Advances in Chemical Engineering and Science
Vol.4 No.2(2014), Article ID:44762,6 pages DOI:10.4236/aces.2014.42019
DFT Study of Poly Furfuryl Alcohol-Rhodamine B Blend
Aqeel M. Ali1, Ali H. Al-Mowali2*
1Molecular Physics Lab (MPL), Department of Physics, College of Science, University of Basrah, Basrah, Iraq
2Department of Chemistry, College of Science, University of Basrah, Basrah, Iraq
Email: *ali_almowali1946@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 10 February 2014; revised 10 March 2014; accepted 17 March 2014
ABSTRACT
A theoretical study of poly furfuryle alcohol (PFA), rhodamine B (Rh B) dye and their blends (PFARh B) is carried out by using the density functional theory (DFT). Electronic states and opto-electronic properties are investigated. The electronic states indicate that the oligomers of FA are insulators and Rh B is a wide band gap semiconductor. Their blends have a narrow band gap of about 0.75 eV. The optoelectronic properties are studied using TDDFT, which indicates that the chain length of poly furfuryl alcohol is an effective parameter to control both energies and intensities of absorption in which longer chain causes absorption with high intensity within long wavelength. A single broad baned of electron excitations is more like 8-oligomer-RhB case, which centers at wavelengths about 599 nm and 625 nm. This band of absorption covers the whole visible region of spectrum.
Keywords
Poly Furfuryle Alcohol, Rhodamine B, DFT, Electron Excitations

1. Introduction
In 1977, the discovery that polymers could be made to be electrically conductive, created the new field of plastic electronics [1] [2] . In 1990, electroluminescence was demonstrated for the first time in a conjugated-polymer system, shifting the emphasis to optoelectronic properties [3] . The first commercial applications of monochrome polymer light-emitting displays (pLEDs) are now a fact. Of course, the final aim is a full-color polymer display. However, due to a wide distribution of HOMO and LUMO levels of polymers, their emission bands are rather broad, giving rise to mixed-color light emission. Furthermore, the processing of three different polymers for the three principal colors is rather costly. One of the recent ideas to circumvent these problems is to use a small fraction (~1%) of dye molecules in a polymer matrix and to exploit the mechanism of energy transfer from polymer to dye [4] -[7] . By changing the dye rather than the polymer, it should be possible to cover the whole visible-light spectrum. Recent electroluminescence experiments have shown the possibility of fine-tuning the emission spectrum of dipyrrolomethane dyes, commercially known as BODIPY dyes [8] [9] , by chemical modification of specific sidegroups. The idea behind the use of these sidegroups is to manipulate the HOMO and LUMO energies by extending the π conjugation and adding an electron-donating methoxy group. The fact that they have relatively small bandwidths makes these dyes good candidates for the further development of full-color displays. Rhodamine B (RhB) is a chromophore from a family of xanthene dyes that is commonly used as an active medium in tunable lasers [10] -[12] due to its high fluorescence quantum yield. It also shows a wide range of applications in materials science, chemistry and biology as sensitizer in solar cells [13] , molecular probe [14] , electrochemical luminescence sensitizer [15] , water-tracing agent [16] and as a biological stain [17] .
A very important aspect for the understanding of the functioning of such polymer-dye blends is the positions of the HOMO and LUMO energy levels of the dye with respect to the polymer. If both the HOMO and LUMO energies of the dye are between those of the polymer, both electrons and holes can be trapped on the dye molecules. If both LUMO and HOMO energies of the dye are lower than the corresponding energies of the polymer, trapping may happen successively: first electrons are trapped by the dye molecules and next the holes by their Coulomb interaction with the electrons. In the reverse situation, the roles of electrons and holes would be interchanged. In all situations, these trapping processes compete with the formation of exciton in the polymer and subsequent Forster/Dexter transfer of the exciton to the dye. This later process can of course only happen if the optical gap of the dye is smaller than that of the polymer. In this work, we will calculate the HOMO and LUMO energies of the dyes shown in Figure 1 and the polymers poly furfuryl alcohol by Density-functional theory in the local density approximation (DFTLDA). The HOMO energy is given by the highest Kohn-Sham energy of the occupied states in a DFT calculation. In principle, DFT cannot be used to calculate LUMO energies, due to the well-known band-gap problem. In particular, energies of unoccupied states are generally under estimated in DFT [18] [19] . However, we will concentrate here not on absolute values but on trends in HOMO and LUMO energies and assume that these are well described in DFT. We will especially pay attention to the trends in the calculated values for the HOMO and LUMO energies of the dyes. We will finally discuss the relative positions of the HOMO and LUMO levels of the dyes and those of the polymers and the consequences for charge and energy transport.
2. Methodology
We have used a linear scaling, fully self-consistent density functional method for performing first-principles Calculations, as implemented in Amsterdam density functional ADF electronic structure program [20] . In our calculations we have used Density Functional Theory (DFT) based on the work by Hohenberg and Kohn [21] and by Kohn and Sham [22] . We have used local density approximation (LDA) [18] for the exchange correlation potential. The double zeta (DZ) basis set was used for all calculations. In this study, we have calculated frontier electronic states and electron excitation spectra of the monomer, oligomers, dye and oligomers-dye complex of furfuryl alcohol with Rhodamine B. To obtain the poly furfuryl alcohol structures, we started from the optimized structures of the neutral form. The geometries structures of molecules and complex molecules were optimized under no constraint. We have examined the electronic states of HOMO and LUMO levels; the energy gap (Eg) evaluated the difference between the HOMO and LUMO energies. The optoelectronic properties are investigated by using the TDDFT (LDA/DZ) calculations on the fully optimized geometries that interpreted in form of oscillator strengths as a function of wavelength.
3. Results and Discussion
Table 1 lists the theoretical electronic parameters of the studied conjugated compounds. The calculated values for the HOMO and LUMO energies of all the oligomers and dyes are shown in Figure 1 and Table 1. The following observations can be made By increasing the repeating units, furfuryl alcohol oligomers have small destabilizations in HOMO and LUMO levels that may change with magnitude of about 0.5 eV. The energy gap has the same destabilization, and still has large values because the adding of a repeating unit does not alter the π-system of the furfuryl alcohol polymer and the polymer skeleton which is build up by σ-bonds which keep the 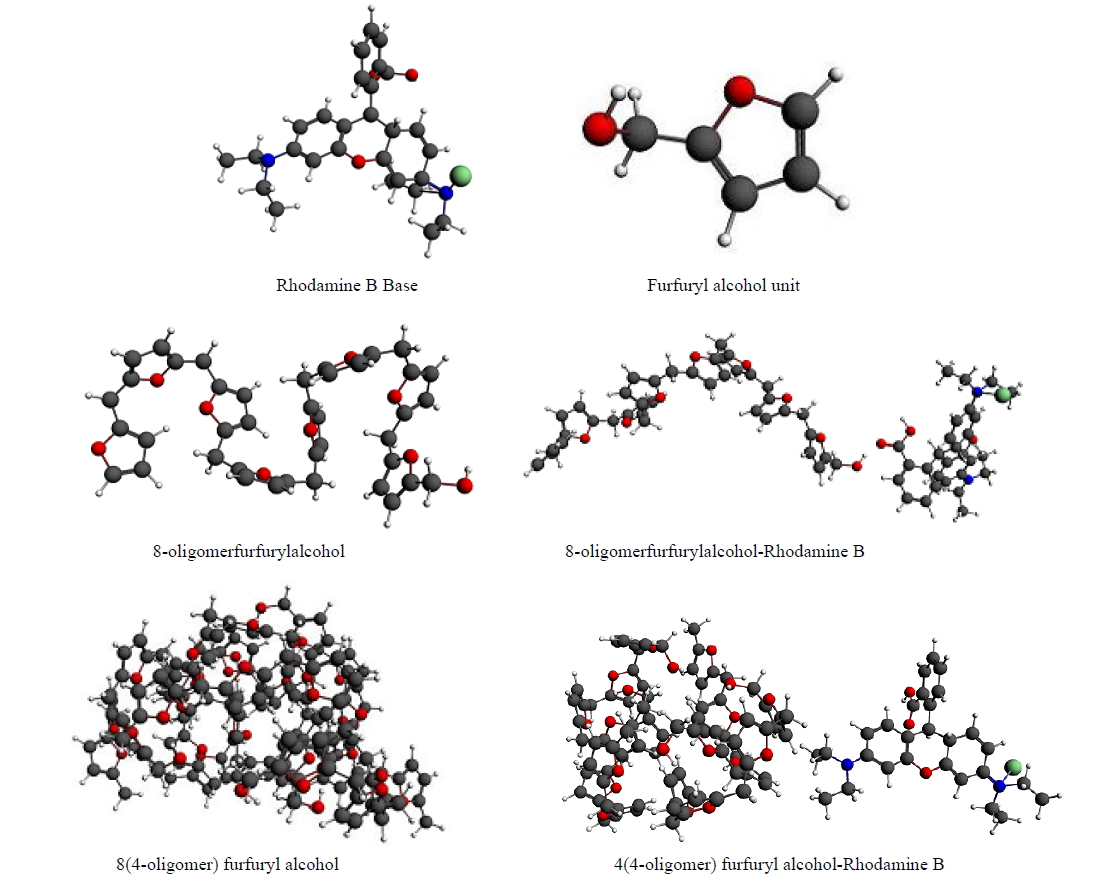
Figure 1. The optimized structures of PFA, Rodium-B and oligmer furfuryl alcohol 8-oligomer furfuryl alcohol.

Table 1. Values of HOMO (eV), LUMO (eV) and Eg (eV) energies calculated for furfuryl alcohol oligomers and dye obtained by LDA/DZ.
energy difference of occupied and unoccupied states. 2). The dye molecule has a semiconductor energy gap, and the HOMO level is higher than that of furfuryl alcohol oligomers by 0.83 - 1.41 eV, while the LUMO level is lower than that of furfuryl alcohol oligomers by 0.5 - 0.78 eV. 3) The dye-dimer has a significant change in HOMO level toward higher energy, also the LUMO level is lowered by 0.26 eV. The energy gap of dye dimer is very small which is in the range of attractive semiconductor values. The dipole moments of furfuryl alcohol oligomers, dye and dye-dimer have high values that reflect the localized states of electron clouds over the molecule skeletons, and this may explain the high energy gaps. Although the dye-dimer has a high dipole moment, the energy gap is very narrow, which may be caused by the nature of interaction between the two dye molecules. The energy levels of dye-dimer are distributed between these two molecules alternatively, where HOMO and LUMO + 1 exist on one molecule and HOMO-1 and LUMO exist on the other. The complex of 8-oligomer + dye is a very interested material, which has a small energy gap and a very high dipole moment. The complex blend case significantly affects dipole moments due to the interaction that changes the positive and negative charge-center distance, and also affects the charge-center positions.
The frontier levels (HOMO-1, HOMO, LUMO, and LUMO + 1) are distributed over the dye molecule as that in the gas phase, but the strong change in the energy gap of complex occurs by the strong interaction between the 8-oligomer and dye molecule. The long range electrostatic interaction in complex case has the value of −2.29 eV which indicates the attractive force between the two kinds of molecules at blend operation.
Figure 2 shows the theoretical prediction of the electronic transitions spectra of furfuryl alcohol oligomers. Calculations of molecular orbital geometry show that the visible absorption maxima of this kind of poly furfuryl alcohol corresponds to the electron transition from HOMO to LUMO. The λmax is a function of the electron availability. This band was due to electronic transition of the cyclic group (transition of π – π* type). The excitation energies are red shifted in all cases as the furfuryl alcohol chain becomes longer.TD-LDA/DZ predicted three absorption peaks for monomeric dye in gas phase as shown in Figure 3: at 450 nm (S0 – S2 transition with the oscillator strength f = 0.021), at 400 nm (S0 – S6 with f = 0.02) and 330 nm (S0 – S12 transition with the. oscillator strength f = 0.078). For comparison, the dimer geometry was fully optimized with the LDA/DZ method in gas phase, which results in an intermolecular distance of 15 A. As expected, the use of fully optimized geometries may lead to noticeable calculated excitation spectra. The electronic transitions of dimer are fully different from one of a single molecule, where it is shifted toward longer wavelength and has a single broad band of excitations (over 450 nm to 800 nm) which are centered on singlet states of 532 and 575 nm. These excitations occur via transfer of one electron between the monomer units that results in the (RhB)1−–(RhB)1+ configuration. In xanthene dyes, the fluorescence quenching may occur as a result of a bimolecularphotoredox reaction that involves intermolecular electron transfer in the excited states. Currently, it is widely accepted that the quenching occurs via internal conversion and intersystem crossing in the excited states of aggregates (e.g., dimers) of dyes in solution. Indeed, xanthene dyes can form aggregates in polar solution due to the strong electrostatic and dispersion interactions between the dye molecules. They are capable of strong optical absorption that cannot be realized in the monomeric-species.
It is suggested that the polymer-RhB blend may be an attractive material for electric and optoelectronic purposes. Figure 4 shows the electronic spectra of three types of blends, first 4-oligomer-RhB, second 8-oligomer-
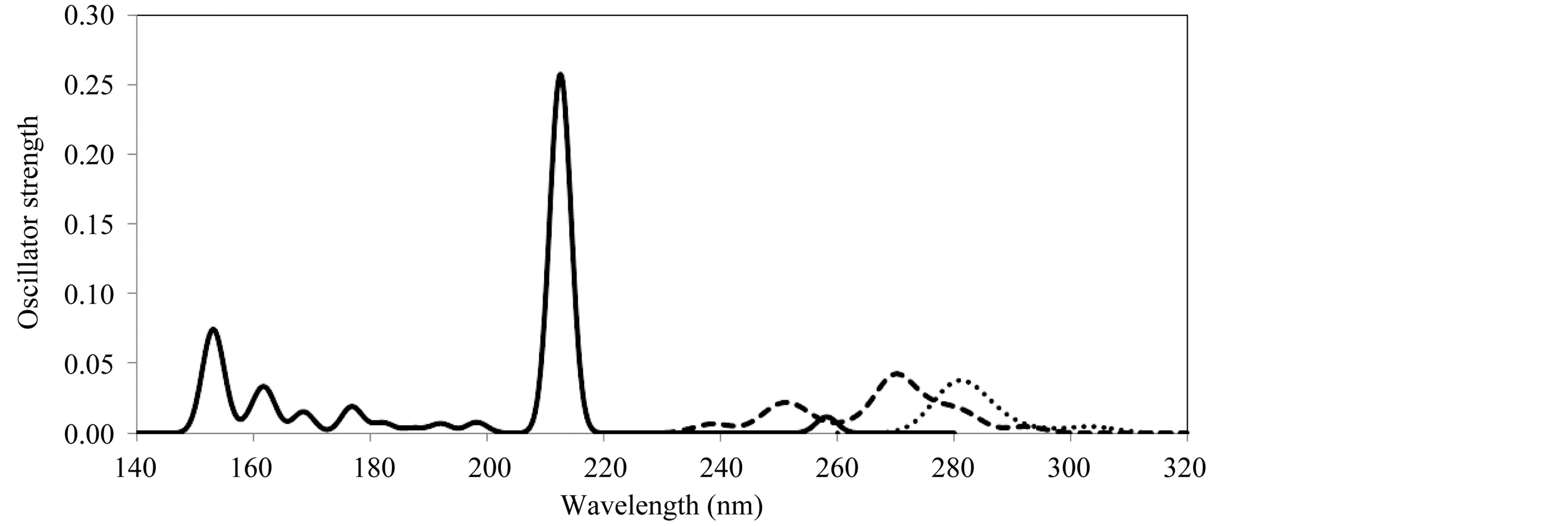
Figure 2. TD-DFT (LDA/DZ) electronic transitions of polyfurfuryl alcohol (PFA) as a function of wavelength. 1-uint (solid line), 4-unit (dashed line) and 8-unit (dotted line).
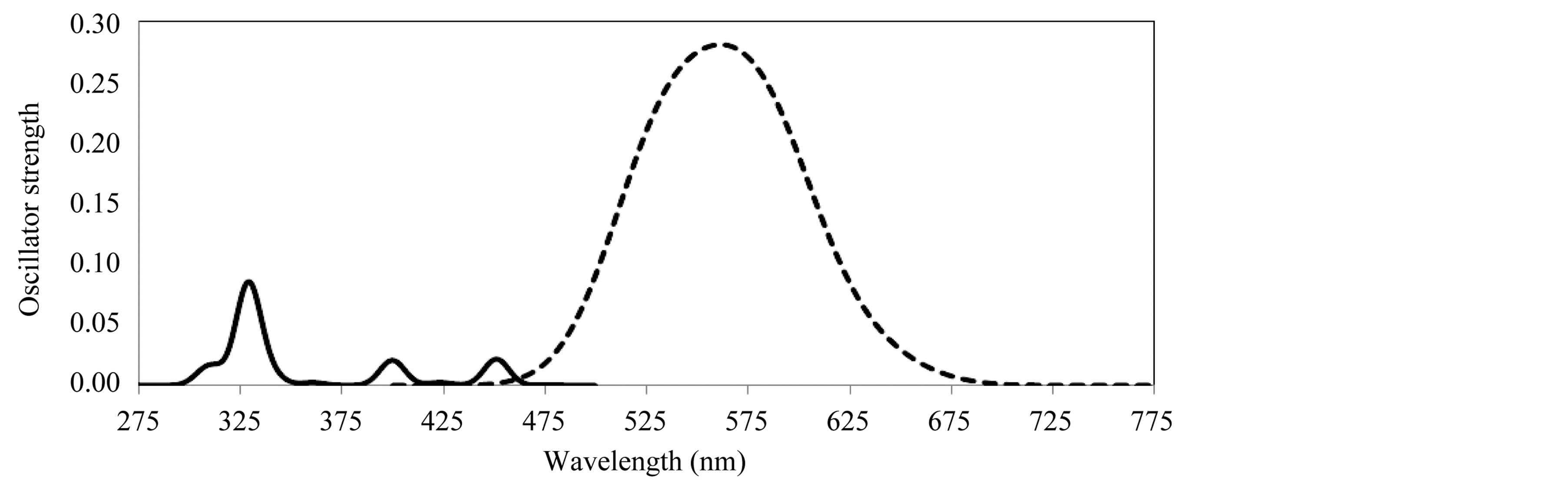
Figure 3. Electronic transitions of dye. Monomer (solid) and dimer (dashed).
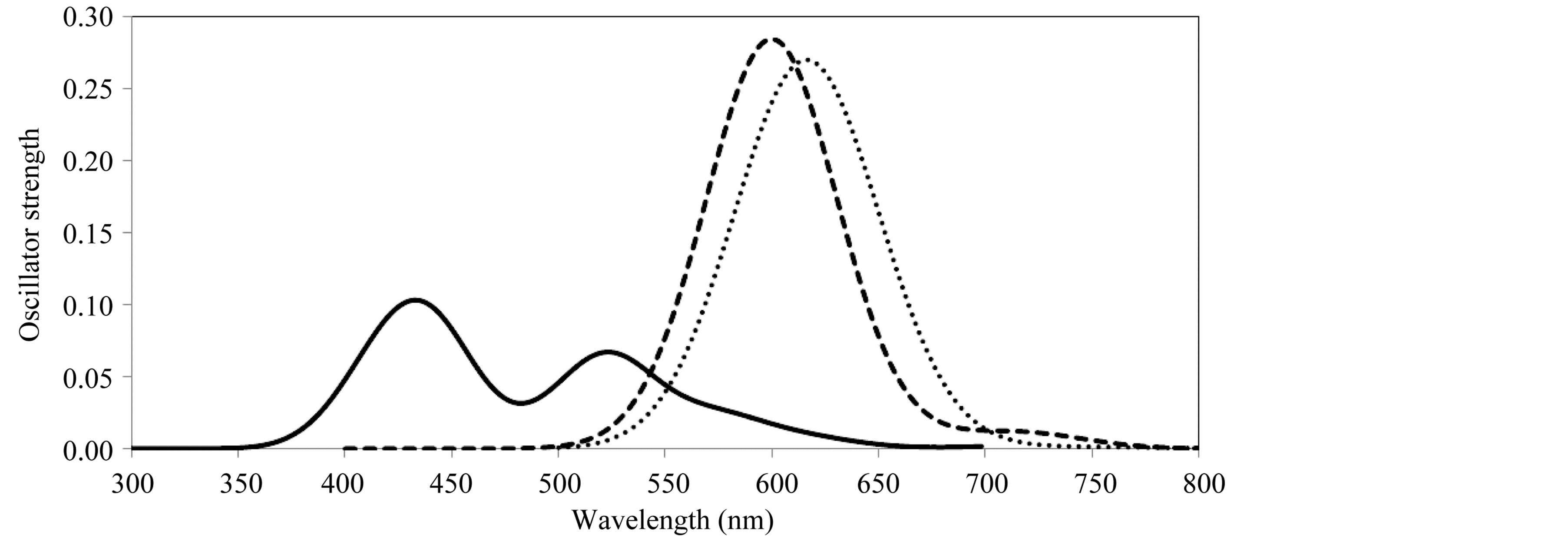
Figure 4. Electronic transitions of polyfurfuryl alcohol-dye blends (4-units-solid, 8-unitsdashed and 4(4-units)-dottted).
RhB and the other is 4-chains of 4-oligomers-RhB. The spectra are very complex, where in the first case the spectrum is similar to that of RhB single molecule but within a different range of wavelength (red shift). The second one is very similar to that of RhB dimer, with the same values of oscillator strength and range of energies. So, the chain length of poly furfuryal alcohol is an effective parameter to control both the energies and intensities of absorption, longer chain causes absorption with high intensity within long wavelength. The longer chain of polymer may make strong electrostatic interaction with dye molecule which affects the electron transfer from the dye to polymer chain. The case of four chains of 4-units is studied here to reflect the properties of poly furfuryl alcohol colloidal when it interacts with RhB molecule. A single broad band of electron excitations is more like 8-oligomer-RhB case, which is centered on wavelengths about 599 nm (f = 0.24) and 625 nm (f = 0.26). This band of absorption covers the whole visible region of spectrum. The colloidal of PFA makes a strong interaction with the RhB molecule even in the case of short chains.
4. Conclusion
The density functional theory shows that the poly furfuryl alcohol becomes semiconductor when mixed with rhodamine-B as reflected from the relation of calculated oscillator strength and wavelength. This behavior is generated from strong electrostatic interaction of rhodamine-B with different chain units of poly furfuryl alcohol.
References
- Chiang, C.K., Fischer, C.R., Park, Y.W., Heeger, A.J., Shirakawa, H., Louis, E.J., Gau, S.C. and MacDiarmid, A.G. (1977) Electrical Conductivity in Doped Polyacetylene. Physical Review Letters, 39, 1096. http://dx.doi.org/10.1103/PhysRevLett.39.1098
- Chiang, C.K., Druy, M.A., Gau, S.C., Heeger, A.J., Louis, E.J., MacDiarmid, A.G., Park, Y.W. and Shirakawa, H.J. (1977) Synthesis of Highly Conducting Films of Derivatives of Polyacetylene, (CH)x. Journal of the American Chemical Society, 100, 1013-1015. http://dx.doi.org/10.1021/ja00471a081
- Giuliani, A., Walker, I. C., Delwiche, J., Hoffmann, S. V., Vieira, P. L., Masonb, N. J., Heyne, B., Hoebeke, M. and Hubin-Franskinc, M. J. (2003) A Potential Energy Surface Construction Scheme for Accurate Reaction Rate Calculations: General Approach and a Test for the H + CH4→H2 + CH3 Reaction. The Journal of Chemical Physics, 119, 14. http://dx.doi.org/10.1063/1.1577328
- Brunner, K., van Haare, J.A.E.H., Langeveld-Voss, B.M.W., Schoo, H.F.M., Hofstraat, J.W. and van Dijken A. (2002) Mechanistic Study of Excitation Energy Transfer in Dye-Doped PPV Polymers. The Journal of Physical Chemistry B, 106, 6834-6841. http://dx.doi.org/10.1021/jp0140419
- List, E.J.W., Creely, C., Leising, G., Schulte, N., Schleuter, A.D., Scherf, U., Mullen, K. and Graupner, W. (2000) Excitation Energy Migration in Highly Emissive Semiconducting Polymers. Chemical Physics Letters, 325, 132-138. http://dx.doi.org/10.1016/S0009-2614(00)00635-7
- Virgili, T., Lidzey, D.G. and Bradley, D.D.C. (2000) Efficient Energy Transfer from Blue to Red in Tetraphenylporphyrin-Doped Poly(9,9-dioctylfluorene) Light-Emitting Diodes. Advanced Materials, 12, 58-62. http://dx.doi.org/10.1002/(SICI)1521-4095(200001)12:1<58::AID-ADMA58>3.0.CO;2-E
- Barsberg, S. and Berg, R.W. (2006) Combined Raman Spectroscopic and Theoretical Investigation of Fundamental Vibrational Bands of Furfuryl Alcohol (2-Furanmethanol). The Journal of Physical Chemistry A, 110, 9500-9504. http://dx.doi.org/10.1021/jp061642x
- Andrade, C.A., Zavaglia, A.G., Reva, I.D. and Fausto, R. (2012) Conformers, Infrared Spectrum and UV-Induced Photochemistry of Matrix-Isolated Furfuryl Alcohol. The Journal of Physical Chemistry A, 116, 2352-2365. http://dx.doi.org/10.1021/jp212169b
- Haugland, R.P. (1996) Handbook of Fluorescent Probes and Research Chemicals. 6th Edition, Molecular Probes, Eugene.
- Andrews, D.L. (1997) Lasers in Chemistry. Springer-Verlag, Berlin. http://dx.doi.org/10.1007/978-3-642-60635-9
- Schafer, F.P. (1977) Dye Lasers. 2nd Edition, Springer-Verlag, Berlin.
- Yahia, I.S., Rammah, Y.S. and Khaled, K.F. (2013) Fabrication of an Electrochemical Cell Based on Rhodamine B Dye for Low Power Applications. Journal of Materials and Environmental Science, 4, 442-447.
- Mottram, L.F., Forbes, S., Ackley, B.D. and Peterson, B.R. (2012) Hydrophobic Analogues of Rhodamine B and Rhodamine 101: Potent Fluorescent Probes of Mitochondria in Living C. elegans. Beilstein Journal of Organic Chemistry, 8, 2156-2165. http://dx.doi.org/10.3762/bjoc.8.243
- Kato, E. and Murakami, T. (1998) Anti-Stokes Fluorescence Imaging of Diffusion Process of Probe Molecules in Poly(N-isopropylacrylamide) Gels. Polymer Gels and Networks, 6, 179. http://dx.doi.org/10.1016/S0966-7822(98)00010-0
- Zhang, C., Zhou, G., Zhang, Z. and Aizawa, M. (1999) Highly Sensitive Electrochemical Luminescence Determination of Thiamine. Analytica Chimica Acta, 394, 165-170.
- Liu, S.P., Liu, Z.F. and Luo, H.Q. (2000) Resonance Rayleigh Scattering Method for the Determination of Trace Amounts of Cadmium with Iodide-Rhodamine Dye Systems. Analytica Chimica Acta, 407, 255-260. http://dx.doi.org/10.1016/S0003-2670(99)00816-8
- Bakkialakshmi, S. and Menaka, T. (2012) Study on the Inclusion Interactions of β-Cyclodextrin with Rhodamine B Base. International Journal of ChemTech Research, 4, 223-231.
- Jones, R.O. and Gunnarson, O. (1989) The Density Functional Formalism, Its Applications and Prospects. Reviews of Modern Physics, 61, Article ID: 689. http://dx.doi.org/10.1103/RevModPhys.61.689
- Godby, R.W., Schleuter, M. and Sham, L.J. (1988) Self-Energy Operators and Exchange-Correlation Potentials in Semiconductors. Physical Review B, 37, Article ID: 10159. http://dx.doi.org/10.1103/PhysRevB.37.10159
- Amsterdam Density Functional Modeling Suite, Scientific Computing and Modeling, ADF.
- Hohenberg, P. and Kohn, W. (1964) Inhomogeneous Electron Gas. Physical Review, 136, Article ID: B864. http://dx.doi.org/10.1103/PhysRev.136.B864
NOTES

*Corresponding author.