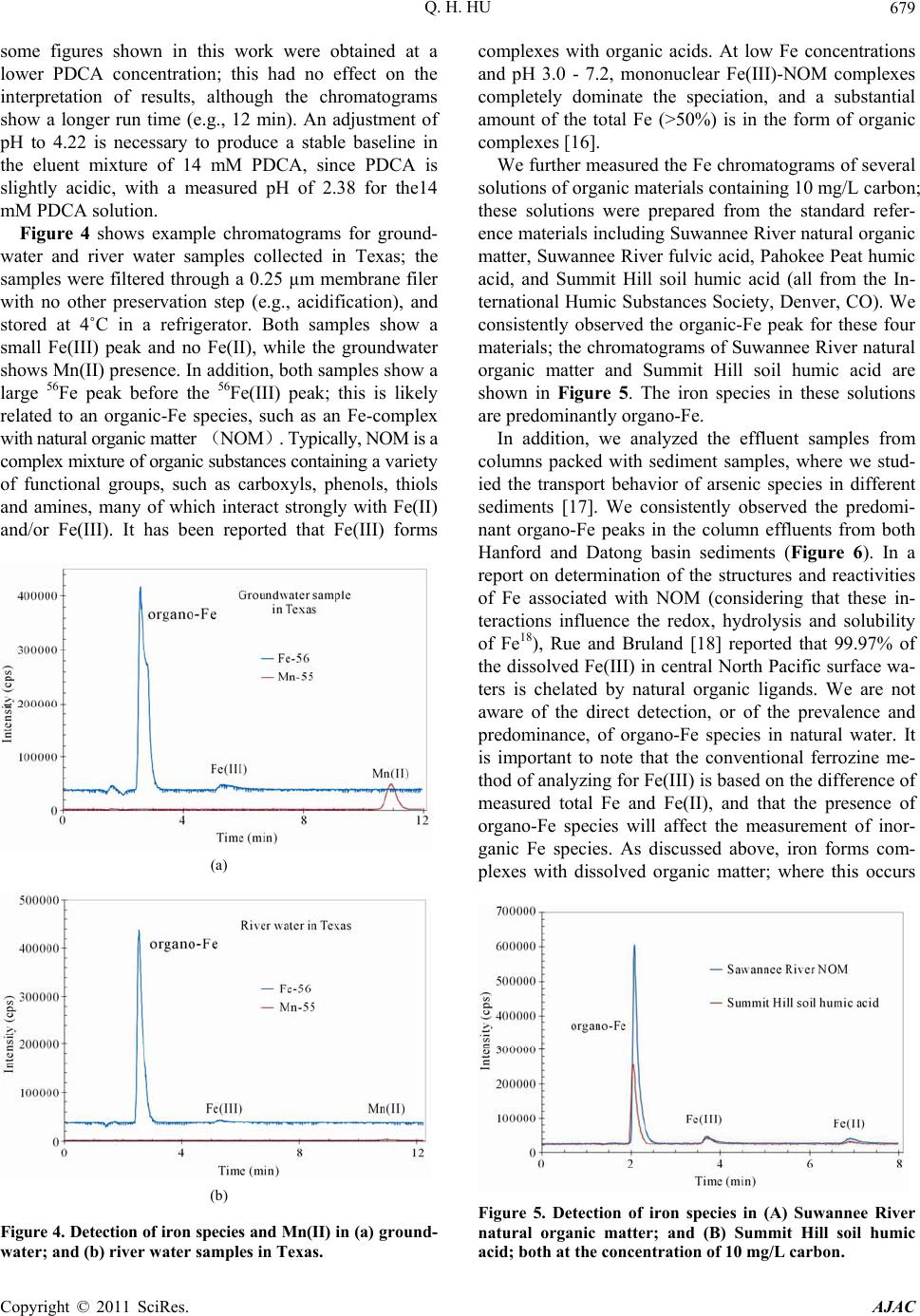 Q. H. HU679 some figures shown in this work were obtained at a lower PDCA concentration; this had no effect on the interpretation of results, although the chromatograms show a longer run time (e.g., 12 min). An adjustment of pH to 4.22 is necessary to produce a stable baseline in the eluent mixture of 14 mM PDCA, since PDCA is slightly acidic, with a measured pH of 2.38 for the14 mM PDCA solution. Figure 4 shows example chromatograms for ground- water and river water samples collected in Texas; the samples were filtered through a 0.25 µm membrane filer with no other preservation step (e.g., acidification), and stored at 4˚C in a refrigerator. Both samples show a small Fe(III) peak and no Fe(II), while the groundwater shows Mn(II) pr esence. In add ition, both samples sh ow a large 56Fe peak before the 56Fe(III) peak; this is likely related to an organic-Fe species, such as an Fe-complex with natural organic matter (NOM). Typically, NOM is a complex mixture of organic substances containing a va riet y of functional groups, such as carboxyls, phenols, thiols and amines, many of which interact strongly with Fe(II) and/or Fe(III). It has been reported that Fe(III) forms (a) (b) Figure 4. Detection of iron species and Mn(II) in (a) gr oun d- water; and (b) river water samples in Texas. complexes with organic acids. At low Fe concentrations and pH 3.0 - 7.2, mononuclear Fe(III)-NOM complexes completely dominate the speciation, and a substantial amount of the total Fe (>50%) is in the form of organic complexes [16]. We further measured the Fe chromatograms of several solutions of organic materials containing 10 mg/L carbon; these solutions were prepared from the standard refer- ence materials including Suwannee River natural organic matter, Suwannee River fulvic acid, Pahokee Peat humic acid, and Summit Hill soil humic acid (all from the In- ternational Humic Substan ces Society, Denver, CO). We consistently observed the organic-Fe peak for these four materials; the chromatograms of Suwannee River natural organic matter and Summit Hill soil humic acid are shown in Figure 5. The iron species in these solutions are predomi nantl y organo-Fe. In addition, we analyzed the effluent samples from columns packed with sediment samples, where we stud- ied the transport behavior of arsenic species in different sediments [17]. We consistently observed the predomi- nant organo-Fe peaks in the column effluents from both Hanford and Datong basin sediments (Figure 6). In a report on determination of the structures and reactivities of Fe associated with NOM (considering that these in- teractions influence the redox, hydrolysis and solubility of Fe18), Rue and Bruland [18] reported that 99.97% of the dissolved Fe(III) in central North Pacific surface wa- ters is chelated by natural organic ligands. We are not aware of the direct detection, or of the prevalence and predominance, of organo-Fe species in natural water. It is important to note that the conventional ferrozine me- thod of an alyzing f or Fe(III ) is ba sed on the diff erence of measured total Fe and Fe(II), and that the presence of organo-Fe species will affect the measurement of inor- ganic Fe species. As discussed above, iron forms com- plexes with dissolved organic matter; where this occurs Figure 5. Detection of iron species in (A) Suwannee River natural organic matter; and (B) Summit Hill soil humic acid; both at the concentration of 10 mg/L carbon. Copyright © 2011 SciRes. AJAC 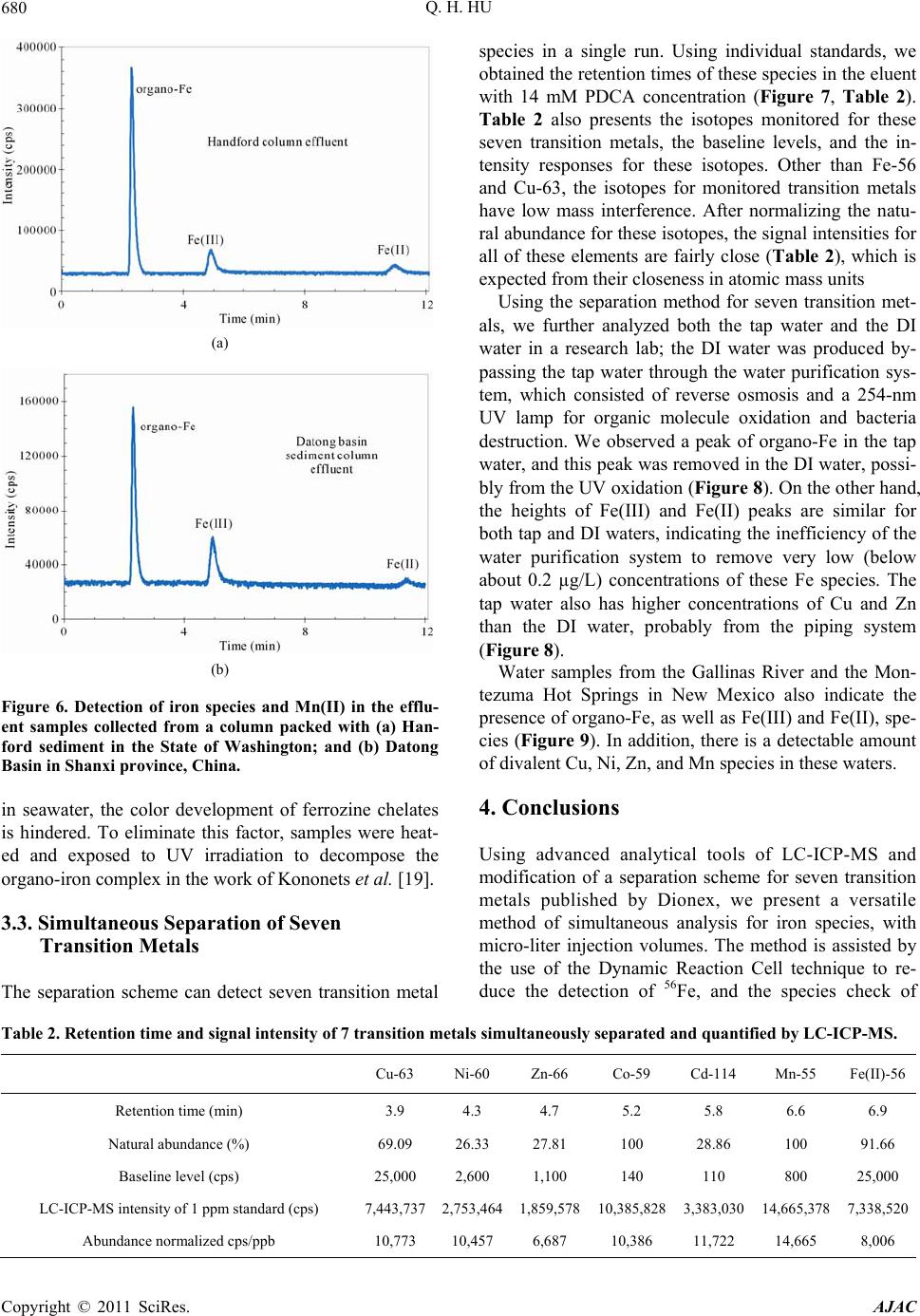 Q. H. HU Copyright © 2011 SciRes. AJAC 680 species in a single run. Using individual standards, we obtained the retention times of these species in the eluent with 14 mM PDCA concentration (Figure 7, Table 2). Table 2 also presents the isotopes monitored for these seven transition metals, the baseline levels, and the in- tensity responses for these isotopes. Other than Fe-56 and Cu-63, the isotopes for monitored transition metals have low mass interference. After normalizing the natu- ral abundance for these isotopes, the signal intensities for all of these elements are fairly close (Table 2), which is expected from their closeness in atomic mass units Using the separation method for seven transition met- als, we further analyzed both the tap water and the DI water in a research lab; the DI water was produced by- passing the tap water through the water purification sys- tem, which consisted of reverse osmosis and a 254-nm UV lamp for organic molecule oxidation and bacteria destruction. We observed a peak of organo-Fe in the tap water, and this peak was removed in the DI water, possi- bly from the UV oxidation (Figure 8). On the other hand, the heights of Fe(III) and Fe(II) peaks are similar for both tap and DI waters, indicating the in efficiency of the water purification system to remove very low (below about 0.2 µg/L) concentrations of these Fe species. The tap water also has higher concentrations of Cu and Zn than the DI water, probably from the piping system (Figure 8). (a) Water samples from the Gallinas River and the Mon- tezuma Hot Springs in New Mexico also indicate the presence of organo-Fe, as well as Fe(III) and Fe(II), spe- cies (Figure 9). In addition, there is a detectable amount of divalent Cu, Ni, Zn, and Mn species in these waters. (b) Figure 6. Detection of iron species and Mn(II) in the efflu- ent samples collected from a column packed with (a) Han- ford sediment in the State of Washington; and (b) Datong Basin in Shanxi province, China. 4. Conclusions in seawater, the color development of ferrozine chelates is hindered. To eliminate this factor, samples were heat- ed and exposed to UV irradiation to decompose the organo-iron complex in the work of Kononets et al. [19]. Using advanced analytical tools of LC-ICP-MS and modification of a separation scheme for seven transition metals published by Dionex, we present a versatile method of simultaneous analysis for iron species, with micro-liter injection volumes. The method is assisted by the use of the Dynamic Reaction Cell technique to re- duce the detection of 56Fe, and the species check of 3.3. Simultaneous Separation of Seven Transition Metals The separation scheme can detect seven transition metal Table 2. Retention time and signal intensity of 7 transition metals simultaneously separated and quantified by LC-ICP -M S. Cu-63 Ni-60 Zn-66 Co-59 Cd-114 Mn-55 Fe(II)-56 Retention time (min) 3.9 4.3 4.7 5.2 5.8 6.6 6.9 Natural abundance (%) 69.09 26.33 27.81 100 28.86 100 91.66 Baseline level (cps) 25,000 2,600 1,100 140 110 800 25,000 LC-ICP-MS intensity of 1 ppm standard (cps ) 7,443,7372,753,4641,859,57810,385,8283,383,030 14,665,3787,338,520 Abundance normalized cps/ppb 10,773 10,457 6,687 10,386 11,722 14,665 8,006 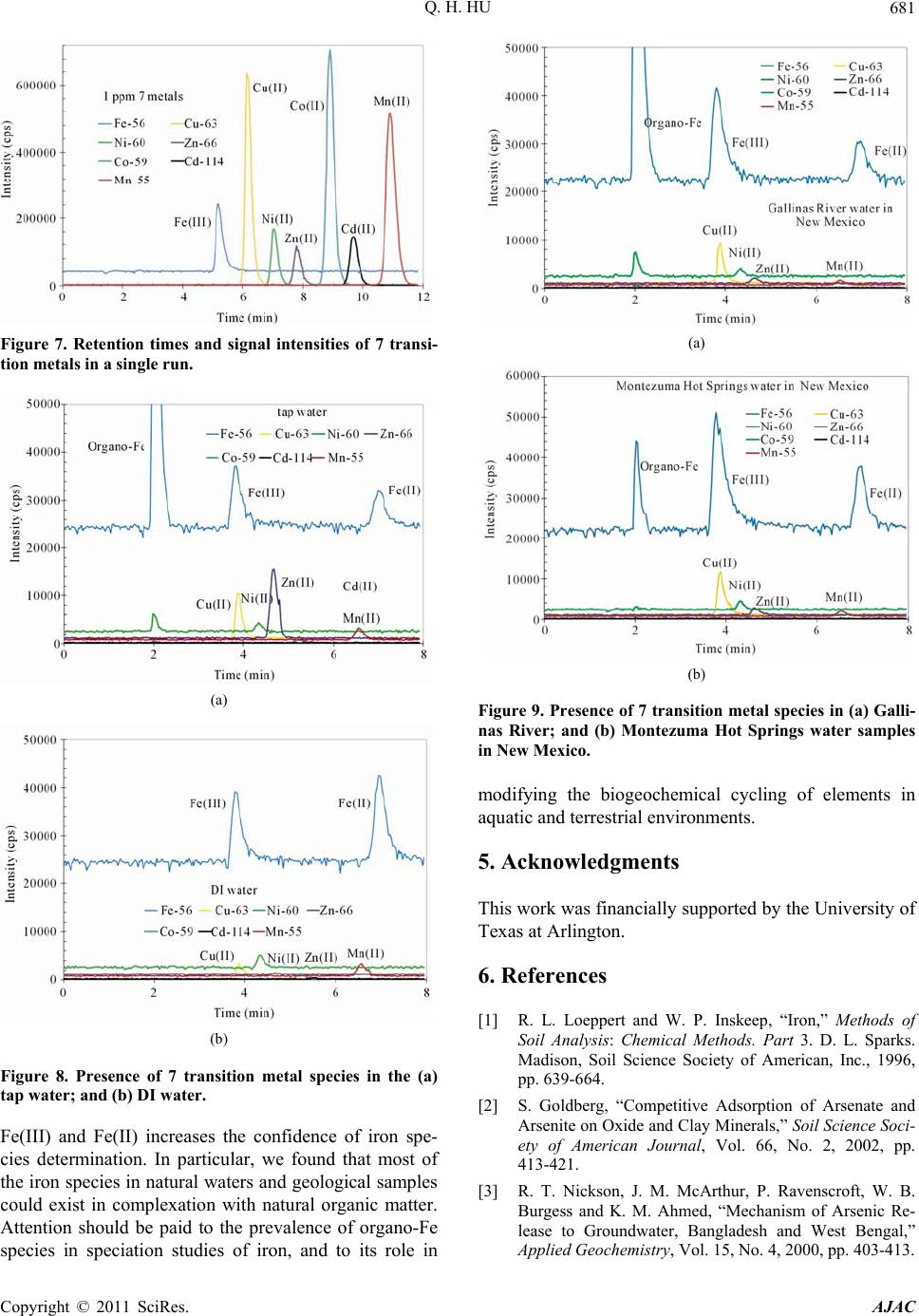 Q. H. HU Copyright © 2011 SciRes. AJAC 681 Figure 7. Retention times and signal intensities of 7 transi- tion metals in a single run. (a) (b) Figure 8. Presence of 7 transition metal species in the (a) tap water; and (b) DI water. Fe(III) and Fe(II) increases the confidence of iron spe- cies determination. In particular, we found that most of the iron species in natural waters and geological samples could exist in complexation with natural organic matter. Attention should be paid to the prevalence of organo-Fe species in speciation studies of iron, and to its role in (a) (b) Figure 9. Presence of 7 transition metal species in (a) Galli- nas River; and (b) Montezuma Hot Springs water samples in New Mexico. modifying the biogeochemical cycling of elements in aquatic and terrestrial environments. 5. Acknowledgments This work was financially supported by the University of Texas at Arlington. 6. References [1] R. L. Loeppert and W. P. Inskeep, “Iron,” Methods of Soil Analysis: Chemical Methods. Part 3. D. L. Sparks. Madison, Soil Science Society of American, Inc., 1996, pp. 639-664. [2] S. Goldberg, “Competitive Adsorption of Arsenate and Arsenite on Oxide and Clay Minerals,” Soil Science Soci- ety of American Journal, Vol. 66, No. 2, 2002, pp. 413-421. [3] R. T. Nickson, J. M. McArthur, P. Ravenscroft, W. B. Burgess and K. M. Ahmed, “Mechanism of Arsenic Re- lease to Groundwater, Bangladesh and West Bengal,” Applied Geochemistry, Vol. 15, No. 4, 2000, pp. 403-413.  Q. H. HU 682 doi:10.1016/S0883-2927(99)00086-4 [4] P. L. Smedley and D. G. Kinniburgh, “A Review of the Source, Behaviour and Distribution of Arsenic in Natural Waters,” Applied Geochemistry, Vol. 17, No. 5, 2002, pp. 517-568. doi:10.1016/S0883-2927(02)00018-5 [5] E. Viollier, P. W. Inglett, K. Hunter, A. N. Roychoudhury and P. Van Cappellen, “The Ferrozine Method Revisited: Fe(II)/Fe(III) Determination in Natural Waters,” Applied Geochemistry, Vol. 15, No. 6, 2000, pp. 785-790. doi:10.1016/S0883-2927(99)00097-9 [6] X. P. Yan, N. J. Hendry and R. Kerrich, “Speciation of Dissolved Iron(III) and Iron(II) in Water by On-Line Coupling of Flow Injection Separation and Preconcentra- tion with Inductively Coupled Plasma Mass Spectrome- try,” Analytical Chemistry, Vol. 72, No. 8, 2000, pp. 1879-1884. doi:10.1021/ac9909655 [7] L. L. Stookey, “Ferrozine a New Spectrophotometric Re- Agent for Iron Determination,” Analytical Chemistry, Vol. 42, No. 7, 1970, pp. 779-784. doi:10.1021/ac60289a016 [8] L. S. Huang and K. C. Lin, “Detection of Iron Species Using Inductively Coupled Plasma Mass Spectrometry under Cold Plasma Temperature Conditions,” Spectro- chim Acta Part B-Atomic Spectroscopy, Vol. 56, 2001, pp. 123-128. doi:10.1016/S0584-8547(00)00292-5 [9] D. L. Giokas, E. K. Paleologos and M. I. Karayannis, “Speciation of Fe(II) and Fe(III) by the Modified Ferro- Zine Method, FIA-Spectrophotometry, and Flame AAS after Cloud-Point Extraction,” Analytical and Bioana- lytical Chemistry, Vol. 373, No. 4-5, 2002, pp. 237-243. doi:10.1007/s00216-002-1326-7 [10] Dionex, “Determination of Transition Metals by Ion Chromatography,” Technical Note 10, 2011. http://wwwsearch.dionex.com/search?site=www&client= www_frontend&proxystyle sheet=www_frontend&output =xml_no_dtd&q=tn10 [11] F. Vanhaecke, L. Balcaen, G. De Wannemacker and L. Moens, “Capabilities of Inductively Coupled Plasma Mass Spectrometry for the Measurement of Fe Isotope Ratios,” Journal of Analytical and Atomic Spectrometry, Vol. 17, No. 8, 2002, pp. 933-943. doi:10.1039/b202409h [12] K. Kawabata and Y. Kishi, “DRC-ICP-MS Analysis of Various Chemicals Used in the Semiconductor Industry (Reprinted with Permission from the Semiconductor Pure Water & Chemicals Conference 2001),” Atomic Spec- trometry, Vol. 24, No. 2, 2003, pp. 66-72. [13] C. F. Yeh and S. J. Jiang, “Speciation of V, Cr and Fe by Capillary Electrophoresis-Bandpass Reaction Cell Induc- tively Coupled Plasma Mass Spectrometry,” Journal of Chromatography A, Vol. 1029, No. 1-2, 2004, pp. 255- 261. doi:10.1016/j.chroma.2003.12.070 [14] S. D’Ilio, N. Violante, M. Di Gregorio, O. Senofonte and F. Petrucci, “Simultaneous Quantification of 17 Trace Elements in Blood by Dynamic Reaction Cell Inductively Coupled Plasma Mass Spectrometry (DRC-ICP-MS) Equipped with a High-Efficiency Sample Introduction System,” Analytical Chimica Acta, Vol. 579, No. 2, 2006, pp. 202-208. doi:10.1016/j.aca.2006.07.027 [15] G. G. Rao and G. Somidevamma, “Volumetric Determi- Nation of Iron (III) with Hydroxylamine as a Reducing Agent,” Fresenius’ Journal of Analytical Chemistry, Vol. 165, No. 6, 1959, pp. 432-436. [16] T. Karlsson and P. Persson, “Coordination Chemistry and Hydrolysis of Fe(III) in a Peat Humic Acid Studied by X-Ray Absorption Spectroscopy,” Geochimica et Cosmo- chimica Acta, Vol. 74, 2010, pp. 30-40. doi:10.1016/j.gca.2009.09.023 [17] Q. H. Hu, G. X. Sun and X. B. Gao, “Conversion, Sorp- tion, and Transport of Arsenic Species in Geological Me- dia,” Applied Geochemistry, 2011 (under revision). [18] E. L. Rue and K. W. Bruland, “Complexation of Iron(III) by Natural Organic-Ligands in the Central North Pacific as Determined by a New Competitive Ligand Equilibra- tion Adsorptive Cathodic Stripping Voltammetric Me- thod,” Marine Chemistry, Vol. 50, No. 1-4, 1995, pp. 117-138. doi:10.1016/0304-4203(95)00031-L [19] M. Y. Kononets, S. V. Pakhomova, A. G. Rozanov and M. A. Proskurnin, “Determination of Soluble Iron Spe- cies in Seawater Using Ferrozine,” Journal of Analytical Chemistry, Vol. 57, No. 7, 2002, pp. 586-589. doi:10.1023/A:1016269832258 Copyright © 2011 SciRes. AJAC
|