Open Journal of Genetics
Vol.3 No.1(2013), Article ID:29367,6 pages DOI:10.4236/ojgen.2013.31007
Tay-Sachs carrier screening in the genomics age: Gene sequencing versus enzyme analysis in non-Jewish individuals
![]()
Department of Genetics, Quest Diagnsotics Nichols Institute, San Juan Capistrano, USA
Email: *Charles.M.Strom@QuestDiagnostics.com
Received 30 January 2013; revised 2 March 2013; accepted 14 March 2013
Keywords: Tay-Sachs Screening; Gene Sequencing; Hexosaminidase A
ABSTRACT
Purpose: To compare the sensitivity of Hexosaminidase A (HexA) enzyme-based testing to gene sequencing for carrier detection in non-Jewish individuals. Methods: Blood samples were obtained from parents and relatives of affected patients at an annual TaySachs and Allied Diseases Foundation meeting. A family history was taken for each individual. Samples were analyzed for leukocyte HexA activity, serum HexA activity and subjected to extensive gene sequencing. The results from these analyses were combined with our previously published data describing 34 obligate Tay-Sachs disease (TSD) carriers. Results: Twelve additional TSD carriers were detected in this study. Gene sequencing successfully identified all 12 carriers whereas enzyme analysis identified 11 of 12 carriers. This individual is a carrier of the B1 variant that is known to cause false negative results with enzyme testing. Combined data from 46 non-Jewish TSD carriers revealed that gene sequencing had a higher sensitivity rate than HexA enzyme-based testing (94% versus 87%) in non-Jewish TSD carriers. In our series, approximately 4% of non-Jewish TSD carriers have this mutation. Conclusions: HexA gene sequencing provides a higher sensitivity for TSD carrier detection than HexA based enzyme analysis in non-Jewish patients primarily due to the presence of individuals with the B1 variant.
1. INTRODUCTION
As automation and technical improvements continue to drive down the costs of DNA sequencing, it has now become feasible to perform Hex A gene sequencing as a primary screening test for Tay-Sach’s Disease (TSD) carrier detection. This study enlarges upon an earlier study that demonstrated that sequencing and enzyme analysis had identical sensitivities in identifying TSD carriers to determine whether DNA sequencing would be an appropriate initial screen for non-Ashkenazi Jewish (AJ) individuals.
Tay-Sachs disease, or GM2 Gangliosidosis, is an autosomal recessive neurodegenerative disorder caused by a deficiency of beta-hexosaminidase A (HexA), resulting in lysosomal accumulation of GM2 ganglioside. There are several forms of TSD with the most common being the classic infantile form that leads to death between 2 and 5 years of age. Infrequently Tay-Sachs disease can first manifest symptoms in adulthood and is called late onset TSD or LOTS. However, even in LOTS there is a progressive of symptoms are progression eventually resulting in profound disability [1].
Certain ethnic groups have an increased carrier frequency for Tay-Sachs disease with the highest being Ashkenazi Jews (AJ) who have a carrier frequency of approximately 1:30. French Canadians, Cajuns and Pennsylvania Dutch also have increased carrier frequencies relative to other ethnic groups. Low risk populations have a carrier frequency of approximately 1:300 [1].
Throughout most of the developed world, high risk populations are offered carrier screening for Tay-Sachs Disease. In the United States and Canada carrier screening was begun in the 1970’s using an enzymatic approximation of HexA activity [1,2]. Enzyme based screening has resulted in an approximate 90% reduction in new cases of TSD in these countries. [1] Because at risk couples are usually screened, most new cases of Tay-Sachs disease in the US and Canada are born to parents where at least one parent belongs to non-high risk group. The American College of Obstetricians and Gynecologists recommends carrier screening for couples of Ashkenazi Jewish, French-Canadian, and Cajun descent as well as for couples in whom only one member of the couple belongs to a high risk ethnic group [3].
Enzyme based HexA carrier screening is performed using an artificial fluorogenic substrate. Unfortunately there is a second enzyme, Hexosaminidase B (HexB) that acts on the same substrate. Discrimination between HexA and HexB activity heat inactivation; takes advantage of the observation that HexA is relatively more heat labile than HexB. The enzyme assay is run twice, both before and after a period of heat inactivation. The percentage of HexA activity is calculated and this is used to determine if an individual is a carrier or noncarrier [2]. The assay can be performed on serum, white blood cells and platelets. The serum assay cannot be used during pregnancy because of the presence of the HexP isozyme which is heat stable and artificially reduces the apparent percentage of HexA. Therefore during pregnancy, only white cells and platelet assays are reliable [4,5]. Regardless of sample type there is a significant overlap between noncarrier and carrier ranges for percentage HexA with between as many as 10% of patients results falling in the indeterminate or “gray zone”. Often even after repeated analyses are performed on multiple samples, certain individuals will be unable to be classified in terms of their TSD carrier status [6].
After population based screening was instituted, it was observed that certain unaffected individuals who presented for carrier detection actually had HexA levels in the affected range. This finding eventually led to the discovery that there are specific DNA mutations, now called pseudodeficiency alleles that cause the enzyme to be deficient in the metabolism of the artificial substrate in vitro, but have normal in vivo activity against the native substrate. Therefore all individuals who are apparent carriers by enzyme analysis require molecular testing to rule out the presence of a pseudodeficiency allele. Approximately 2% of AJ and 3% of non-AJ individuals carry a pseudodeficiency allele [7,8]. Thus all individuals regardless of ethnic background who are apparent TSD carriers by enzyme testing require followup with molecular testing to investigate the presence of a pseudofediciency allele.
There are also at least 5 known mutations that cause a phenotype called the B1 variant where Hex A shows normal or near normal activity against the artificial substrate but is deficient in vivo against the naturally occurring substrate [9-11]. Consequently, individuals heterozygous for this genotype will have false negative or indeterminate enzymatic carrier tests but are still carriers for TSD. Since these individuals may have false negative enzyme testing they would remain undetected without molecular testing.
Although reasonably accurate and cost effective, the enzyme assay suffers from some inherent limitations. As noted above, there is significant overlap between the ranges of non-carrier and carriers, the presence of a pseudodeficiency allele causes false positive results, and the presence of a B1 allele can cause false negative results. Enzyme analysis can be affected by medications including oral contraceptives which can cause false positive results [12,13].
The enzyme assay also cannot differentiate between carriers of the severe infantile form of TSD from the milder forms. Finally, HexA enzyme analysis requires special logistics because the samples must be received in the laboratory in a timely fashion at appropriate temperatures in order to obtain reasonably accurate results. Once the samples are received in the laboratory they must be processed rapidly leading some laboratories to only accept specimens on certain days and times.
In Ashkenazi Jews, 3 mutations account for 92% - 98% of all carriers of TSD. Many authors have suggested that for AJ individuals, a panel consisting of these 3 mutations and the 2 pseudo deficiency alleles is preferable to enzyme testing for a carrier screening program [6,14]. However, for non Jewish individuals, this panel would detect only about 50% of TSD carriers [6,14,15].
Previously we collected samples from 34 obligate or self reported non-AJ TSD carriers at a National TaySachs and Allied Diseases meeting. We compared 4 modalities of testing for sensitivity for detection of TSD: enzyme analysis of serum, enzyme analysis of leucocytes, common mutation analysis, and HexA gene sequencing. As expected, the common AJ mutation panel had a sensitivity of only 52% in this population. The enzyme analysis and DNA sequencing analysis had equal sensitivities of 91%. Specificities could not be determined since these individuals were known TSD carriers. The current study represents and extension of the original study [15]. In addition we examined our data from 64 samples submitted for clinical HexA sequencing to determine the prevalence of novel mutations and variants of unknown clinical significance (VUCS).
2. MATERIALS AND METHODS
2.1. Clinical Samples
Volunteers were requested at the 2009 Annual Meeting of the Tay-Sachs and Allied Diseases Association.
Inclusion Criteria:
Any individual affected with TSD and any individual who was a first or second degree relative of a patient with TSD, with Tay-Sachs disease were offered enrollment in the study.
Exclusion Criteria: Patients and individuals related to patients with diseases other than TSD were excluded from the study.
Patients were consented in a protocol approved by the Western Institutional Review Board. For this study, only samples obtained from 23 individuals who were self identified as being of non-Jewish ancestry and were related to patients affected with infantile TSD were used. Each individual was tested for serum Hex A activity, white cell Hex A activity, and complete gene sequencing.
2.2. DNA Extraction
DNA from most of the samples were extracted using Gentra Autopure LS (Minneapolis, MN) exactly as described by the manufacturer, and some samples were extracted using Qiagen Biorobot 9604 platform using MegAttract® DNA blood M96 kit (Valencia, CA). Whole blood was used for DNA extraction in most cases, and in some cases, leukocytes were used. We observed no differences in sequencing quality between different extraction platforms and different starting materials.
2.3. HEXA Gene Sequencing
DNA sequencing and PCR were performed essentially as described by Huang et al. [16] except that DNA purification post-PCR was done using exonuclease (USB Corporation, Cleveland, OH)/calf intestinal alkaline phosphatase (Promega, Medison, WI) digestion, and post dye terminator reaction DNA was purified by ethanol precipitation and resuspended in Hi-Di formamide (ABI, Foster City, CA). Fourteen amplicons, each amplifying one of 14 exons, were generated through individual PCR reactions. Amplification efficiency and correctness of sizes were verified by agarose gel electrophoresis. Sequencing covered exons 1 through 14 of chromosome 15 and at least 20 bp flanking sequences. Samples were resolved on an ABI 3730 automated DNA analyzer and data were analyzed using SeqScape software (both from Applied Biosystems).
2.4. HEXA DNA Common Mutation Test
We purchased xTAG, Ashkenazi Jewish Reagents from Tm Biosciences (Toronto, Canada) and used them according to manufacturer’s instructions as described previously [17]. Mutation analysis was performed for 5 common mutations and 2 pseudoalleles: 1278insTATC; 1421 + 1G ® C (+1 IVS12 G ® C); 805G ® A (GLY269SER [G269S]); 1073 + 1G ® A (+1 IVS9 G ® A); 7.6-KB DEL, EX1; 739C ® T (ARG247TRP [R247W]); and 745C ® T (ARG249TRP [R249W]). This is a multiplex assay that analyzes 31 mutations in various genes using multiplex PCR followed by allele specific primer extension, Luminex bead conjugation, and analysis on a Luninex 200 instrument (Luninex Corporation, Houston, TX).
2.5. HEXA Enzyme Assay
Serum collected from subjects was separated from blood clots by centrifugation and kept frozen at −20˚C until analysis. Leukocytes were extracted by the method of Snyder and Brady [18]. with some minor modifications and protein concentration of the leukocyte cell pellets was determined by the method of Lowry [19].
Total HEXA activity was determined by incubation of 1 mcg/ml protein in citrate-phosphate consisting of, pH 4.4 (0.01 M Di-Sodium hydrogen phosphate, 6mM Citric acid), pH 4.4 containing 4-Methylumbelliferyl-2-acetamido-beta-D-glucopyranoside (Sigma-Aldrich, St Louis, MO) as a substrate [20]. Reactions were incubated at 37 deg for 40 minutes and fluorescence measured on a Perkin Elmer (Waltham, MA) Model 1420 fluorimeter.
The Hex-B enzyme is heat stable, but Hex-A is heat labile. The amount of Hex-B was determined by measuring total hexosaminidase activity, both before and after heat inactivation. The percent HEX-A is determined by the equation: (HEX (total) – HEXB)/HEX (total). Our established reference intervals are non-carriers: percent HEXA > 57% and <80%, TSD carriers percent HEXA >= 25% and <52%, with indeterminate (gray zone) percent HEXA 52% - 57%.
2.6. Statistical Analysis
Chi square analysis was performed using the software program Epistat.
3. RESULTS
Of the 23 tested individuals, 11 were determined to be non-carriers by both enzymatic and molecular techniques. Table 1 tabulates the results for the 12 carriers in this series. The most illuminating case is an individual with a B1 phenotype and the genotype of c.533G > A (p.Arg178His). This individual is a TSD carrier but has indeterminate results on leucocyte testing and a false negative non-carrier result on serum testing. In the previous series we had detected another individual with the same mutation who had false negative non-carrier by both the leukocyte and serum enzyme assay. Combining the data from this study and the previous study [11]. demonstrates that for the individuals tested in these 2 studies, extensive gene sequencing has a higher sensitivity than enzyme analysis for TSD carrier detection. DNA sequencing had a sensitivity of 94% versus 87% for enzyme analysis in 40 TSD carriers. Although the numbers in this study are too small to reach statistical significance, it is clear that DNA sequencing has at least an equal, if not superior sensitivity to enzyme analysis.
Leukocyte testing performed slightly better than serum testing due to an increased rate of indeterminate results in the serum (25%) versus leukocyte series (8%).
One of the complications of performing gene sequencing for TSD carrier detection is the possibility of detecting variants of unknown clinical significance (VUCS). In the current series, 4 of the 12 carriers had novel variants defined by the fact we could not find any publications or databases containing these mutations. Of these 4, none would be considered a VUCS. The mutation c.1214- 1215delAAinsG causes a frame shift mutation and therefore almost certainly deleterious, as is the c.759- 774dup. The nonsense mutation c.1292G > A (p.W431X) is also clearly pathogenic. The c.253 + 1G > A affects a consensus splice donor sight and therefore would also be predicted to be pathogenic.
In order to investigate the frequency of VUCS that might be encountered should HexA gene sequencing be used as a first line TSD carrier screen, we reviewed the data from our initial 64 clinical sequencing assays. There were 10 patients with novel mutations and are listed in Table 2. The mutation p.Met459Val was seen in 3 unrelated patients. One of these patients is affected with TSD and is a compound heterozygote for a classic TSD mutation. A second individual was a heterozygote was identified as a carrier for TSD by enzyme analysis thus confirming that this allele is almost certainly a deficiency allele. There were 2 individuals in the same TSD family with the novel variant p.Ala479Thr that is predicted to probably damaging by the algorithm of Poly-Phen (http://genetics.bwh.harvard.edu/pph2/) [21]. The allele c.316 + 1G > T alters a consensus splice site and other mutations that alter this site have been demonstrated to be causative mutations [22]. suggesting that this is also almost certainly a TSD allele. The variant c.1A > C is also almost certainly a disease causing allele since it alters the origin of transcription and other mutations
Table 1. Testing results for 12 Tay-Sachs carriers.
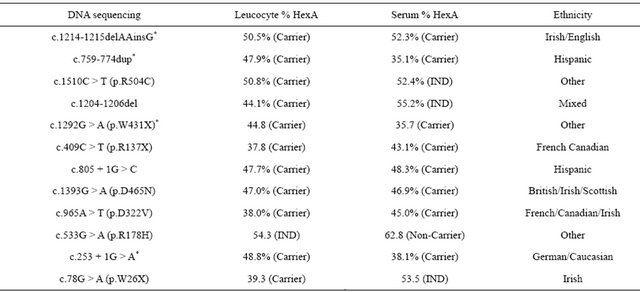
*Novel mutation.
Table 2. Novel variants discovered in HexA sequencing assay.

altering this site are demonstrated to be TSD alleles. The variant p.Gly520Asp is predicted to be benign by the Poly-Phen site. An individual with indeterminant enzyme results had a novel VCUS of p.Glu162Lys (c.484G > A). Therefore this individual’s carrier status could not be resolved.
A second VCUS result was an individual with the variant p.Phe434Leu (c.1302C > G). We were unable to determine if this patient had prior enzyme testing. Thus, 2 of 64 (3%) cases were reported as VCUS. This is comparable or lower than the number of individuals who fall within the gray zone for HexA enzyme testing results in most laboratories.
4. DISCUSSION
Although population based carrier detection using enzyme analysis has reduced the births of affected children in high risk ethnic groups drastically, infants with TSD continue to be born. Since almost all new cases of TSD have at least one parent from a non high risk group, it is important to optimize the sensitivity of carrier detection in those populations. It is clear that a panel consisting of the common AJ and French Canadian-Cajun alleles will only detect approximately 51% of all carriers from other ethnicities. This study has demonstrated that the 4% prevalence of the B1 phenotype, in addition to the 8% indeterminant rate for leukocyte HexA activity limits the sensitivity of the enzyme assay to below 90% in the patients tested in this study. Our observed sensitivity for the enzyme based assay for the 40 confirmed carriers in our combined studies was 87%. Gene sequencing performed better with a sensitivity of 94% although the numbers were to low to reach statistical significance. Since there are so few births of affected children, getting more obligate carriers to enlarge this study would be difficult.
In terms of specificity, the low rate of VCUS makes sequencing equal or better than enzyme analysis. Individuals with VCUS can have confirmatory enzyme testing, similar to the current practice of all TSD carriers on enzyme analysis being reflexed to common mutation analysis to rule out a pseudodeficiency allele. In this manner almost all VCUS will be classified into benign variants and disease causing mutations. As the database expands, progressively fewer VCUS will be encountered. It is possible that for a small number of individuals who have a VCUS, a negative family history and negative or indeterminant enzyme analysis could have an, as yet, undescribed B1 variant and be a carrier for TSD. It is impossible to predict how often this might occur, but the possibility could be discussed in a genetic counseling session.
Using DNA sequencing as the primary screening test for non AJ patients would have several advantages over HexA enzyme analysis. In addition to advantages listed in the introduction, prenatal diagnosis by mutation analysis is immediately available for couples. Pseudodeficiency and LOTS alleles will be identified and the false negative results for individuals with the B1 phenotype will be avoided. Even dried filter paper spots can be used for DNA sequencing analysis allowing samples to submitted from remote areas. Saliva testing and buccal swab testing would also be possible.
Gene sequencing is not 100% sensitive either. In the initial study 3 individuals had negative sequencing results and positive enzyme results. In addition DNA sequencing will not detect the non-Jewsish TSD allele 7.6 kb deletion (7.6 kb delex1). This deletion is detected in our common mutation panel. If DNA sequencing is introduced as a primary screen for non-Jewish individuals, a separate assay would need to be performed to detect this mutation.
Since neither enzyme based testing nor molecular based testing is capable of detecting all TSD carriers, the combination of both modalities will provide the highest sensitivity. Although not appropriate for all patients, this should certainly be considered for a spouse of a known carrier in order to optimize the negative predictive value of the testing.
However, for routine carrier testing, the data suggests that for an AJ, a common mutation analysis is sufficient. For a non-AJ, DNA HexA sequencing with a 7.6 kb del assay could replace the enzyme based assay and yield equal, if not higher sensitivity and specificity for carrier detection. Therefore, now that DNA sequencing can be performed at a comparable cost and turn around time to HexA enzyme analysis it may be time to consider molecular testing as first line approach to TSD carrier screening in all populations.
![]()
![]()
REFERENCES
- Kaback, G., Lim-Steele, J., Dabholkar, D., Brown, D., Levy, N. and Zeiger K. (1993) Tay-Sachs disease—Carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. The international TSD data collection network. JAMA, 270, 2307- 2315. doi:10.1001/jama.1993.03510190063028
- O’Brien, J.S., Okada, S., Chen, A. and Fillerup, D.L. (1970) Tay-Sachs disease. Detection of heterozygotes and homozygotes by serum hexosaminidase assay. The New England Journal of Medicine, 283, 15-20. doi:10.1056/NEJM197007022830104
- ACOG Committee on Genetics. (2005) Screening for Tay-Sachs disease. Obstetrics & Gynecology, 106, 893- 894.
- Navon, R., Mark, Z., Mashiah, S. and Padeh, B. (1973) Determination of Tay-Sachs genotypes in pregnant women. Clinical Genetics, 4, 286-287. doi:10.1111/j.1399-0004.1973.tb01158.x
- Nitowsky, H.M., Davis, J., Nakagawa, S. and Fox, D. (1979) Human hexosaminidase isozymes. IV. Effects of oral contraceptive steroids on serum hexosaminidase activity. American Journal of Obstetrics & Gynecology, 134, 642-647.
- Bach, G., Tomczak, J., Risch, N. and Ekstein, J. (2001) Tay-Sachs screening in the Jewish Ashkenazi population: DNA testing is the preferred procedure. American Journal of Medical Genetics, 99, 70-75. doi:10.1002/1096-8628(20010215)99:1<70::AID-AJMG1120>3.0.CO;2-0
- Triggs-Raine, B.L., Mules, E.H., Kaback, M.M., LimSteele, J.S., Dowling, C.E., Akerman, B.R., Natowicz, M.R., Grebner, E.E., Navon, R. and Welch, J.P. (1992) A pseudodeficiency allele common in non-Jewish TaySachs carriers: Implications for carrier screening. The American Journal of Human Genetics, 51, 793-801.
- Cao, Z., Natowicz, M.R., Kaback, M.M., Lim-Steele, J.S., Prence, E.M., Brown, D., Chabot, T. and Triggs-Raine, B.L. (1993) A second mutation associated with apparent b-hexosaminidase A pseudodeficiency: Identification and frequency estimation. The American Journal of Human Genetics, 53, 1198-1205.
- Kytzia, H.J. and Sandhoff, K. (1985) Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. Journal of Biological Chemistry, 260, 7568- 7572.
- Rozenberg, R., Kok, F. and Burin, M.G. (2006) Diagnosis and molecular characterization of non-classic forms of Tay-Sachs disease in Brazil. Journal of Child Neurology, 21, 540-544.
- Grebner, E.E. and Tomczak, J. (1991) Distribution of three a-chain b-hexosaminidase A mutations among TaySachs carriers. The American Journal of Human Genetics, 48, 604-607.
- Tutor, J.C. (2004) Biochemical characterization of the GM2 gangliosidosis B1 variant. Brazilian Journal of Medical and Biological Research, 37, 777-783. doi:10.1590/S0100-879X2004000600001
- Whitley, C.B., Anderson, R.A. and McIvor, R.S. (1992) Heterozygosity for the “DN allele” (G533->A) of the bhexosaminidase a subunit gene identified by direct DNA sequencing in a family with the B1 variant of GM2-gangliosidosis. Neuropediatrics, 23, 96-101. doi:10.1055/s-2008-1071320
- Triggs-Raine, B.L., Feigenbaum, A.S., Natowicz, M., Skomorowski, M.A., Schuster, S.M., Clarke, J.T., Mahuran, D.J., Kolodny, E.H. and Gravel, R.A. (1990) Screening for carriers of Tay-Sachs disease among Ashkenazi Jews. A comparison of DNA-based and enzyme-based tests. The New England Journal of Medicine, 323, 6-12. doi:10.1056/NEJM199007053230102
- Park, N.J., Morgan, C., Sharma, R., et al. (2010) Improving accuracy of Tay-Sachs carrier screening of the non-Jewish population: Analysis of 34 carriers and six late-onset patients with HEXA enzyme and DNA sequence analysis, Pediatric Research, 67, 217-220. doi:10.1203/PDR.0b013e3181c6e318
- Huang, D., Chen, C., Sun, W., Strom, C.M. and Bender, R.A. (2004) High-throughput gene sequencing assay development for hereditary nonpolyposis colon cancer. Clinical Colorectal Cancer, 4, 275-279. doi:10.3816/CCC.2004.n.027
- Strom, C.M., Janeczko, R.A., Anderson, B., et al. (2005) Technical validation of a multiplex platform to detect thirty mutations in eight genetic diseases prevalent in individuals of Ashkenazi Jewish descent. Genetics in Medicine, 7, 633-639. doi:10.1097/01.gim.0000187120.93597.16
- Snyder, R.A. and Brady, R.O. (1969) The use of white cells as a source of diagnostic material for lipid storage diseases. Clinica Chimica Acta, 25, 331-338. doi:10.1016/0009-8981(69)90275-7
- Lowry, O.H., Rosebrough, N.J., Farral, A.L. and Randall, R.J. (1951) Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry, 193, 265-275.
- O’Brien, J.S., Okada, S., Chen, A. and Fillerup, D.L. (1970) Tay-Sachs disease: Detection of heterozygotes and homozygotes by serum hexosaminidase assay. The New England Journal of Medicine, 283, 15-20.
- PolyPhen-2. http://genetics.bwh.harvard.edu/pph2/
- Chelly, A. S., Lacorte, J.-M., Poenara, L. and Kahn, A. (2004) Seven novel Tay-Sachs mutations detected by chemical mismatch cleavage of PCR-amplified DNA fragments. Genomics, 11, 124-134.
NOTES
*Corresponding author.