Journal of Biomedical Science and Engineering
Vol.5 No.7(2012), Article ID:20534,3 pages DOI:10.4236/jbise.2012.57051
A simplified protocol for the semi-large scale recovery of plasmids from Escherichia coli grown on agar plates
![]()
1Section of Gene Expression Regulation, Frontier Science Research Center, Kagoshima University, Kagoshima, Japan
2Department of Pediatric Dentistry, Graduate School of Medical and Dental Sciences, Kagoshima University, Kagoshima, Japan
3Division of Basic Molecular Science and Molecular Medicine, School of Medicine, Tokai University, Tokyo, Japan
4Department of Surgery II, National Defense Medical College, Tokorozawa, Japan
5Department of Organ Regeneration, Graduate School of Medicine, Shinshu University, Matsumoto, Japan
6Animal Genome Research Unit, Division of Animal Science, National Institute of Agrobiological Sciences, Tsukuba, Japan
Email: masasato@ms.kagoshima-u.ac.jp
Received 9 May 2012; revised 8 June 2012; accepted 28 June 2012
Keywords: Agar Plate; DNA Purification; E. coli; Gene Engineering; Plasmid
ABSTRACT
Semi-large scale liquid cultivation of transformed Escherichia coli (E. coli) in medium (100 - 200 ml) has been widely used for the acquisition of relatively large amounts of plasmid DNA (50 - 300 μg). However, this method requires an expensive high-speed centrifugation apparatus to precipitate E. coli before lysis, which is both laborious and time-consuming. Here, we demonstrate a method for agar plate-based cultivation of bacteria that does not employ a high-speed centrifugation apparatus. This procedure proves to be simple and reproducible, yielding an average of 82 μg of plasmid DNA per experiment. It may therefore be valuable for cloning/transfection experiments under limited financial backgrounds.
1. INTRODUCTION
Purification of plasmid DNA from transformed Escherichia coli (E. coli) is an important tool in molecular biology. In the past 2 decades, several protocols for this approach have been established. Liquid cultivation of E. coli cells in the presence of antibiotics is one of the most employed methods for isolation of large amounts of plasmids [1,2]. Specifically, cultivation in a 100-ml volume with gentle shaking for semi-large scale isolation of plasmids (50 - 300 μg) is often performed but requires that E. coli cells be precipitated by centrifugation with an expensive high-speed centrifugation apparatus. These collected cells are then lysed to release the plasmid, and the lysed debris is then separated from the plasmid-containing supernatant by centrifugation with the above apparatus, a process that takes approximately 40 - 60 min. The collected supernatant is then subjected to further purification by using column [1,2] or density-gradient ultracentrifugation of a cesium chloride/ethidium bromide (EtBr) mixture [3].
Growth of E. coli on agar plates is commonly used for the preparation of MiniPrep DNA [3], but rarely used for isolation of plasmids at a semi-large scale level. This procedure does not require the above-mentioned centrifugation apparatus, but requires ultra-rapid centrifugation equipment, which is generally used for precipitation of plasmid DNA in microcentrifuge tubes of 1.5 - 2-ml volumes. Briefly, E. coli grown on agar plates is picked by a scraper and transferred to a 2-ml centrifuge tube for lysis. After centrifugation of the lysate by ultra-rapid centrifugation, the resulting supernatant can be processed similarly to liquid culture-derived supernatant.
In this study, we examined whether sufficient amounts (>50 μg) of plasmid DNA could be recovered from E. coli grown in agar plates and compared the DNA quantity and quality with that produced by liquid culture.
2. MATERIALS AND METHODS
The manuscript does not contain experiments using animals as well as human studies.
2.1. Preparation of Plasmid-Containing E. coli
The plasmid pCE-29 [4], consisting of a CAG promoter [5], a portion of the 2nd intron and 3rd exon of the rabbit b-globin gene [6], enhanced green fluorescent protein (EGFP) cDNA (isolated from pEGFP-N1; Invitrogen Co., Carlsbad, CA), poly(A) sites of the SV40 gene, pBluescript SK(-) plasmid backbone (Stratagene Inc., La Jolla, CA), and an ampicillin (Amp) resistance gene, was used for transformation of E. coli. TaKaRa E. coli HB101 competent cells v.06 (#9051; Takara Bio Inc., Shiga, Japan) were transformed with the pCE-29 vector according to the manufacturer’s protocol (Takara Bio Inc.). These transformed E. coli-containing solutions were plated onto agar containing 100 μg/ml of Amp (Invitrogen Co.) and subsequently cultured for 16 h (overnight) in a 37˚C incubator. Colonies were selected and picked using a sterile toothpick and dissolved in 100 μl of sterile Plusgrow (#08246-44; Nacalai-tesque Inc., Kyoto, Japan).
Half of the E. coli-containing solution (50 μl) was mixed with 100-ml Plusgrow containing 100 μg/ml of Amp in a 500-ml glass flask and subjected to cultivation (for 16 h at 37˚C) with gentle shaking (liquid culture group). The remaining half of the E. coli-containing solution (50 μl) was mixed with 950 μl of Plusgrow containing 100 μg/ml of Amp, and a 500-μl aliquot was plated onto agar [containing 2% solid agar (#01028-85; Nacalai-tesque Inc.), 100 μg/ml of Amp, and Plusgrow] in a 100-mm bacteriological petri dish (Eiken Kizai Co., Tokyo, Japan), as depicted in Figure 1A. A total of 2 agar plates were used, and these plates were then subjected to static culture for 16 h in a 37˚C incubator (agar plate culture group).
2.2. Recovery of E. coli and Cell Lysis
For the liquid culture group, the cell suspension (100 ml) was transferred to a 400-ml centrifuge bottle (330437A; Hitachi, Ibaraki, Japan) and then subjected to centrifugation for 10 min at 5000 ´ g at 4˚C, using a Hitachi HighSpeed Refrigerated Centrifuge Himac CF9RX. After centrifugation, the supernatant was discarded and the pellet was lysed using a Qiagen Mid DNA isolation kit (#12143; Hilden, Germany) according to the manufacturer’s instruction with some modifications. Briefly, the pellet was first dissolved in 2 ml of chilled Buffer P1, and then mixed with 2 ml of Buffer P2 (alkaline buffer) to disrupt the cellular structure. Approximately 5 min after mixing, the lysate was added with 2 ml of chilled Buffer P3 (neutral buffer). The resulting lysate was cleared by centrifugation using the Himac CF9RX centrifuge for 20 - 30 min at 6000 ´ g at 4˚C to remove any bacterial debris.
For the agar plate culture group, E. coli cells grown on the surface of solid agar were collected using Cell Scraper-L (#MS-93300; Sumitomo Bakelite Co. Ltd., Tokyo, Japan) and transferred to four 2-ml micro centrifuge tubes (#111568; Eppendorf AG, Hamburg, Germany), as depicted in Figure 1(a). Lysis of E. coli was performed using a Qiagen Mid DNA isolation kit according to a modified protocol of the manufacturer. Briefly, 330 μl of chilled Buffer P1 was added to each tube, and cells were resuspended by vigorous vortexing. The tube was then mixed with 330 μl of Buffer P2, left for 5 min, and then 330 μl of chilled Buffer P3 was added to the reaction. This mixture was then subjected to centrifugation on an ultra-rapid centrifuge (MX-100; Tomy Seiko Ltd., Tokyo, Japan) for 10 min at 16,000 ´ g at 4˚C to precipitate the bacterial debris, as depicted in Figure 1(a).
2.3. Purification of Plasmid DNA
Plasmid was purified using the Qiagen Mid DNA isolation kit according to the manufacturer’s instruction with some modifications. After equilibration of the column with Buffer QBT, the supernatant (approximately 4 ml) was separated from the bacterial debris, and washed with 4 ml of Buffer QC. The plasmid DNA bound to the resin of the column was then eluted in 5 ml of Buffer QF. The eluate was next mixed with 3.5 ml of isopropanol, and centrifuged at 16,000 ´ g for 15 min on an MX-100 centrifuge to precipitate DNA. The resulting DNA pellets were finally redissolved in 250 μl of sterile water.
2.4. Measurement of DNA Concentration and Restriction Enzyme Analysis
DNA concentration was measured using a B10967 Jenway Genova UV spectrophotometer (Jenway Ltd., Dunmow, Essex, UK) at a wavelength of 260 nm.
Restriction enzyme analysis was performed according to the protocol of Sambrook et al. [3]. Briefly, 200 ng of DNA per tube was digested with the appropriate enzymes and subjected to 0.8% agarose gel electrophoresis. The gel was stained with EtBr for visualization of DNA bands. Lambda DNA digested with Hin dIII and EcoRI (Toyobo, Tokyo, Japan) was used as molecular weight markers.
3. RESULTS AND DISCUSSION
The aims of this study were to describe that agar platebased culture of bacteria confers the semi-large scale recovery of plasmids. We first compared the quantity and quality of the contents of plasmid DNA extracted from E. coli cultured on liquid culture and agar plate culture groups. The DNA isolation was repeated 3 times on different days for each method. The average total DNA content in the agar plate culture group was 82 μg, which was roughly comparable to that (980 μg) obtained from the liquid culture group (Figure 1(b)). The procedure took 60 - 65 min to completion for plasmid purification in the agar plate culture group, while it took 90 - 100 min in the liquid culture group. The gel-electrophoretic pattern of the DNA digested with several enzymes was the same for both groups (Figure 1(c)).
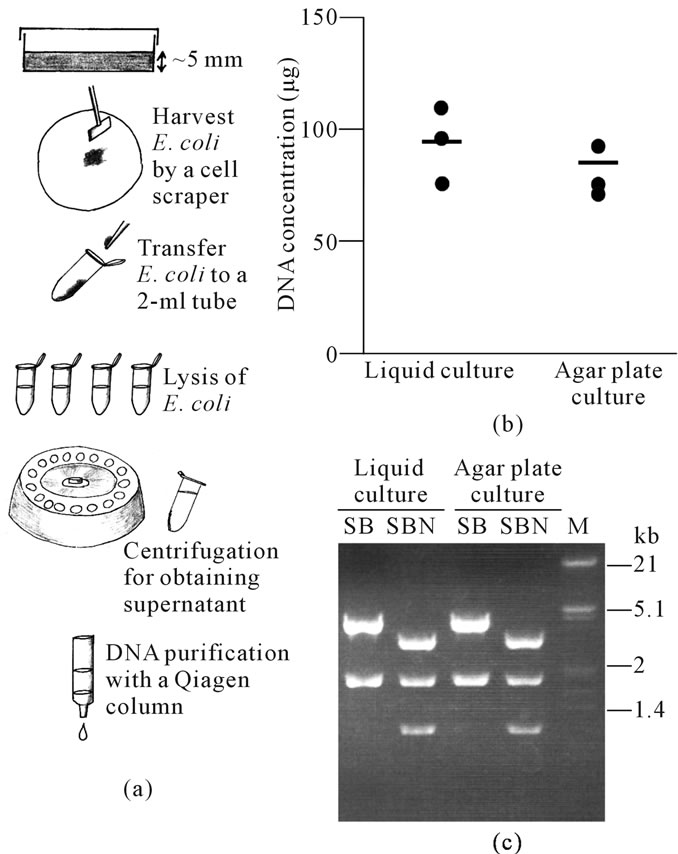
Figure 1. (a) Schematic representation of agar plate-based plasmid recovery; (b) DNA from plasmids extracted from liquid culture or agar plate culture. Each dot corresponds to the amount of DNA obtained from each culture. Bars indicate the average amount of DNA from 3 samples; (c) Restriction enzyme analysis of pCE-29 plasmid DNA [digested with Sal I (S)/Bam HI (B) or S/B/Not I (N)] recovered from liquid culture or agar plate culture. M, lambda phage DNA digested with Eco RI and Hin dIII.
Our present results clearly indicate that agar platebased culturing of E. coli can be employed for the semilarge scale production of plasmid DNA. As previously mentioned, few previous protocols for plasmid DNA purification have been based on bacterial growth in liquid culture. In this paper, the collection of bacteria by centrifugation is necessary for this method, requiring the use of an expensive high-speed centrifuge, such as the Himac CF9RX centrifuge that allows for centrifugation of 100 - 200 ml volumes. However, agar plate-based culturing of E. coli does not require such an apparatus. Furthermore, this method can take less time to complete DNA purification, compared with previous protocols based on liquid culture of bacteria (60 - 65 min vs. 90 - 100 min, respectively).
4. CONCLUSION
Our present agar plate-based culturing protocol appears to be a convenient method compared with liquid-based culturing for purifying plasmid DNA at a semi-large scale level. This technique may therefore be valuable for cloning/transfection experiments under limited financial backgrounds. This study investigated whether agar platebased culture of gene-engineered bacteria can confer semi-large scale preparation of plasmid DNA, which does not employ an expensive high-speed centrifuge apparatus.
![]()
![]()
REFERENCES
- DNA Purification in Promega Home Page. http://www.promega.com/resources/product-guides-and-sel ectors/protocols-and-applications-guide/dna-purification/
- QIAGEN Plasmid Purification System in Qiagen Home Page. http://www.ebiotrade.com/buyf/productsf/qiagen/QIAGEN_plasmid_purification_system.htm
- Sambrook, J., Fritsche, E. and Maniatis, T. (1989) Molecular cloning: A laboratory manual. Cold Spring Harbor Press, New York.
- Sato, M., Ishikawa, A. and Kimura, M. (2002) Direct injection of foreign DNA into mouse testis as a possible in vivo gene transfer system via epididymal spermatozoa. Molecular Reproduction and Development, 61, 49-56. doi:10.1002/mrd.1130
- Niwa, H., Yamamura, K. and Miyazaki, J. (1991) Efficient selection for high-expression transformants with a novel eukaryotic vector. Gene, 108, 193-200. doi:10.1016/0378-1119(91)90434-D
- van Ooyen, A., van den Berg, J., Mantei, N. and Weissmann, C. (1979) Comparison of total sequence of a cloned rabbit beta-globin gene and its flanking regions with a homologous mouse sequence. Science, 206, 337- 344. doi:10.1016/0378-1119(91)90434-D