Materials Sciences and Applications
Vol.3 No.12(2012), Article ID:25410,11 pages DOI:10.4236/msa.2012.312126
FP-LMTO Calculations of Structural and Electronic Properties of Alkaline-Earth Chalcogenides Alloys AY: A = Ca, Sr, Ba; Y = S
![]()
1Department of Physics, University Djillali Liabes, Sidi Bel Abbes, Algeria; 2Department of Mathematics, University of Sidi Bel Abbes, Sidi Bel Abbes, Algeria; 3Laboratory of Statistics and Stochastic Processes, University Djillali Liabes, Sidi Bel Abbes, Algeria; 4Nono Institute of Electronic Engineering, University Malaysia Perlis, Kangar, Malaysia
Email: *lttnsameri@yahoo.fr.
Received August 28th, 2012; revised September 27th, 2012; accepted October 26th, 2012
Keywords: SrS; BaS; CaS; Semiconductors; FP-LMTO; Alloys; Bowing Parameter
ABSTRACT
The structural and the electronic properties of the ternary SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys have been calculated using the full-potential linear muffin-tin-orbital (FP-LMTO) method based on density functional theory, within both local density approximation (LDA) and generalized gradient approximation (GGA). The calculated equilibrium lattice constants and bulk modulus are compared with previous results. The concentration dependence of the electronic band structure and the direct and indirect band gaps are investigated. Using the approach of Zunger and co-workers, the microscopic origins of the band gap bowing are investigated also. A reason is found from the comparison of our results with other theoretical calculations.
1. Introduction
The II-VI compound semiconductors (AY: A = Ca, Sr, Ba; Y = S) have recently received considerable interest from the blue to the near-ultraviolet spectral region of both experimental and theoretical points of view [1]. This was due to their potential technological applications, in light-emitting diodes (LEDs) and laser diodes (LDs) [2]. Many experimental and theoretical works were reported for II-VI chalcogenides, calcium chalcogenides [3,4], strontium chalcogenides, and beryllium chalcogenides [5]. The alkaline earth chalcogenides form a closed-shell ionic system crystallized in the rocksalt type structuretype (B1) at ambient conditions. They are technologically important materials, with applications in the area of luminescent devices, radiation dosimetry, fast high-resolution optically stimulated luminescence imaging, and infrared sensitive devices [6-8]. Group II-VI semiconductors and their heterostructures are well known to form ternary alloys with a direct fundamental band gap over most of the alloy composition range with high absorption coefficients. They are used as which can be used as materials for fabricating thin film heterojunction photovoltaic (PV) devices. The principal energy gaps of ternary II-VI semiconductor alloys can cover various light spectra over the entire composition, and lattice parameters can be tailored independently to fabricate photovoltaic devices on suitable lattice matched substrates [9]. The use of II-VI semiconductor alloys for device applications requires: better controlling of the process conditions and parameters during the material growth and device fabrication, reliable and accurate modeling simulation of structural and electronic properties of heterostructures as a function of interface strain and composition [10]. The principal energy gaps of ternary II-VI semiconductor alloys can cover various light spectra over the entire composition, and lattice parameters can be tailored independently to fabricate photovoltaic devices on suitable lattice matched substrates [11]. Therefore, the goal of this work is to calculate the structural and the electronic properties by using the full-potential linear muffin-tin-orbital (FP-LMTO) method within the local density approximation (LDA), and two newly developed refinements, named the generalized gradient approximation (GGA) of Perdew et al. The physical origins of gap bowing are calculated following the approach of Zunger et al. (and workers) [12]. In this approach, the alloys are is studied in an ordered structure (we use here a cubic super cell of eight atoms) designed to reproduce the most important pair-correlation functions of random disordered alloys and where the chemical and structural effects are captured very well. The paper is organized as follows. The computational method that we have adopted for the calculation is described in Section 2. We present our results in Section 3. Finally, conclusions are given in Section 4.
2. Method of Calculations
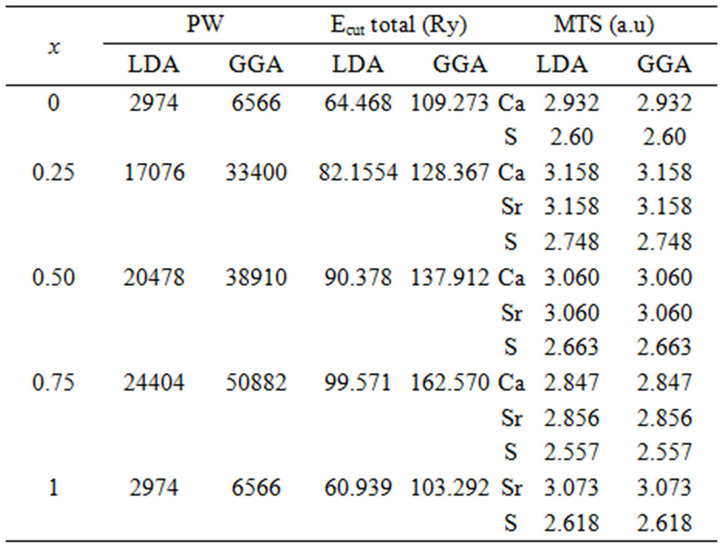
The calculations reported here were carried out using the ab-initio full-potential linear muffin-tin orbital (FPLMTO) method [13-16] as implemented in the Lmtart code [17]. The exchange and correlation potential were calculated using the local density approximation (LDA) [18] and the generalized approximation (GGA) [19]. This is an improved method compared to previous (LMTO) methods. The FP-LMTO method treats muffin-tin spheres and interstitial regions on the same footing, leading to improvements in the precision of the eingen-values. At the same time, the FP-LMTO method, in which the space is divided into an interstitial regions (IR) and non overlapping muffin-tin spheres (MTS) surrounding the atomic sites use more complete basis than its predecessors. The space in this method is divided into non-overlapping muffin-tin (MT) spheres centered at the atomic sites separated by an interstitial region (IR). In the IR regions, the basis set consists of plane waves. Inside the MT spheres, the basis sets is described by radial solutions of the one particle Schrödinger equation (at fixed energy) and their energy derivatives multiplied by spherical harmonics. The charge density and the potential are represented inside the MTS by spherical harmonics up to lmax = 6. The integrals over the Brillouin zone are performed up to 35 special k-points for binary compounds and 27 special k-points for the alloys in the ireducible Brillouin zone (IBZ), using the Blöchl’s modified tetrahedron method [20]. The self-consistent calculations are considered to be converged when the total energy of the system is stable within 10–5 Ry. In order to avoid the overlap of atomic spheres, the MTS radius for each atomic position is taken to be different for each composition. We point out that the use of the full-potential calculation ensures that the calculation is not completely independent of the choice of sphere radii. Both plane waves cut-off are varied to ensure the total energy convergence. The values of the sphere radii (MTS) number of plane waves (NPLW), used in our calculation are summarized in Table 1.
3. Results and Discussions
3.1. Structural Properties
The structural properties of CaS, SrS and BaS binary compounds and their SrxCa1-xS, BaxCa1-xS, BaxSr1-xS alloys in the cubic structure by means of the full-potential LMTO method are calculated. As for the ternary semiconductor alloys with the type of BxA1-xC, we have started our FP-LMTO calculations of the structural properties with the rocksalt structure and let the calculation
 (a)
(a)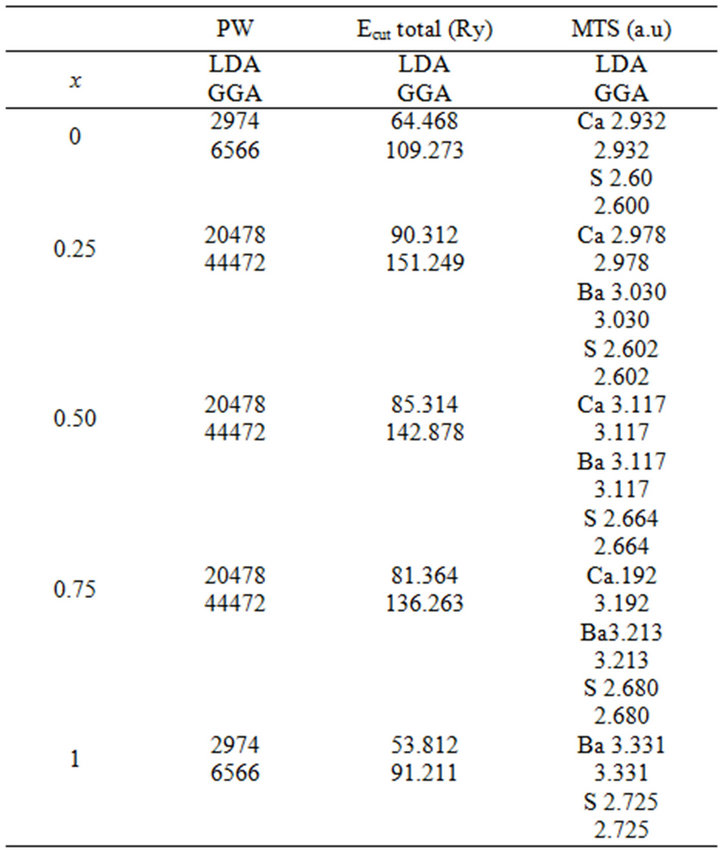 (b)
(b)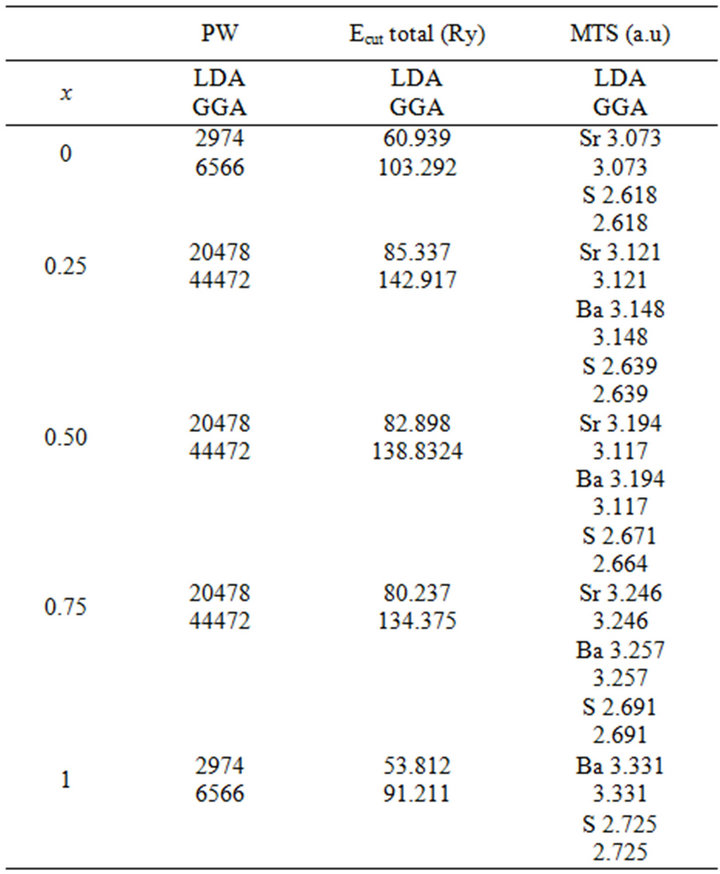 (c)
(c)
Table 1. (a) The plane wave number PW, energy cutoff (in Ry) and the muffin-tin radius (MTS) (in a.u.) used in calculation for binary SrS and CaS and their SrxCa1-xS, BaxCa1-xS, BaxSr1-xS, alloy in the rocksalt structure (B1);(b) binary BaS and CaS and their BaxCa1-xS alloy in in the rocksalt structure (B1); (c) binary BaS and CaS and their BaxSr1-xS alloy in in the rocksalt structure (B1).
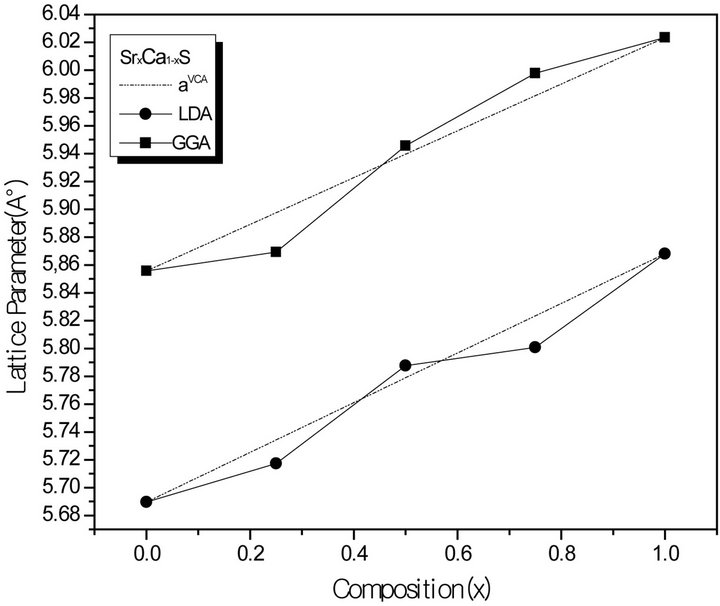
forces move the atoms to their equilibrium positions. We have chosen the basic cubic cell as the unit cell. In the unit cell, there are four C anions, three A and one B, two A and two B, one A and three B cations, for x = 0.25, 0.50 and 0.75, respectively. For the considered structures, we have performed the structural optimization by calculating the total energies for different volumes around the equilibrium cell volume V0 of CaS, SrS and BaS binary compounds and their alloys. The calculated total energies are fitted to the Murnaghan’s equation of state [21] to determine the ground state properties such as the equilibrium lattice constant a and the bulk modulus B. The calculated equilibrium parameters (a and B) are given in Table 2 that also contains results of previous calculations as well as the available experimental data. As a whole, our calculated structural parameters are in good agreement with those obtained by first-principles methods within different approximations. The calculated lattice parameter for CaS, SrS and BaS binary compound is in good agreement with other data, which ensures the reliability of the present first-principles computations. The computed lattice parameter slightly underestimates and overestimates the other data by using LDA and GGA, respectively, which are consistent with the general trend of these approximations. As it can be seen, the calculated lattice parameter for BaS is larger than those of SrS and CaS. As the anion atom is the same in the compounds, this result can be easily explained by considering the

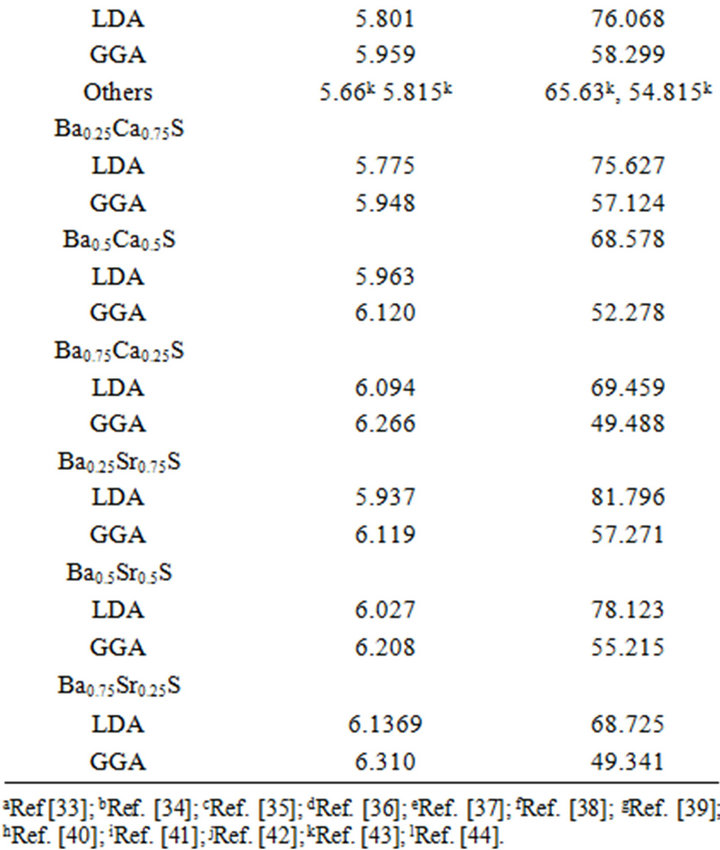
Table 2. Calculated lattice parameter a and bulk modulus B compared to experimental and other theoretical results of CaS, SrS and BaS and their SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys in the rocksalt structure (B1).
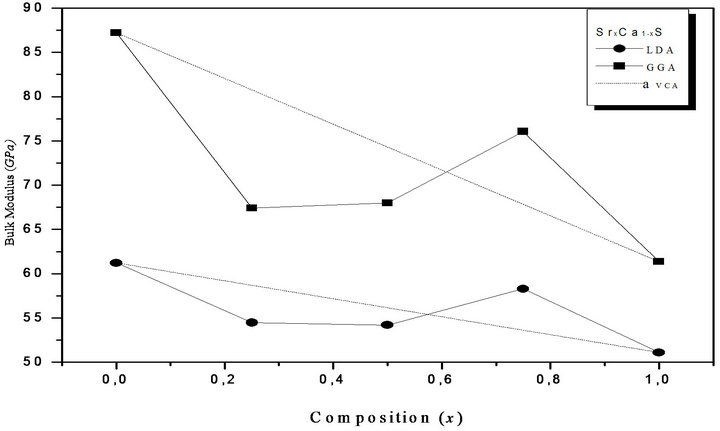
atomic radii of Ca, Sr, and Ba: R(Ca) = 1.80 Å, R(Sr) = 2.00 Å, R(Ba) = 2.15 Å; i.e. the lattice constant increases with increasing atomic size of the cation element. The bulk modulus value for CaS is larger than those of SrS and BaS; B (CaS) > B (SrS) > B (BaS); i.e. in inverse sequence to a0 in agreement with the well-known relationship between B and the lattice constants: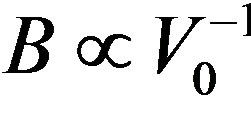 , where
, where 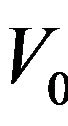 is the unit cell volume. Furthermore, the values of the calculated bulk modulus using both approximations decrease in going from CaS, to SrS, and to BaS suggesting that the compressibility increases from CaS, to SrS, and to BaS. Usually, in the treatment of alloys where the experimental data are rare, it is assumed that the atoms are located at the ideal lattice sites and the lattice constant varies linearly with composition x according to the Vegard’s law [22].
is the unit cell volume. Furthermore, the values of the calculated bulk modulus using both approximations decrease in going from CaS, to SrS, and to BaS suggesting that the compressibility increases from CaS, to SrS, and to BaS. Usually, in the treatment of alloys where the experimental data are rare, it is assumed that the atoms are located at the ideal lattice sites and the lattice constant varies linearly with composition x according to the Vegard’s law [22].
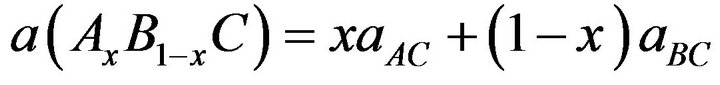 (2)
(2)
where aAC and aBC are the equilibrium lattice constants of the binary compounds AC and BC respectively, a(Ax B1-x C) is the alloy lattice constant. However, variation of Vegard’s law has been observed in semiconductor alloys both experimentally [23] and theoretically [24-28]. Hence, the lattice constant can be written as:
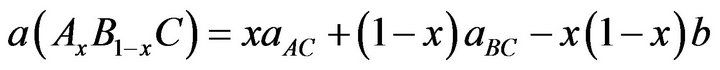 (3)
(3)
where the quadratic term b is the bowing parameter. Figures 1(a)-(c) and 2(a)-(c) show the variation of the calculated equilibrium lattice constant and bulk modulus as a function of concentrations x for SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys. The obtained results for the composition dependence of the calculated equilibrium lattice parameter exhibit an agreement with Vegard’s law [22]. In going from CaS, SrS and BaS; when the Sr and Bacontent increases, the values of the lattice parameters of the SrxCa1-xS, BaxCa1-xS, BaxSr1-xS alloys increases. Oppositely, one can see from Figure 2 that the value of the bulk modulus increases with the decrease of Sr and Ba concentrations. The bowing parameters are determined by a polynomial fit. From Tables 3(a)-(c), both approxiations follow the tendency demonstrated by both experimental measurement and theoretical calculations.
3.2. Electronic Properties
The calculated band structure energies for binary compounds as well as their investigated alloys using FPLMTO within both LDA and GGA schemes yields indirect band gap (Γ→X) for the binary compounds of CaS, SrS and BaS. When the composition x varies, the valence band maximum (VBM) and the conduction band minimum (CBM) occur at the Γ-point, resulting in direct band gap (Γ→Γ) for the studied alloys. The calculated direct band gap (Γ→Γ) values for SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys are listed in Table 4, along with the available theoretical and experimental results. The computed direct band gaps for the binary compounds are in good agreements with other results. For SrS and CaS compounds, the calculated band gap is smaller than the experimental value but for the BaS is the opposite. To the
 (a)
(a)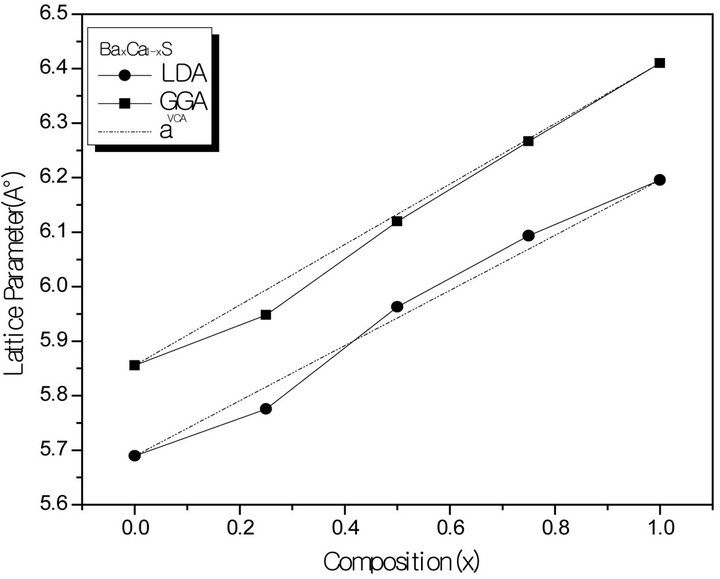 (b)
(b)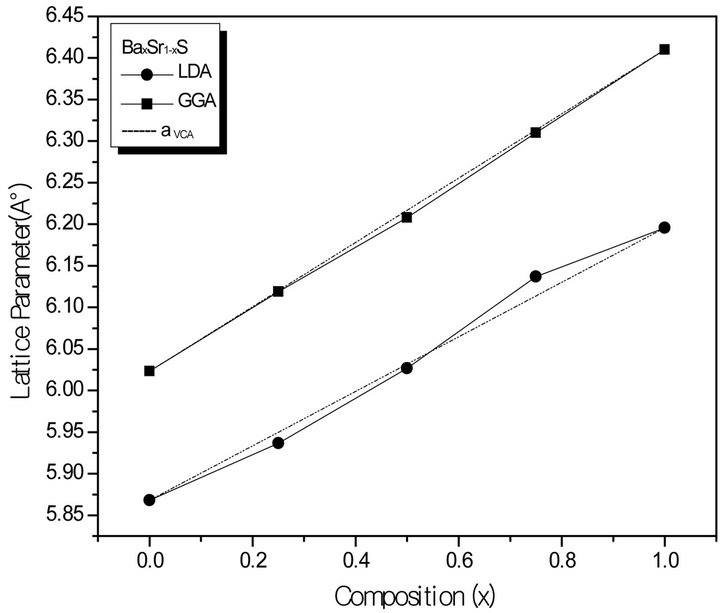 (c)
(c)
Figure 1. Composition dependence of the calculated lattice constants within GGA (solid squares) and LDA (solid circle) of (a) SrxCa1-xS; (b) BaxCa1-xS, (c) BaxSr1-xS alloys compared with Vegard’s prediction (dot line).
 (a)
(a)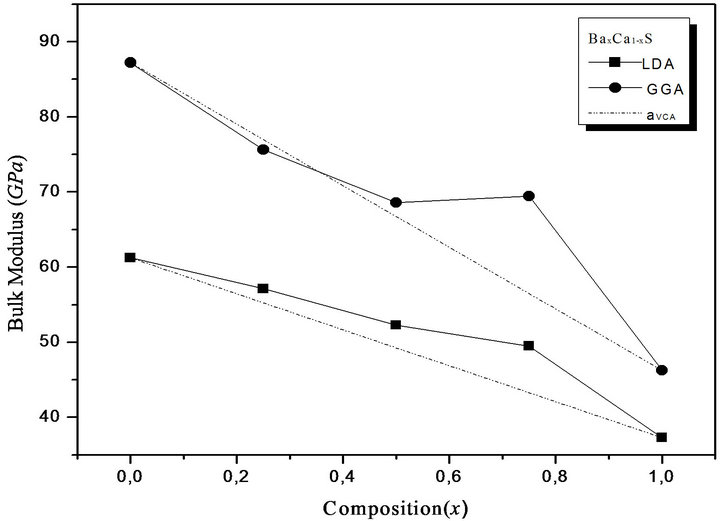 (b)
(b)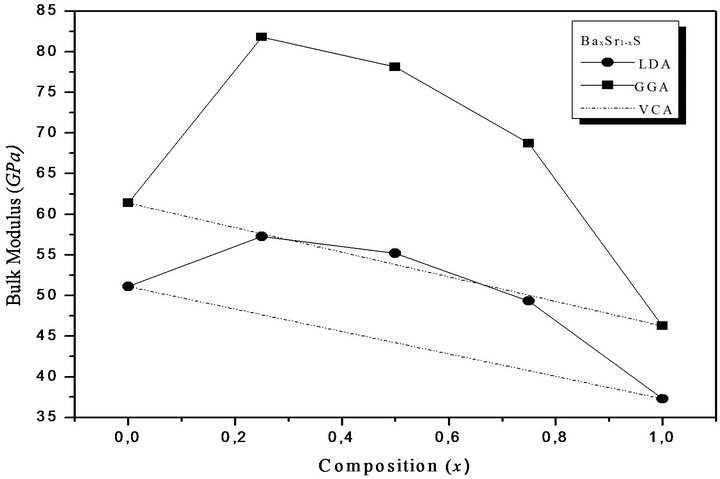 (c)
(c)
Figure 2. Composition dependence of the calculated bulk modulus within GGA (solid circle) and LDA (solid squares) of (a) SrxCa1-xS; (b) BaxCa1-xS; (c) BaxSr1-xS alloys compared with Vegard’s prediction (dot line).
best of our knowledge, there are no theoretical or experimental data on the energy band gaps for x = 0.25, 0.5 and 0.75 available in literature to make a meaningful comparison. It is worthy to mention here that both LDA and GGA do not reproduce a perfect agreement with the experimental results because they do not take into account the quasi particle self energy correctly [32] that make them not sufficiently flexible to accurately reproduce both the exchange-correlation energy and its charge derivative. It is important to note that the density functional formalism is limited [30] and the derived band
 (a)
(a)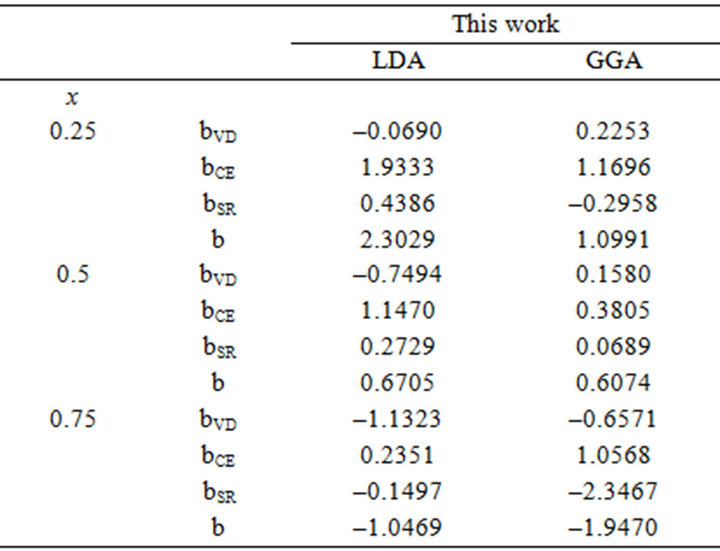 (b)
(b)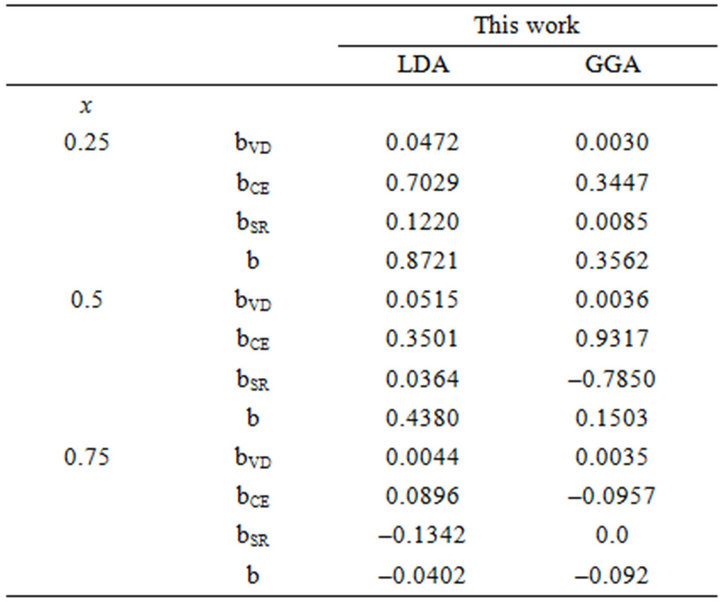 (c)
(c)
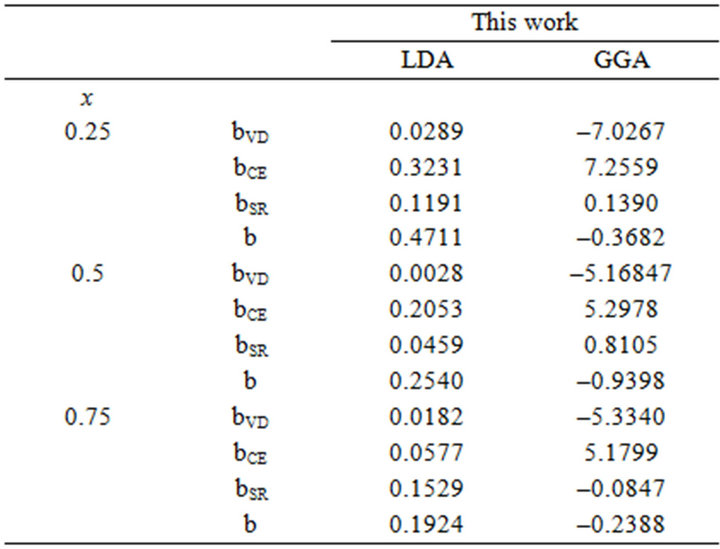
Table 3. Decomposition of optical bowing into volume deformation (VD), charge exchange (CE) and structural relaxation (SR) contributions (all values in eV) for (a) SrxCa1-xS; (b) BaxCa1-xS; (c) BaxSr1-xS.
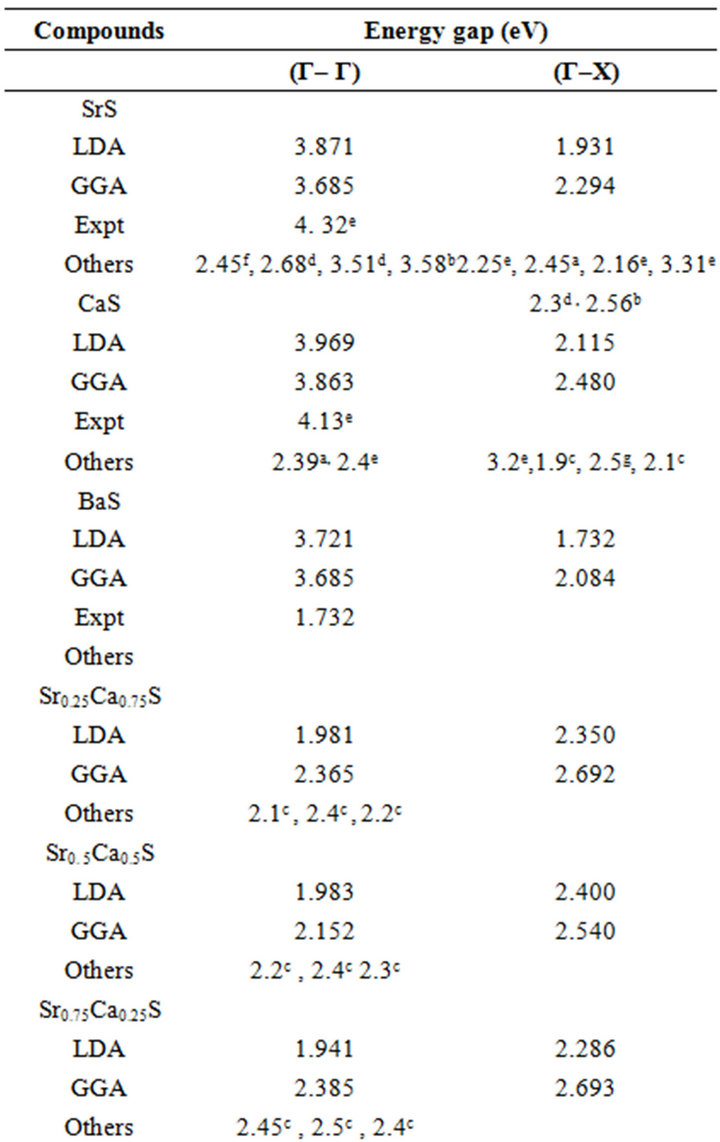
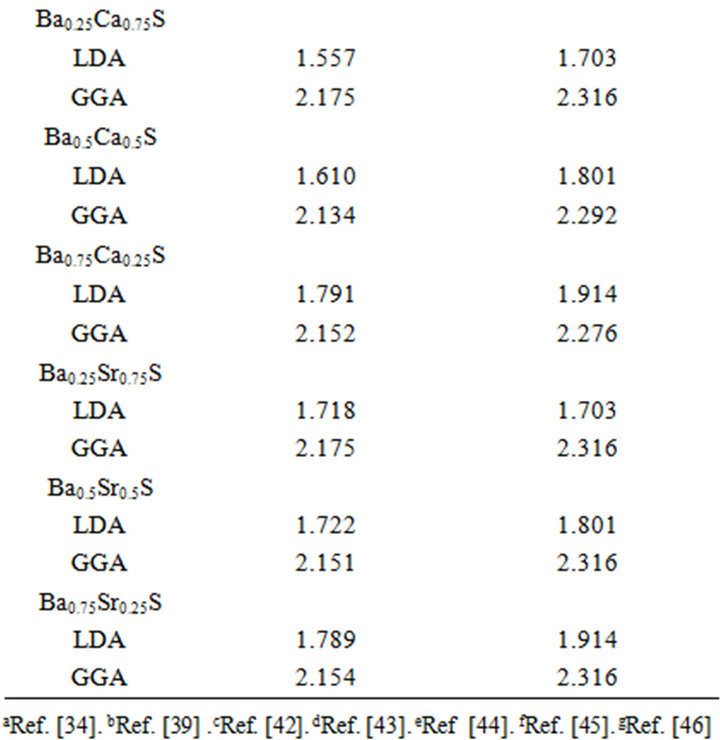
Table 4. Direct and indirect band gap energy of SrxCa1-xS, BaxCa1-xS and BaxSr1-xS at different CaS, SrS and BaS concentrations (all values are in eV).
structure can’t be used directly for comparison with experiment. We also mention, it is far to say that the experimental data are well reproduced by calculations. On For this reason for this that the theory holds applies pertains at T = 0 K, whereas the experimental measurements were performed at room temperature where the lattice vibrations are present.
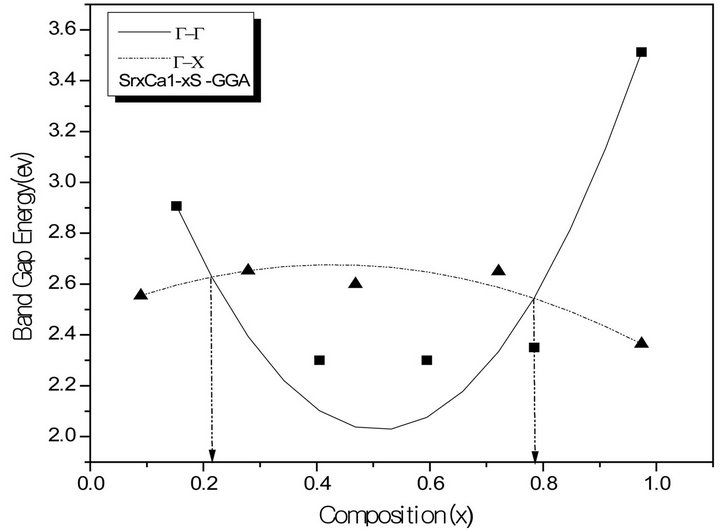
The variation of the composition (x) versus the direct (Γ→Γ) and indirect (Γ→X) band gaps with both LDA and GGA approximations is shown in Figures 3(a)-(c). According to this figure we notice that for concentrations (x) ranging from x = 0.224 to 0.769 LDA, x = 0.216 to 0.784 GGA the alloy exhibits a direct band gap (Γ→Γ) for both approximations SrxCa1-xS, from x = 0.26 to 0.73 LDA, x = 0.25 to 0.73 GGA the alloy exhibits a direct band gap (Γ→Γ) for both approximations BaxSr1-xS, from x = 0.27 to 0.73 LDA, x = 0.25 to 0.73 GGA the alloy exhibits a direct band gap (Γ→Γ) for both approximations BaxSr1-xS,. It is also seen that the direct and indirect band gaps show a nonlinear behaviour with increasing Strontium, Calcium and Baryum concentrations providing a positive and negative band gap bowing for the direct (Γ→Γ) and indirect (Γ→X) band gaps, respectively. Indeed, it is a general trend to describe the band gap of AxB1-xC alloys in terms of the binary compounds energy gaps EAC and EBC by the following semi-empirical formula:
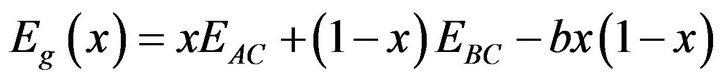 (4)
(4)
where EAC and EBC corresponds to the energy gap of SrS and CaS for the SrxCa1-xS alloy. From Table 4, it is clear that Eg of LDA is slightly larger than those of GGA.
The calculated band gap versus concentration was fitted by a polynomial equation. The results are shown in Figure 3 and are summarized as follows:
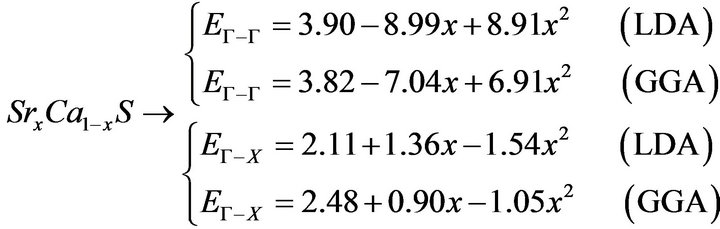 (5)
(5)
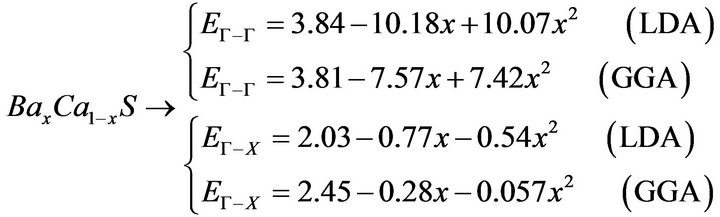 (6)
(6)
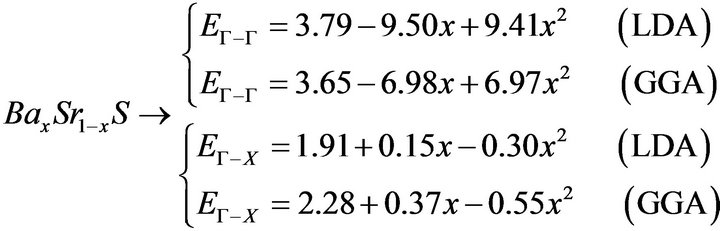 (7)
(7)
 (a)
(a)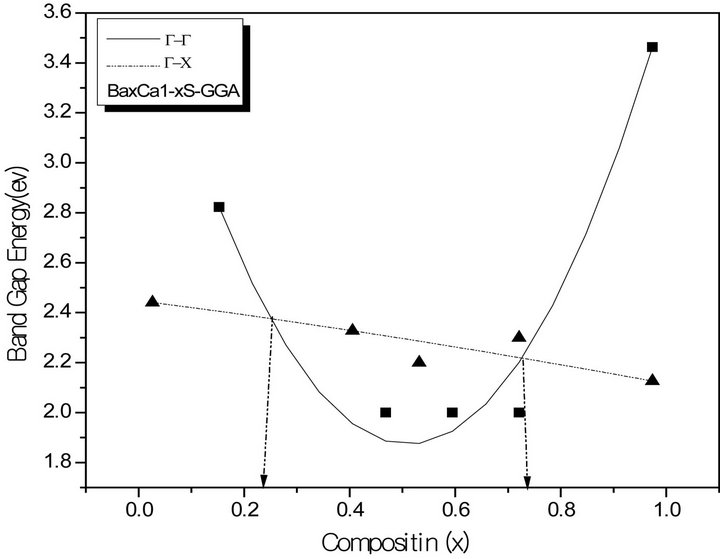 (b)
(b)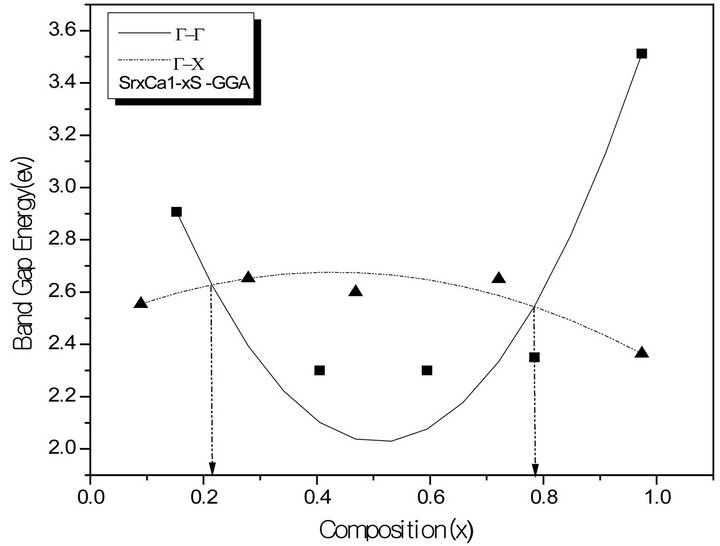 (c)
(c)
Figure 3. The calculated direct (G→G) and indirect (G→X) band gaps of (a) SrxCa1-xS alloy as a function of Sr, Ca, and concentrations using LDA (a) and GGA (b) schemes; (b) BaxCa1-xS, as a function of Ba and Ca concentrations using LDA (a) and GGA (b) schemes; (c) BaxSr1-xS alloy as a function of Ba and Sr concentrations using LDA (a) and GGA (b) schemes.
In order to understand the physical origins of bowing parameters in AxB1-xC alloy, we follow the procedure of Bernard and Zunger [31], and decompose the total bowing parameter b into physically distinct contributions. The overall bowing coefficient at a given average composition x measures the change in the band gap according to the formal reaction.
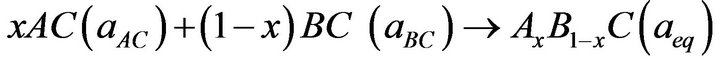 (6)
(6)
where 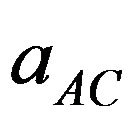 and
and 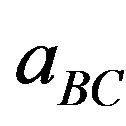 are the equilibrium lattice constants of the binary compounds and
are the equilibrium lattice constants of the binary compounds and 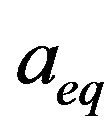 is the equilibrium lattice constant of the alloy with the average composition x.
is the equilibrium lattice constant of the alloy with the average composition x.
Equations (5)-(7) are decomposed into three steps:
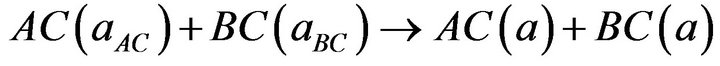 (8)
(8)
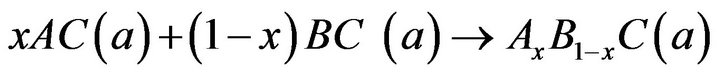 (9)
(9)
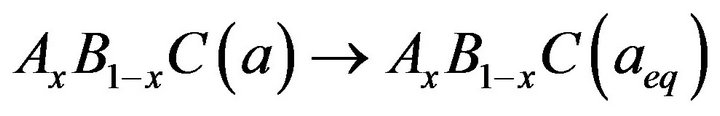 (10)
(10)
The first step attributes the volume deformation (VD) effect on the bowing. The corresponding contributions bVD to the bowing parameter represents the relative response of the band structure of the binary compounds AC and BC to hydrostatic pressure that arises from the change of their individual equilibrium lattice constants to the alloy value a = a(x). The second contribution, the charge exchange (CE) contribution bCE, reflects a charge transfer effect which is due to different (averaged) bonding behaviour of at the lattice constant a. The final step, the structural relaxation (SR), reflects changes in passing from the unrelaxed to the relaxed alloy by bSR. Consequently, the total bowing parameter is defined as.
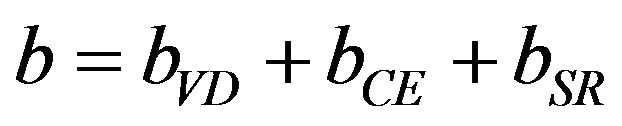 (11)
(11)
The general representation of the composition-dependent band gap of the alloys in terms of binary compounds energy gaps of the, EAC(aAC) and EBC(aBC), and the total bowing parameter b is
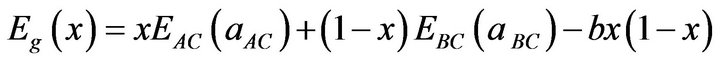 (12)
(12)
This allows a division of the total bowing b into three contributions according to:
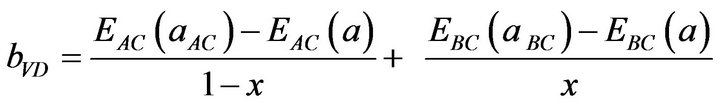 (13)
(13)
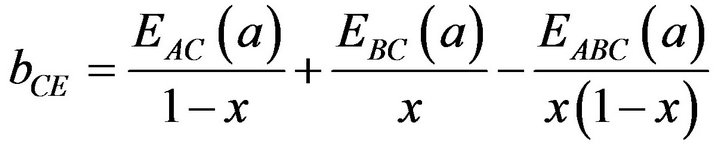 (14)
(14)
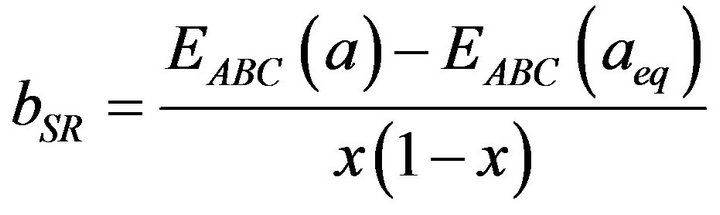 (15)
(15)
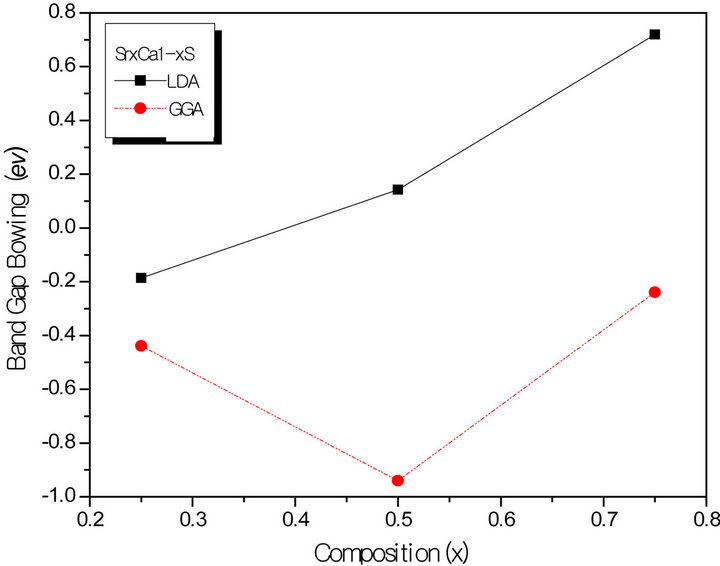
All these energy gaps mentioned in (13)-(15) have been calculated for the indicated atomic structures and lattice constants. Table 3(a) shows the calculated optical band gap bowing b for SrxCa1-xS for three different molar fractions (x = 0.25, 0.5 and 0.75). The LDA (GGA) calculated band gap bowing (b) is found to be equal to –0.3185 (–0.7746) eV, 0.078 (0.0214) eV and 4.0222 (3.6608) eV for x = 0.25, 0.5 and 0.75 respectively. From Table 3(a) we notice that the structural relaxation effect is negligible for the studied compositions x = 0.25, 0.50 and 0.75 and the main contribution to the band gap bowing is essentially due to the charge exchange (CE) contribution, for x = 0.75 the band gap bowing is due to nearly equal contribution of the volume deformation (SR) effects. Table 3(b) shows the calculated optical band gap bowing b for BaxCa1-xS for three different molar fractions (x = 0.25, 0.5 and 0.75). The LDA (GGA) calculated band gap bowing (b) is found to be equal to 2.3029 (1.0991) eV, 0.6705 (0.6074) eV and –1.0469 (–1.947) eV for x = 0.25, 0.5 and 0.75 respectively. From Table 3(b) we notice that the structural relaxation effect is negligible for the studied compositions x = 0.25, 0.50 and 0.75 and the main contribution to the band gap bowing is essentially due to the charge exchange (CE) contribution, for x = 0.25 and 0.5. For x = 0.75 the band gap bowing is due to nearly equal contribution of the volume deformation (VD) effects. Table 3(c) shows the calculated optical band gap bowing b for BaxSr1-xS for three different molar fractions (x = 0.25, 0.5 and 0.75). The LDA (GGA) calculated band gap bowing (b) is found to be equal to 0.8721 (0.3562) eV, 0.438 (0.1503) eV and –0.0402 (–0.0922) eV for x = 0.25, 0.5 and 0.75 respectively. From Table 3(c) we notice that the structural relaxation effect is negligible for the studied compositions x = 0.25, 0.50 and 0.75 and the main contribution to the band gap bowing is essentially due to the charge exchange (CE) contribution for x = 0.25 and 0.5. For x = 0.75 the band gap bowing is due to nearly equal contribution of the volume deformation (SR) effects. Figure 4(a) shows the variation of the band gap bowing versus concentration. It is shown that the optical bowing remains linear and increases slowly in going from 0.25 to 0.75, for LDA and for GGA it remains linear varies rapidly in going from 0.25 to 0.5, and beyond 0.5 it increases rapidly too, which confirms the evaluation of the band gap according to the concentration,. Figure 4(b) for BaxCa1-xS shows the variation of the band gap bowing versus concentration. It is shown that the optical bowing remains linear (in going) from 0.25 to 0.75 for LDA and for GGA it remains linear with a tow slope varying slowly (in going) from 0.25 to 0.5, and beyond 0.5 it decreases rapidly. Figure 4(c) for BaxSr1-xS shows the variation of the band gap bowing versus concentration. It is shown that the optical bowing remains linear, it monotonicly decreases from 0.25 to 0.75 both for LAD, GGA with two slopes. To the best of our knowledge, there are no theoretical or experimental data on the band gap bowing to compare with our predicted results.
3.3. Effective Masses
Knowledge of the electron and hole effective mass values is indispensable for the understanding of transport phenomena, exciton effects and electron-hole in semiconductors. Excitonic properties are of great interest for semiconductor materials; therefore, it is worthwhile to estimate the electron and hole effective mass values for SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys at different compositions. Experimentally, the effective masses are usually determined by cyclotron resonance, electro reflectance measurements or from the analysis of transport data or transport measurements [32]. Theoretically, the effective masses can be estimated from the energy band curvatures. Generally, the effective mass is a tensor with nine components, however for the much idealized simple case, where the E-k diagram can be fitted by a parabola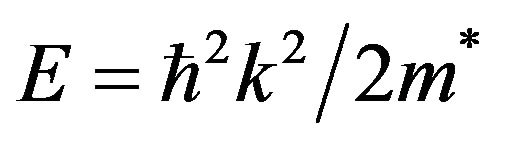 , the effective mass becomes a scalar at high symmetry point in Brillouin zone. We have computed the electron effective mass at the conduction band minima (CBM) and the hole effective mass at the valence
, the effective mass becomes a scalar at high symmetry point in Brillouin zone. We have computed the electron effective mass at the conduction band minima (CBM) and the hole effective mass at the valence
 (a)
(a)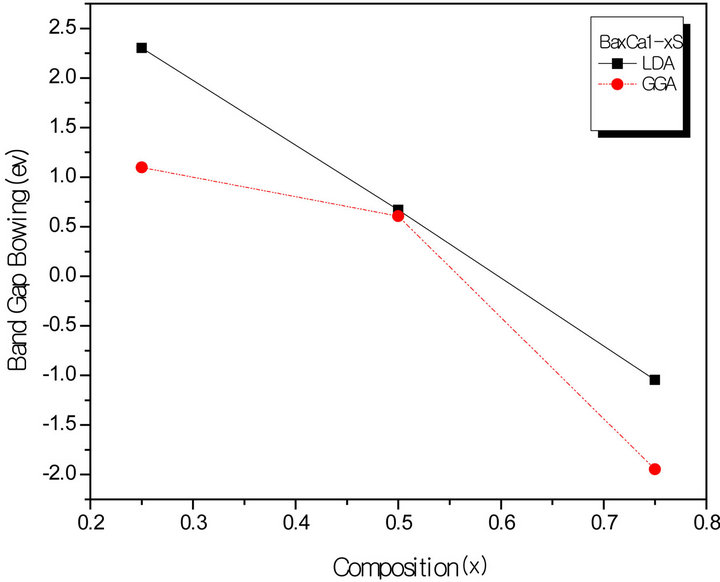 (b)
(b)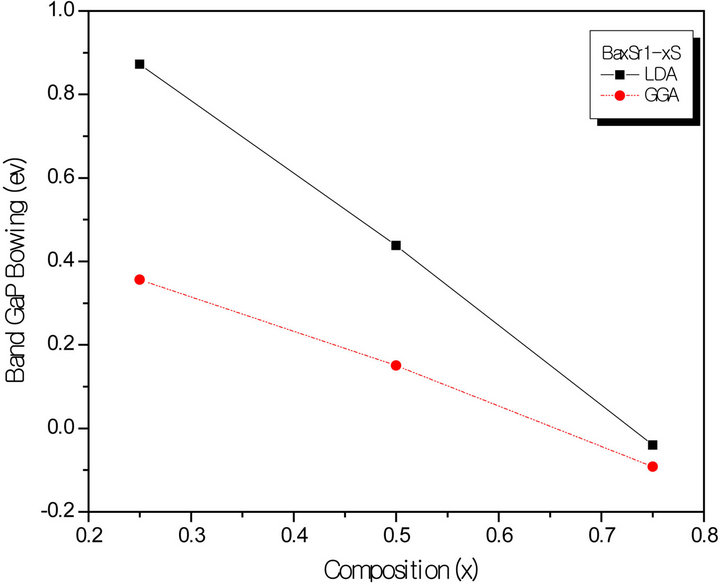 (c)
(c)
Figure 4. The calculated optical bowing parameters as a function of (a) Sr and Ca; (b) Ba and Ca; (c) Ba and Sr, concentrations within LDA and GGA schemes.
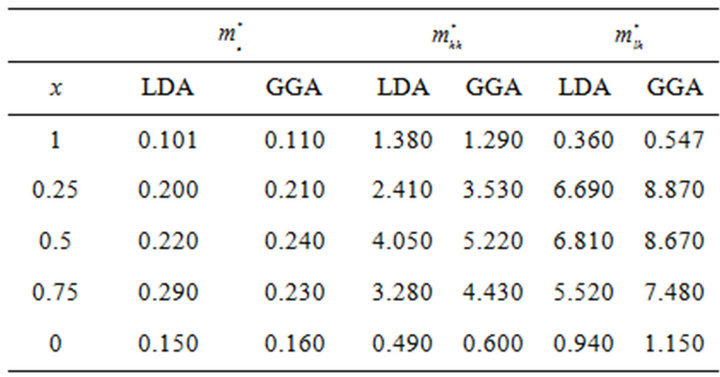
band maxima (VBM) for the composition ranging from 0 to 1.0 for SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys. The electron effective mass value are obtained from the curvature of the conduction band near the X-point for SrS ,BaS and CaS and near the Γ-point at the CBM for X = 0.25, 0.5 and 0.75. The hole effective masse value is calculated from the curvature near the Γ-point at the VBM for all concentration. The calculated electron and hole effective mass values for for SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys are mentioned in Tables 5(a)-(c).
 (a)
(a)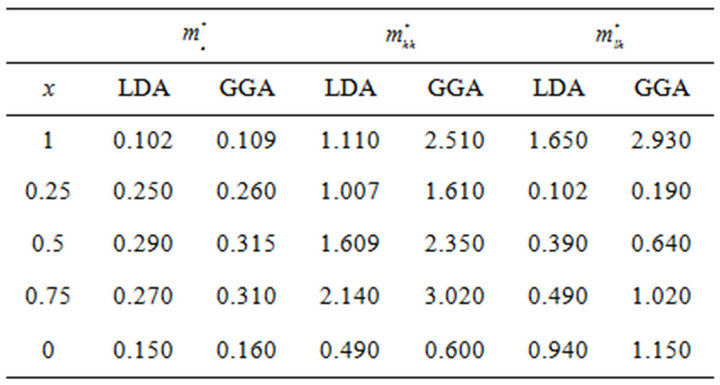 (b)
(b)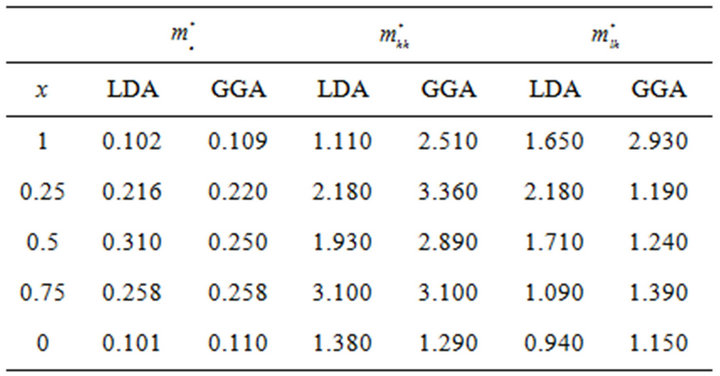 (c)
(c)
Table 5. Electron (me*), heavy hole (mhh*) and light hole (mlh*) effective masses (in units of free electron mass m0) of the ternary (a) SrxCa1-xS; (b) BaxCa1-xS; (c) BaxSr1-xS alloy using LDA and GGA schemes.
4. Conclusions
The (FP-LMTO) method is used to calculate the structural and electronic properties of the rocksalt SrS, CaS, BaS and their SrxCa1-xS, BaxCa1-xS and BaxSr1-xS alloys.From this, we may conclude that:
1) The variation of the structural parameters with Sr, Ca and Ba concentrations obeys Vegard’s law.
2) The electronic band structure shows a non-linear variation of the fundamental band gaps versus Sr, Ca and Ba concentrations. The deviation from linearity is characterized by a calculated optical bowing parameter. The main contribution to the optical bowing is essentially due to volume deformation effects.
3) The effective masses indicate that the charge carriers in these alloys should be dominated by electrons.
REFERENCES
- S. Labidia, H. Meradji, M. Labidia, S. Ghemid, S. Drablia and F. El H. Hassan, “First Principles Calculations of Structural, Electronic and Thermodynamic Properties of SrS,SrSe,SrTe Compounds and SrS1-xSex Alloy”, Physics Procedia, Vol. 2, No. 3, 2009, pp. 1205-1212. doi:10.1016/j.phpro.2009.11.083
- M. A. Haase, J. Qiu, J. M. DePuydt and H. Cheng, “BlueGreen Laser Diodes,” Applied Physics Letters, Vol. 59, No. 11, 1991, p. 1272.
- A. Yamasaki and T. Fujiwara, “Electronic Structure of Oxides MO (M = Mg, Ca, Ti, V) in the GW Approximation,” Physical Review B, Vol. 66, 2002, Article ID: 245108.
- Z. Charifi, H. Baaziz, F. El H. Hassan and N. Bouarissa, “High Pressure Study of Structural and Electronic Properties of Calcium Chalcogenides,” Journal of Physics: Condensed Matter, Vol. 17, No. 26, 2005, p. 4083. doi:10.1088/0953-8984/17/26/008
- V. Wagner, et al., “Lattice Dynamics and Bond Polarity of Be-Chalcogenides A New Class of II-VI Materials,” Physica Status Solidi B, Vol. 215, No. 1, 1999, pp. 87-91. doi:10.1002/(SICI)1521-3951(199909)215:1<87::AID-PSSB87>3.0.CO;2-D
- Y. Nakanishi, T. Ito, Y. Hatanaka and G. Shimaoka, “First-Principles Study of Structural, Electronic and Optical Properties of SrS1−xSex Alloys,” Applied Surface Science, Vol. 65-66, 1992, pp. 515-519. doi:10.1016/0169-4332(93)90712-K
- S. Asano, N. Yamashita and Y. Nakao, “Luminescence of the Pb2+-Ion Dimer Center in CaS and CaSe Phosphors,” Physica Status Solidi B, Vol. 89, No. 2, 1978, pp. 663- 673. doi:10.1002/pssb.2220890242
- R. Pandey and S. Sivaraman, “Spectroscopic Properties of Defects in Alkaline-Earth Sulfides,” Journal of Physics and Chemistry of Solids, Vol. 52, No. 1, 1991, pp. 211- 225.
- Ö. Akinci, H. H. Gürel and H. Ünlü, “Semi-Empirical Tight Binding Modelling of CdSTe/CdTe, ZnSSe/ZnSe and ZnSSe/CdSe Heterostructures,” Thin Film Chalogenide Photovoltaic Materials, Vol. 517, No. 7, 2009, pp. 2431-2437.
- H. Morko, C. H. UnlU and G. Ji, “Principles & Technology of MODFETs,” Wiley, New York, 1991.
- “Thin Film Chalcogenide Photovoltaic Materials,” In: R. Scheer, J.-F. Guillemoles, A. N. Tiwari, et al., Eds., Society Symposium Proceedings on Ferekides European Materials Research, 2008.
- A. Zunger, S.-H. Wei, L. G. Ferreira and J. E. Bernard, “Special Quasirandom Structures,” Physical Review Letters, Vol. 65, No. 3, 1990, pp. 353-356. doi:10.1103/PhysRevLett.65.353
- S. Savrasov and D. Savrasov, “Full-Potential LinearMuffin-Tin-Orbital Method for Calculating Total Energies and Forces,” Physical Review B, Vol. 46, 1992, pp. 2181-2195.
- S. Y. Savrasov, “Linear-Response Theory and Lattice Dynamics: A Muffin-Tin-Orbital Approach,” Physical Review B, Vol. 54. 1996, pp. 16470-16486.
- P. Hohenberg and W. Kohn, “Inhomogeneous Electron GaS,” Physical Review, Vol. 136, 1964, pp. B864-B871
- W. Kohn and L. J. Sham, “Self-Consistent Equations Including Exchange and Correlation Effects,” Physical Review, Vol. 140, No. 4A, 1965, pp. 1133-1138. doi:10.1103/PhysRev.140.A1133
- S. Y. Savrasov, “Program LMTART for Electronic Structure Calculations”, Zeitschrift fur Kristallographie, Vol. 220, No. 5-6, 2005, pp. 555-557. doi:10.1524/zkri.220.5.555.65067
- J. P. Perdew and Y. Wang, “Pair-Distribution Function and Its Coupling-Constant Average for the Spin-Polarized Electron Gas,” Physical Review B, Vol. 46, No. 20, 1992, pp. 12947-12954. doi:10.1103/PhysRevB.46.12947
- J. P. Perdew, S. Burke and M. Ernzerhof, “Generalized Gradient Approximation Made Simple,” Physical Review Letters, Vol.77, No. 18, 1996, pp. 3865-3868.
- P. Blochl, O. Jepsen and O. K. Andersen, “Improved Tetrahedron Method for Brillouin-Zone Integrations,” Physical Review B, Vol. 49, No. 23, 1994, pp. 16223-16233. doi:10.1103/PhysRevB.49.16223
- F. D. Murnaghan, “The Compressibility of Media under Extreme Pressures,” Proceedings of the National Academy of Sciences USA, Vol. 30, 1944, pp. 244-247. doi:10.1073/pnas.30.9.244
- L. Vegard, “Formation of Mixed Crystals by Solid-Phase Contact”, Zeitschrift fur Kristallographie, Vol. 5, No. 5, 1921, pp. 393-395. doi:10.1007/BF01327675
- B. Jobst, D. Hommel, U. Lunz, T. Gerhard and G. Landwehr, “E0 Band-Gap Energy and Lattice Constant of Ternary Zn1−xMgxSe as Functions of Composition,” Applied Physics Letters, Vol. 69, No. 1, 1996, pp. 97-100. doi:10.1063/1.118132
- F. El H. Hassan and H. Akbardadeh, “First-principles Investigation of BNxP1-x , BNxAs1-x and BPxAs1-x Ternary Alloys,” Materials Science and Engineering B, Vol. 121, No. 1-2, 2005, pp. 170-177. doi:10.1016/j.mseb.2005.03.019
- Y. Al-Douri, “Structural Phase Transition of Boron Nitride Compound,” Solid State Communications, Vol. 132, No. 7, 2004, pp. 465-470. doi:10.1016/j.ssc.2004.08.020
- Y. Al-Douri, “Electronic and Positron Properties of ZincBlende Structure of GaN, AlN, and Their Alloy Ga1−xAlxN,” Journal of Applied Physics, Vol. 93, No. 12, 2003, pp. 9730-9736. doi:10.1063/1.1573739
- Y. Al-Douri, “Electronic and Optical Properties of ZnxCd1−xSe,” Materials Chemistry and Physics, Vol. 82, No. 1, 2003, pp. 49-54.
- Y. Al-Douri, S. Mecabih, N. Benosman and H. Aourag, “Pressure Effect on Electronic and Positron Charge Densities of Zn0.5Cd0.5Se,” Physica B, Vol. 325, No. 1-4, 2003, pp. 362-371.
- S. N. Rashkeev and W. R. L. Lambrecht, “Second-Harmonic Generation of I-III-VI2 Chalcopyrite Semiconductors: Effects of Chemical Substitutions,” Physical Review B, Vol. 63, No. 16, 2001, Article ID: 165212.
- G. Onida, L. Reining and A. Rubio, “Electronic Excitations: Density-Functional versus Many-Body Green’s-Function Approaches,” Reviews of Modern Physics, Vol. 74, No. 2, 2002, pp. 601-659.
- J. E. Bernard and A. Zunger, “Electronic Structure of ZnS, ZnSe, ZnTe, and Their Pseudobinary Alloys,” Physical Review B: Condens Matter, 1987, pp. 3199-3228.
- O. Zakharov, A. Rubio, X. Blase, M. L. Cohen and S. G. Louie, “Quasiparticle Band Structures of Six II-VI Compounds: ZnS, ZnSe, ZnTe, CdS, CdSe, and CdTe,” Physical Review B, Vol. 50, No. 15, 1994, pp. 10780-10787. doi:10.1103/PhysRevB.50.10780
- K. Syassen, “Pressure-Induced Structural Transition in SrS,” Physica Status Solidi (A), Vol. 91, No. 1, 1985, pp. 11-15. doi:10.1002/pssa.2210910102
- R. Khenata, H. Baltache, M. Rerat, M. Driz, M. Sahnoun, B. Bouhafs and B. Abbar, “First-Principle Study of Structural, Electronic and Elastic Properties of SrS, SrSe and SrTe under Pressure,” Physica B: Physics of Condensed Matter, Vol. 339, No. 4, 2003, pp. 208-215. doi:10.1016/j.physb.2003.07.003
- P. Cartona and P. Masri, “Cohesive Properties and Behaviour under Pressure of CaS, CaSe, and CaTe: Results of ab Initio Calculations,” Journal of Physics: Condensed Matter, Vol. 10, No. 40, 1998, pp. 8947-8955. doi:10.1088/0953-8984/10/40/003
- Z. Charifi, H. Baaziz, F. El H. Hassan and N. Bouarissa, “High Pressure Study of Structural and Electronic Properties of Calcium Chalcogenides,” Journal of Physics: Condensed Matter, Vol. 17, No. 26, 2005, p. 4083. doi:10.1088/0953-8984/17/26/008
- A. Bouhemadou, R. Khenata, F. Zegrar, M. Sahnoun, H. Baltache and A. H. Reshak, “Ab Initio Study of Structural, Electronic, Elastic and High Pressure Properties of Barium Chalcogenides,” Computational Materials Science, Vol. 38, No. 2, 2006, pp. 263-270. doi:10.1016/j.commatsci.2006.03.001
- S. Drablia, H. Meradji, S. Ghemid, G. Nouet and F. El H. Hassan, “First Principles Investigation of Barium Chalcogenide Ternary Alloys,” Computational Materials Science, Vol. 46, No. 2, 2009, pp. 376-382. doi:10.1016/j.commatsci.2009.03.013
- I. B. S. Banu, M. Rajagopalan, B. Palanivel, G. Kalpana, P. Shenbagaraman and J. L. Temp, “Structural and Electronic Properties of SrS, SrSe, and SrTe under Pressure,” Physics, Vol. 112, No. 3-4, 1998, pp. 211-226.
- S. Labidi, H. Meradji, S. Ghemid, M. Labidi and F. El H. Hassan, “FP-LAPW Investigations of SrS1−xSex, SrS1−xTex and SrSe1−xTex Ternary Alloys,” Journal of Physics: Condensed Matter, Vol. 20, No. 44, 2008, Article ID: 445213. doi:10.1088/0953-8984/20/44/445213
- S. Ekbundit, A. Chizmeshya, R. LaViolette and G. H. Wolf, “Experimental and Theoretical Investigation of the High-Pressure Behavior of CaS and MgS,” Journal of Physics: Condensed Matter, Vol. 8, No. 43, 1996, pp. 8251-8265. doi:10.1088/0953-8984/8/43/018
- A. Shaukat ,Y. Saeed, S. Nazir, N. Ikram and M. Tanveer, “Ab Initio Study of Structural, Electronic and Optical Properties of Ca1−xSrxS Compounds,” Physica B: Physics of Condensed Matter, Vol. 404, No. 21, 2009, pp. 3964- 3972. doi:10.1016/j.physb.2009.07.147
- Y. Kaneko and T. Koda, “New Developments in IIa-VIb (Alkaline-Earth Chalcogenide) Binary Semiconductors,” Journal of Crystal Growth, Vol. 86, No. 1-4, 1990, pp. 72-78. doi:10.1016/0022-0248(90)90701-L
- S. Yamaoka, O. Shimomura, H. Nakazawa and O. Fukunaga, “Pressure-Induced Phase Transformation in BaS,” Vol. 33, No. 1, 1980, pp. 87-89.
- G. B. Bachelet and N. E. Christensen, “Relativistic and Core-Relaxation Effects on the Energy Bands of Gallium Arsenide and Germanium,” Physical Review B, Vol. 31, No. 2, 1985, pp. 879-887. doi:10.1103/PhysRevB.31.879
- V. S. Stepanyuk, A. Szasz, O. V. Farberovich, A. A. Grigorenko, A. V. Kozlov and V. V. Mikhailin, “An Electronic Band Structure Calculation and the Optical Properties of Alkaline-Earth Sulphides,” Physica Status Solidi B, Vol. 155, No. 1, 1989, pp. 215-220. doi:10.1002/pssb.2221550121
NOTES
*Corresponding author.