Open Journal of Nephrology
Vol.3 No.1(2013), Article ID:29072,3 pages DOI:10.4236/ojneph.2013.31007
Remission of Relapsing Immunotactoid Glomerulonephritis with Fludarabine*
1Department of Medicine, Staten Island University Hospital, New York, USA
2Division of Nephrology, Staten Island University Hospital, New York, USA
3Division of Renal Pathology, Department of Pathology and Cell Biology, Columbia University Medical Center, New York Presbyterian Hospital, New York, USA
Email: #mehtasuchita@gmail.com
Received December 9, 2012; revised February 4, 2013; accepted March 17, 2013
Keywords: Glomerulonephritis; Fludarabine; ITGN
ABSTRACT
Immunotactoid gloemrulonephritis is a glomerular disease characterized by organized microtubular deposits of monoclonal immunoglobulin. These deposits are Congo red-negative, have hollow centers, measure >30 nm, and are arranged in stacked or parallel arrays. Treatment of immunotactoid glomerulonephritis is not well-defined, with poor outcomes seen in native kidneys. In fewer than 10% of the cases, trials with steroids alone or cytotoxic agents with steroids or plasmapheresis with steroids has been associated with clinical remission of proteinuria. 50% of patients with renal impairment progress to end stage renal disease (ESRD) in two to four years. There are three case reports of recurrent ITG in renal allograft; one was treated with pulse steroids plus cyclophosphamide, pulse steroids in second case and use of rituximab in the third case. Recurrence of ITG has been described in the renal allograft but there is no literature on relapse of ITG in the native kidneys. Here, we have a case of ITG that relapsed in the native kidneys eight years after being in remission. Renal function improved with fludarabine both times and the patient had stable renal function at last follow-up.
1. Introduction
Schwartz et al. first described immunotactoid glomerulpathy (ITGN) in 1980 [1]. Fibrillary glomerulonephritis (FGN) and ITGN are generally considered as distinct entities, based upon immunopathologic, ultrastructural and clinical differences [2]. ITGN is characterized by Congo red-negative microtubules that have hollow centers, usually measuring >30 nm, that are arranged in stacked or parallel arrays and which stain with antisera to immunoglobulins by routine immunofluorescence [2]. Fibrillary, immunotactoid and cryoglobulinemic glomerulopathies together account for about 1% of all renal biopsies [3] and ITGN by itself constitutes 0.1% of renal biopsy specimens [4]. In the study by Rosenstock et al., fibrillary glomerulonephritis constituted 6% of the renal biopsies whereas immunotactoid glomerulonephritis was seen in only 0.06% of the total native kidney biopsies [2]. Treatment of immunotactoid glomerulonephritis is not well-defined, with generally poor outcomes seen in both native kidneys and in recurrent disease in renal transplants [5]. In addition, there are no reported cases of ITGN recurring in native kidneys. Here, we report a case of immunotactoid glomerulonephritis associated with chronic lymphocytic leukemia (CLL) that relapsed in the native kidneys eight years later following remission induced by fludarabine therapy, and which subsequently responded again to fludarabine with a complete remission.
2. Case Report
An 83-year-old woman with a history of chronic lymphocytic leukemia (CLL) presented in January 2002 with a serum creatinine of 3.7 mg/dl (baseline creatinine 1.0 mg/dl) and a 24-hour urine protein of 7.8 gm/day. Her white blood cell count was 48,300 with 75% lymphocytes and 12% neutrophils. A renal biopsy was performed showing diffuse endocapillary and focal extracapillary proliferative glomerulonephritis. Immunofluorescence showed focal infiltrates of IgG lambda-bearing lymphocytes, along with segmental glomerular capillary wall deposits of IgG lambda. Electron microscopy showed glomerular electron dense deposits composed of tubular substructures measuring 20 - 30 nm, all consistent with features of immunotactoid glomerulonephritis, likely CLL-related. Chemotherapy was initiated for CLL in the hope that treatment of the underlying malignancy would produce a favorable outcome for the glomerulonephritis. Aside from the renal failure, the patient did not have any other manifestations of CLL that would warrant treatment. The patient was started on fludarabine. Her serum creatinine gradually decreased from 3.8 mg/dl to 1.2 mg/dl over the next nine months, and her 24-hour urinary protein improved from 7.8 to 0.59 g/day. The patient remained in complete remission for eight years. However, in December, 2010 her serum creatinine had increased to 2.66 mg/dl and a spot urine protein to creatinine ratio was 10.8 gm/gm. A repeat renal biopsy was performed, and it again showed segmental endocapillary proliferative and membranous glomerulonephritis with monoclonal IgG lambda deposits, consistent with a relapse of her CLL-associated immunotactoid glomerulonephritis (See figures 1 and 2). Her serum creatinine further increased to 3.5 mg/dl, and she was again treated with fludarabine. She responded well to the treatment with a second remission. Her serum creatinine decreased to 1.53 mg/dl and her urine protein to creatinine ratio was 0.57 g/g as of June 2012.
4. Discussion
ITGN is a glomerular disease characterized by microtubular immunoglobulin deposits occurring in the absence of systemic diseases such as amyloidosis, cryoglobulinemia, and systemic lupus erythematosus [5]. Approximately 60% of patients with ITGN present with nephrotic range proteinuria; 70% have microscopic hematuria and hypertension, and 53% present with renal insufficiency at diagnosis [5-7]. The average age at pre-
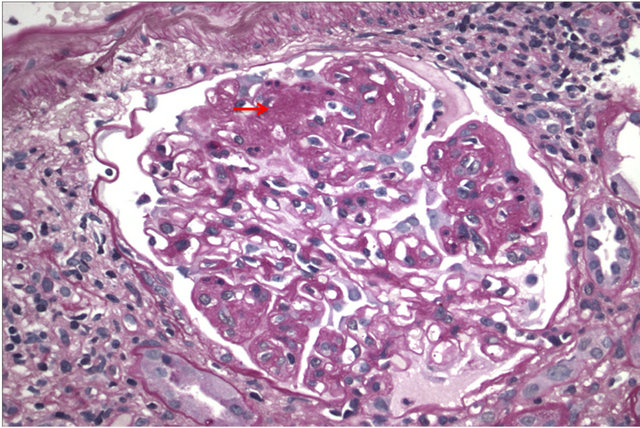
Figure 1. Light microscopy of a glomerulus showing segmental endocapillary hypercellularity. Red arrow points out the hypercellularity.

Figure 2. Electron microscopy showing subepithelial and mesangial electron dense deposits with organizes microtubular substructure with the fibril size ranging between 20 - 30 nm. Red arrow points to the microfibril deposition.
sentation is 60 years [8]; 90% patients are white with the distribution being equal in men and women [7]. Many immunotherapies have been tried, but the response has generally been poor, although the experience with these has been limited [5]. Fewer than 10% of patients treated with steroids alone, or in combination with cytotoxic agents, and/or plasmapheresis, have achieved a remission of proteinuria [5]. About 50% of patients with renal impairment at diagnosis progress to ESRD within two to four years [6,7]. As patient survival at 1 year is 100%, and over 80% of patients are alive after 5 yr [6], the question of whether renal transplantation is a reasonable option for these patients is quite relevant. Sathyn [9], Carles [10], and Mak [11], described a total of three patients in whom ITGN recurred in renal transplants, with proteinuria or renal insufficiency beginning from 7 weeks to 10 months post-transplant. Rituximab [9] had no effect on proteinuria, although renal function improved; pulse steroids [11] improved neither proteinuria nor renal function, and a combination of plasma exchange, and monthly IV cytoxan × 6 doses [10], resulted in a complete remission. All patients remained on their usual maintenance immunosuppression. As such, the advisability of transplanting patients with ITG remains an open question.
A total of twelve patients with CLL-associated ITGN have been described in the literature. Six such patients were reported by Bridoux et al., but relapse was frequent and individual results for serum creatinine, urinary protein excretion, and length of follow up were not reported [12]. In the remaining six cases of ITGN with CLL, there was a good response to both fludarabine and rituximab as noted in the cases described below. This case is the first report of relapse of ITGN following successful remission induced by fludarabine therapy.
The first patient reported by Rosenstock et al. is the same patient reported by us in the present case; this was initially reported in 2003 when the patient had her first complete remission with fludarabine [2]. The second patient with CLL-associated ITGN was reported by Bhat et al., and was treated with rituximab, but nevertheless progressed to end stage renal disease [13]. Vilayur et al. reported a patient with CLL-associated glomerulopathy in whom a course of cyclophosphamide and steroids failed to induce any remission; four weeks later, because of increasing proteinuria and worsening renal function, two doses of rituximab were given seven months apart. His proteinuria decreased to 0.3 g/day and the serum creatinine returned to baseline at 1.2 mg/dl [14]. The last three patients were reported by Guiard et al.; one patient received no treatment and subsequently died; a second patient received CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) and chlorambucil, but had no response; and the third patient was given CHOP and fludarabine, and had a complete remission, again suggesting that fludarabine may have been of therapeutic benefit.
In summary, we report a patient with two distinct episodes of CLL-associated ITGN and renal failure occurring over a period of eight years, who had a prompt remission of both her proteinuria and renal dysfunction on both occasions after treatment with fludarabine. It is important to report the relapse of this disease as it has not been reported to recur in the native kidneys and thus making it unique. This case report addresses the need for further understanding of pathophysiology of ITGN so as to better define the treatment. Given the rarity of this disease it might be difficult to perform larger studies and compare the different drugs available. However, fludarabine or rituximab may be considered as initial therapy for this disease in order to prevent progression to end stage renal disease.
REFERENCES
- M. M. Schwartz and E. J. Lewis, “The Quarterly Case: Nephrotic Syndrome in a Middle-Aged Man,” Ultrastructural Pathology, Vol. 1, No. 4, 1980, pp. 575-582. doi:10.3109/01913128009140563
- J. L. Rosenstock, G. S. Markowitz, A. M. Valeri, G. Sacchi, G. B. Appel and V. D. D’Agati, “Fibrillary and Immunotactoid Glomerulonephritis: Distinct Entities with Different Clinical and Pathologic Features,” Kidney International, Vol. 63, No. 4, 2003, pp. 1450-1461. doi:10.1046/j.1523-1755.2003.00853.x
- G. A. Herrera and E. A. Turbat-Herrera, “Renal Diseases with Organized Deposits: An Algorithmic Approach to Classification and Clinicopathologic Diagnosis,” Archives of Pathology & Laboratory Medicine, Vol. 134, No. 4, 2010, pp. 512-531.
- R. J. Falk, J. C. Jennette and P. H. Nachman, “Primary Glomerular Disease,” In: B. M. Brenner, Ed., Brenner and Rector’s the Kidney, 7th Edition, Saunders, Philadelphia, 2004.
- M. M. Schwartz, S. M. Korbet and E. J. Lewis, “Immunotactoid Glomerulopathy,” Journal of the American Society of Nephrology, Vol. 13, No. 5, 2002, pp. 1390-1397. doi:10.1097/01.ASN.0000013397.06964.19
- S. M. Korbet, M. M. Schwartz and E. J. Lewis, “Immunotactoid Glomerulopathy,” American Journal of Kidney Diseases, Vol. 17, No. 3, 1991, pp. 247-257.
- P. H. Pronovost, H. R. Brady, M. E. Gunning, O. Espinoza and H. G. Rennke, “Clinical Features, Predictors of Disease Progression and Results of Renal Transplantation in Fibrillary/Immunotactoid Glomerulopathy,” Nephrology Dialysis Transplantation, Vol. 11, No. 5, 1996, pp. 837-842. doi:10.1093/oxfordjournals.ndt.a027409
- D. N. Howell, X. Gu and G. A. Herrera, “Organized Deposits in the Kidney and Look-Alikes,” Ultrastructural Pathology, Vol. 27, No. 5, 2003, pp. 295-312. doi:10.1080/01913120390231555
- S. Sathyan, F. N. Khan and K. V. Ranga, “A Case of Recurrent Immunotactoid Glomerulopathy in an Allograft Treated with Rituximab,” Transplantation Proceedings, Vol. 41, No. 9, 2009, pp. 3953-3955. doi:10.1016/j.transproceed.2009.03.100
- X. Carles, L. Rostaing, A. Modesto, C. Orfila, J. M. Cisterne, M. B. Delisle and D. Durand, “Successful Treatment of Recurrence of Immunotactoid Glomerulopathy in a Kidney Allograft Recipient,” Nephrology Dialysis Transplantation, Vol. 15, No. 6, 2000, pp. 897-900.
- Y. F. Mak, S. H. Wong and T. C. Chow, “A 50-Year-Old Man with Heavy Proteinuria 2 Months Post Transplantation,” American Journal of Kidney Diseases, Vol. 43, No. 3, 2004, pp. 580-585.
- F. Bridoux, V. Hugue, O. Coldefy, J. Goujon, M. Bauwens, A. Sechet, J. Preud’homme and G. Touchard, “Fibrillary Glomerulonephritis and Immunotactoid (Microtubular) Glomerulopathy Are Associated with Distinct Immunological Features,” Kidney International, Vol. 62, 2002, pp. 1764-1775. doi:10.1046/j.1523-1755.2002.00628.x
- P. Bhat, S. Weiss, G. B. Appel and J. Radhakrishnan, “Rituximab Treatment of Dysproteinemias Affecting the Kidney: A Review of Three Cases,” American Journal of Kidney Diseases, Vol. 50, No. 4, 2007, pp. 641-644. doi:10.1053/j.ajkd.2007.05.027
- E. Vilayur, P. Trevillian and M. Walsh, “Monoclonal Gammopathy and Glomerulopathy Associated with Chronic Lymphocytic Leukemia,” Nature Clinical Practice Nephrology, Vol. 5, No. 1, 2009, pp. 54-58.
NOTES
*Conflict of interest: There are no conflicts of interest and the results presented in this paper have not been published previously in whole or part, except in abstract format.
#Corresponding author.