Open Journal of Genetics
Vol.2 No.1(2012), Article ID:17760,6 pages DOI:10.4236/ojgen.2012.21007
Non genetic risk factors of Long-QT Syndrome
![]()
1Department of Biochemistry, Medical University of Silesia, Sosnowiec, Poland
2Department of Biopharmacy, Medical University of Silesia, Sosnowiec, Poland
Email: *ejaniszewska@sum.edu.pl
Received 3 November 2011; revised 17 December 2011; accepted 23 January 2012
Keywords: Long-QT Syndrome Acquired; Congenital; Drug-Induced; Non-Genetic Factors; Environmental Factors
ABSTRACT
The purpose of the present study is to provide guidelines regarding risk factors that may worsen the LongQT Syndrome (LQTS), based on available literature. This review evaluates the current knowledge on these risk factors of acquired LQTS, with an emphasis on non genetic risk factors, including environmental factors. PubMed was searched for literature in English from 1999 to 2011 on the molecular and clinical studies of Long-QT Syndrome. We agree, with recent investigations described in the literature, that variety of factors, inherited or environmental, can influence expression of ion channel proteins with impact on repolarization.
1. INTRODUCTION
The Long-QT Syndrome (LQTS) is a disorder affecting ion channels that is characterized by: long QT interval (> 450 ms) on electrocardiogram (ECG) corrected for heart rate (QTc), occurrence of polymorphic ventricular tachycardia TdP (torsade de pointes), fainting, and sudden cardiac death (SCD). The typical clinical presentation is the occurrence of syncope or cardiac arrest in young and otherwise healthy individuals [1]. Cardiac events can be induced by environmental factors and use of certain drugs. The purpose of the present study is to provide guidelines regarding risk factors that may worsen the syndrome, based on available literature. This review evaluates the current knowledge on the risk factors of acquired LQTS with an emphasis on the various, including environmental, risk factors.
2. EXPERIMENTAL PROCEDURES
PubMed was searched for literature data from 1999 to 2011 on the clinical and molecular studies of Long-QT Syndrome. The papers and their literature references were examined. Additional information was achieved from our study experiences.
3. DISCUSSION OF RESEARCH RESULTS
The symptoms of the Long-QT (LQTS) syndrome include tachycardia, syncope or cardiac arrest, often related to psychological or physical stress in young, usually healthy individuals. The syndrome can have inherited or acquired background and is commonly caused by mutations in the potassium channel or a lack of inactivation of the sodium channels and up to the present 12 candidate genes were identified. The most common causes of acquired conditions are electrolyte abnormalities, intracranial disease, dietary deficiencies, myocardial infarction (MI), dilated cardiomyopathy (DCM), mitral valve prolapse (MVP), bradycardia, severe malnutrition among anorexic patients and the use of medication [1-4].
During the past decade pharmaceutical companies have been faced with the withdrawal of some of their marketed drugs because of rare, yet lethal, postmarketing reports associated with ventricular arrhythmias. The implicated drugs include antiarrhythmics, but also noncardiac drugs, such as histamine blockers, antipsychotics, and antibiotics [3]. These undesired effects involve prolongation of the QT interval, which may lead to characteristic ventricular tachyarrhythmias, known as torsade de pointes (TdP). These clinical symptoms of the acquired LQTS are also found in an inherited form of the disease, called congenital LQTS. Nowadays, a number of environmental (non-genetic) and genetic risk factors of acquired LQTS have been described. Non-genetic factors include age and gender-dependent factors, hypokalemia or hypomagnesemia, and other heart diseases [2,3,5,6].
Furthermore, there is also evidence that interindividual differences in drug metabolism caused by functional polymorphisms in drug-metabolizing enzyme genes may be a risk factor of acquired LQTS, especially if multiple drugs are involved [5].
It is logical that concomitant administration of more than one drug which prolongs repolarization, would increase risk of drug-induced LQTS. In some cases, the mechanism of increased risk is due to drug-drug interactions altering metabolism, rather than by simple additive effects on rapid activated potassium current channel IKr. Many drugs are metabolized by the cytochrome P450 superfamily of proteins, and CYP3A is the predominant cytochrome P450 in human adult liver. Several drugs which are substrates for CYP3A are also IKr blockers, including erythromycin and terfenadine [2-4].
Environmental and genetic factors interact to define susceptibility to drug-induced long QT syndrome. Although erythromycin induces long QT syndrome, substantial variability exists with regard to its incidence. Erythromycin (100 μmol/l) produced no block of I(hERG) at 22˚C but produced significant block at 37˚C. The extent of block of I(hERG) increased linearly (r = 0.46, p < 0.01) as temperature increased between 36˚C and 42˚C. To assess physiologic relevance, action potential duration (APD) was recorded at temperatures between 36˚C and 42˚C in neonatal ventricular myocytes. Significantly greater prolongation of APD by erythromycin was observed at 42˚C compared with 37˚C. To assess whether transmembrane diffusion of erythromycin was the ratelimiting step for block of I(hERG) at 22˚C, erythromycin was applied within the patch pipette. Under these conditions, erythromycin rapidly blocked I(hERG) even at 22˚C. The F656C mutation in the distal S6 of KCNH2 gene completely abrogated block of I(hERG) measured at 37˚C. Progressively greater block of hERG and prolongation of APD by erythromycin was observed at temperatures between 36 and 42 degrees C. Temperaturedependent block of I(hERG) is explained by temperature-dependent access of erythromycin to the intracellular binding site at F656 [7].
The clinical characteristics of patients with different LQTS mutations have many similarities, although the frequency of various triggering factors for cardiac events may differ somewhat in the various forms of LQTS. Exercise and exertion are important triggering factors in LQT1, abrupt loud noise with startle are frequently initiating factors in LQT2, and cardiac events frequently occur during inactivity in LQT3. Emotional stress with sudden or sustained activation of the sympathetic nervous system and accompanying catecholamine stimulation of the heart have been hypothesized as important physiologic mechanisms for triggering cardiac events in patients with LQTS. Antiadrenergic therapies with betablockers or surgical left cervicothoracic sympathetic ganglionectomy have been a mainstay in the management of patients with this disorder [8].
These LQTS-related treatments, as well as other primary, secondary, and tertiary prevention methods, have led to a decline in sudden cardiac death (SCD) due to LQTS, making prospective studies more difficult and expensive. An alternative approach is to focus on arrhythmogenic cardiac events and use each patient as his/her own control, using a method called the “case-crossover” interview. Here the patient describes the circumstances preceding an event of interest as well as those preceding a specified control date in that patient's life. Anger, exertion, and sexual activity have all been shown to increase the risk of acute MI using this methodology. Two previous studies in recipients of implantable cardioverterdefibrillators (ICDs) have examined life-threatening arrhythmias as an outcome and found positive associations with emotional stress and physical exertion. No study has demonstrated an association between positive emotion or other potentially protective factors and any health outcome in any population, and none have been performed in LQTS patients [8].
Happiness is associated with a reduction in the 24- hour risk of cardiac events in patients with LQTS, with stress having an opposite effect. First report, published in 2009 by Lane et al. [8], indicated that positive emotion may have a protective effect on life-threatening ventricular arrhythmias. This study lends further support to the role of emotions in influencing cardiac events in arrhythmia-prone patients.
Research on cardiovascular correlates of positive emotion has been sparse, but current evidence is consistent with findings by Lane et al. [8]. Positive emotion tends to be associated with heart rate decrease as compared to heart rate increase associated with negative emotion, faster recovery times following cardiovascular activation, lower blood pressure, and increases in the high-frequency (vagal) component of heart rate variability. These findings are consistent with greater parasympathetic activity counteracting sympathetic effects. In the context of coronary artery disease (CAD), vagal tone and reflexes are known to be protective against SCD [6,8].
There are at least two other risk factors for acquired arrhythmia that can be mechanistically linked to IKr: gender and hypokalemia. Women are more vulnerable to torsade de pointes caused by IKr-blocking drugs than men, although two recent, apparently contradictory, studies are illustrative of the further complexity of this gender-based susceptibility. Preliminary studies have indicated that sex hormones may contribute to QT shortening in males or the lack of shortening in females [3,4,9].
Hypokalemia is another commonly observed risk factor of drug-induced LQTS. Low extracellular potassium level paradoxically reduces IKr, either by enhanced inactivation or exaggerated competitive blockage by sodium. Thus, even in the absence of a drug, hypokalemia prolongs the QT interval. Furthermore, drug IKr blockage is increased by lowering extracellular potassium. Correction of serum potassium levels to the high-normal range has been shown to correct the QT interval, both in congenital LQT2 and drug-induced LQTS. Recent conversion from atrial fibrillation was noted in early series of drug-induced LQTS, with the prevailing hypothesis that increased risk was secondary to relative bradycardia, another recognized risk factor [2,3].
In several studies of patients with torsade de pointes, hypokalemia was considered a contributory factor in the context of missense mutations in either HERG (S818L and V822M) or MiRP1 (Q9E) genes. This has been attributed to several properties of HERG (Human Ether-á- go-go-Related-Gene); the most well characterized of these being contrary to what one would expect, outward HERG current is decreased by reduction of extracellular potassium near physiological levels despite an increase in electrochemical gradient. Therefore hypokalemic patients receiving IKr blockers such as quinidine and dofetilide are at increased risk for acquired long QT syndrome because of an already reduced repolarization reserve. Increasing serum potassium in patients suffering from hypokalemia-induced torsade de pointes shortens the QT interval and attenuates abnormalities in myocardial repolarization [4,10].
Metabolic conditions that produce electrolyte imbalance, reduced renal and hepatic clearance of drugs, cardiac dysfunction, starvation, and polypharmacy can have effect on cardiac repolarization and also predispose affected persons to TdP [3].
High-level arsenic exposure is consistently associated with QT prolongation, a risk factor of arrhythmia and sudden cardiac death. Arsenic may act on QT by increasing cardiac calcium currents. In 2009 Mordukchovich et al. [11] hypothesized that low-level arsenic exposure would be associated with QT duration and that this effect would be stronger among persons not using calcium channel blockers. They performed a cross-sectional analysis in elderly men from the Normative Aging Study to analyze associations between toenail arsenic and QT and heart rate-corrected QT (QTc) durations and to examine effect modification by calcium channel blocker use, using linear regression and adjusting for potential confounders. Participants were examined in Boston, Massachusetts, between 2000 and 2002 or in 2006. An interquartile range increase in arsenic concentration was associated with a 3.8-millisecond increase in QT (95% confidence interval: 0.82, 6.8) and a 2.5-millisecond increase in QTc (95% confidence interval: 0.11, 4.9). There was no evidence of effect modification by medication use for either QT (P = 0.93) or QTc (P = 0.58). The authors observed positive associations between a biomarker of arsenic exposure and QT duration but found no evidence of effect modification by calcium channel blocker use, possibly because of modest power [11].
Arsenic may prolong QT interval duration by increasing cardiac calcium currents, which regulate the plateau phase of the cardiac action potential. The metal may also act by reducing surface expression of the cardiac potassium channel human ether-a go-go related gene protein product (hERG), which is essential for repolarization of cardiac myocytes. Arsenic interferes with hERG trafficking to the cell surface by inhibiting hERG-chaperone complexes. The mechanism, by which arsenic increases cardiac calcium currents, is unclear but may be the result of direct enzymatic modification of the calcium channel or its accessory proteins. In addition to changes in cardiac ion channels, possible mechanisms of arsenic-induced QT prolongation include alterations in DNA repair and methylation, generation of reactive oxygen species, and induction of cardiomyocyte apoptosis [11].
Several studies have demonstrated that obesity or severe dieting, per se, is associated with specific cardiac changes, including prolongation of QT interval [4,7]. The regional fat distribution may also be an important environmental factor in determining the QTc interval. Several studies have indicated that QTc may be affected by carbohydrate metabolism [6].
Overall, there was a significant association between impaired fasting serum glucose (IFG) and the QTc interval with an increase of 2.6 ms (95% confidence interval (CI): 0.3; 5.0) in those with fasting glucose > 6 mmol/l. Hyperinsulinemia was also associated with QTc prolongation (3.0 ms (0.8; 5.3)) in those with fasting insulin > or = 100 pmol/l. Impaired fasting glucose (IFG) and hyperinsulinemia were significantly associated with a decrease of the RR interval (–33.7 ms (–48.8; –18.6) and –44.4 ms (–58.7; –30.0) respectively). Participants in the fourth quartile of the QTc and QT intervals had a significantly increased risk of sudden cardiac death compared to participants in the first quartile (hazard ratio (HR) 2.87 (95% CI: 2.02 - 4.06); HR 3.05 (1.99 - 4.67) respectively). Furthermore, there was a significant inverse association between the fourth quartile of the RR interval compared to the first quartile and the risk of sudden cardiac death (HR 0.49 (0.34 - 0.80)) [12]. Population-based study, prepared in 2010 by van Noord et al. [12], demonstrated that IFG and hyperinsulinemia are associated with a significantly increased QTc interval and with significant shortening of the RR interval, the latter probably due to an increased sympathetic activity. In addition, they demonstrated that both prolonged QTc interval and shortened RR interval are associated with the increased risk of sudden cardiac death [3,12].
The presence of arterial hypertension, diabetic cardiovascular complications and electrocardiographic abnormalities of left ventricular hypertrophy and conduction disturbances were associated with increased QT dispersion in diabetes mellitus [3,12,13].
Overall measurements of QT dispersion, except for adjacent QT dispersion, were greater among diabetic patients than in the control group. Even the subgroup of diabetic patients with normal ECGs, no arterial hyper-tension, no degenerative complications (microor macro-vascular) and shorter duration of diabetes, had higher QT dispersion measurements when compared to the control group [14].
Clinical hypoxia has been shown to increase QT dispersion (QTd). Hypoxia may alter ventricular repolarization and QT dispersion in several ways: hypoxia severity is not the same over different regions of the ventricle; in some regions cellular adenosine triphosphate (ATP) decrease results in activation of ATP sensitive potassium channels; stress under hypoxia may increase beta-adrenergic signaling and the resulting cyclic adenosine monophosphate dependent (cAMP) phosphorylation of some potassium channels and calcium channels may shorten the action potential and QT interval. Osturk et al. [14]
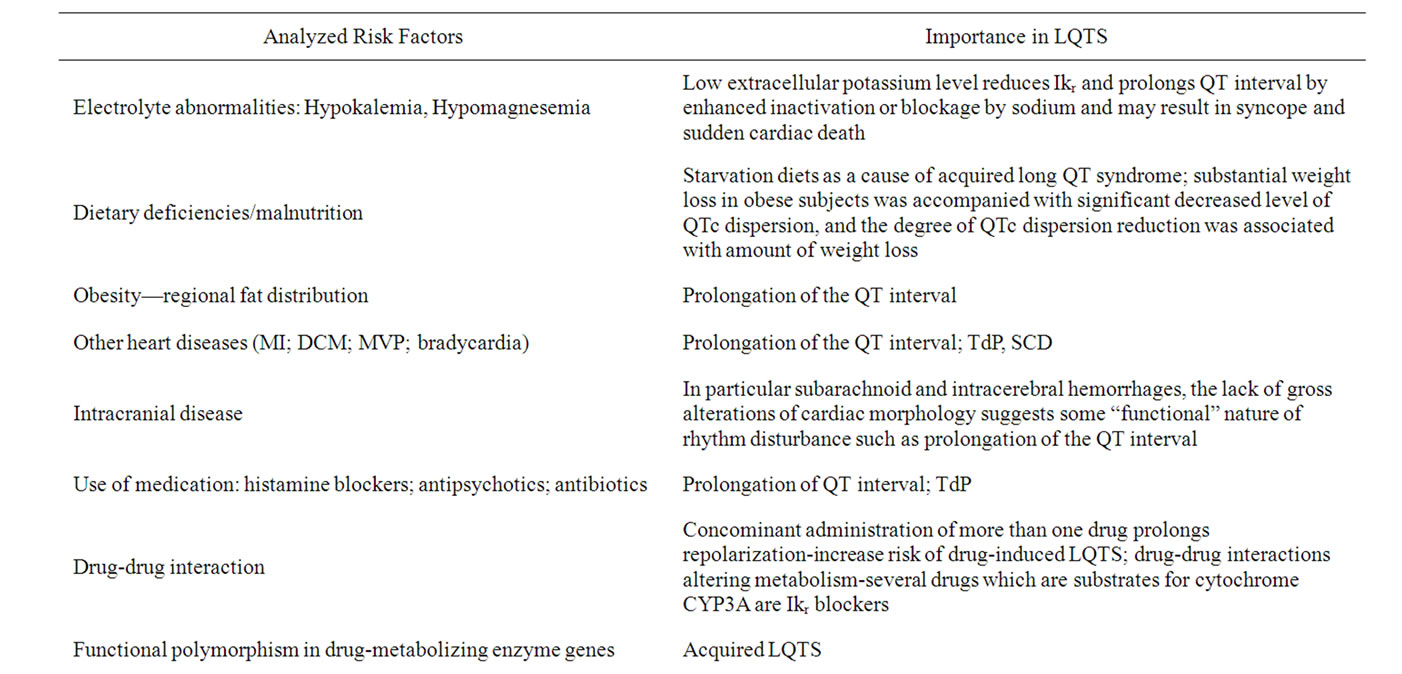

Table 1. Risk factors and their significance in LQTS.
aimed to examine QTd during hypobaric chamber training to observe the effect of hypobaric hypoxia on QT dispersion. QT dispersion values were as follows: prehypoxic 64.09 ± 8.39 ms, hypoxic 50.35 ± 11.06 ms and posthypoxic 59.83 ± 9.06 ms (Median: 64, 50, 60; Mean rank: 2.65, 1.28, 2.07) (p = 0.0001 for prehypoxic-hypoxic, p = 0.046-prehypoxic-posthypoxic, and p = 0.002 for posthypoxic-hypoxic). Heart rate (HR) values were as follows: prehypoxic 74.09 ± 6.43 beats/min, hypoxic 127.1 ± 17.39 beats/min, and posthypoxic 95.17 ± 11.35 beats/min (Median: 75, 122, 92; Mean rank: 1, 3, 2) (p = 0.0001 for prehypoxic-hypoxic, prehypoxic-posthypoxic, and posthypoxic-hypoxic). The change in QTd (QT duration) and HR during hypobaric chamber exposure was statistically significant but the change in QTcd was not (p < 0.001, p < 0.001, p > 0.1, respectively). From the findings of this study, it is not possible to directly comment on the validity of QTd in revealing arrhythmogenic predisposition of healthy subjects exposed to hypobaric hypoxia. The relationship between QT dispersion and hypobaric hypoxic exposure is not clear, particularly when QTd is corrected for the increased heart rate. QT dispersion measurement has not been proven a reliable and practical method to show arrhythmia predisposition during a hypobaric hypoxic exposure in healthy individuals [14].
4. CONCLUSION
We agree, with recent investigations described in the literature, that variety of factors, inherited or environmental, can influence expression of ion channel proteins with impact on repolarization. That environment can impact on genetic determinants of repolarization for intervals of varying duration (as in cardiac memory) or for the life of an individual (when spontaneous mutation occurs) is a concept that is not as generally appreciated as it likely should be [15]. In summary, data presented above indicate that both genetic factors and shared family environment factors play a role in determining the variation in PR interval, QRS duration, QTc interval, and HR (see Table 1). Furthermore, since these results showed that genetic factors and the adjusted covariates contribute no more than 60% to the variance of the ECG parameters, other environmental factors, gene-gene interactions, geneenvironment interactions could also influence these phenotypes and remain to be identified [16].
REFERENCES
- Moric-Janiszewska, E., Głogowska-Ligus, J., Paul-Samojedny, M., Smolik, S., Woźniak, M., Markiewicz- Łoskot, G., Mazurek, U., Węglarz, L. and Szydłowski, L. (2011) Expression of genes KCNQ1 and HERG encoding potassium ion channels lkr, lks in long QT syndrome. Kardiologia Polska, 69, 423-429.
- Kannankeril Prince, J. and Roden, D.M. (2007) Druginduced long QT and torsade de pointes: Recent advances. Current Opinion in Cardiology, 22, 39-43. doi:10.1097/HCO.0b013e32801129eb
- Kunkler, K. (2002) Acquired long QT syndrome: Risk assessment, prudent prescribing and monitoring, and patient education. Journal of the American Academy of Nurse Practitioners, 14, 382-389. doi:10.1111/j.1745-7599.2002.tb00139.x
- Paulussen, A.D.C. and Aerssens, J. (2005) Risk factors for drug-induced long-QT Syndrome. Netherlands Heart Journal, 13, 47-56.
- Aerssens, J. and Paulussen, A.D.C. (2005) Pharmacogenomics and acquired long QT syndrome. Review Pharmacogenomics, 6, 259-270.
- Friedlander, Y., Lapidos, T., Sinnreich, R. and Kark, J.D. (1999) Genetic and environmental sources of QT interval variability in Israeli families: The kibbutz settlements family study. Clinical Genetics, 56, 200-299. doi:10.1034/j.1399-0004.1999.560304.x
- Guo, J., Zhan, S., Lees-Miller, J.P., Teng, G. and Duff, H.J. (2005) Exaggerated block of hERG (KCNH2) and prolongation of action potential duration by erythromycin at temperatures between 37 degrees C and 42 degrees C. Heart Rhythm, 2, 860-866. doi:10.1016/j.hrthm.2005.04.029
- Lane, R.D., Reis, H.T., Peterson, D.R., Zareba, W. and Moss, A.J. (2009). Happiness and stress alter susceptibility to cardiac events in Long-QT Syndrome. Annals of Noninvasive Electrocardiology, 14, 193-200. doi:10.1111/j.1542-474X.2009.00295.x
- Moric-Janiszewska, E., Głogowska-Ligus, J., Paul-Samojedny, M., Węglarz, L., Markiewicz-Łoskot, G. and Szydłowski, L. (2011) Ageand sex-dependent mRNA expression of KCNQ1 and HERG in patients of Long-QT Syndrome Type 1 and Type 2. Archives of Medical Science, 7, 941-947. doi:10.5114/aoms.2011.26604
- Anantharam, A., Markowicz, S.M. and Abbott, G.W. (2003) Pharmacogenetic considerations in diseases of cardiac ion channels. Journal of Pharmacology and Experimental Therapeutics, 307, 831-838. doi:10.1124/jpet.103.054569
- Mordukhovich, I., Wright, R.O., Amarasiriwardena, C.H., Baja, E., Baccarelli, A., Suh, H., Sparrow, D., Vokonas, P. and Schwartz, J. (2009) Association between low-level environmental arsenic exposure and QT interval duration in a general population study. American Journal of Epidemiology, 170, 739-746. doi:10.1093/aje/kwp191
- Van Noord, C., Sturkenboom, M.C., Straus, S.M., Hofman, A., Kors, J.A., Witteman, J.C. and Stricker, B.H. (2010). Serum glucose and insulin are associated with QTc and RR intervals in nondiabetic elderly. European Journal of Endocrinology, 162, 241-248. doi:10.1530/EJE-09-0878
- Cardoso, C., Salles, G., Bloch, K., Deccache, W. and Siqueira-Filho, A.G. (2001) Clinical determinants of increased QT dispersion in patients with diabetes mellitus. International Journal of Cardiology, 79, 253-262.
- Oztürk, C., Sen, A., Açikel, C.H. and Akin, A. (2008) QT dispersion during hypobaric hypoxia. Anadolu Kardiyoloji Dergisi, 8, 266-270. doi:10.1016/S0167-5273(01)00443-0
- Rosen, M.R. and Cohen, I.S. (2006) Molecular/genetic determinants of repolarization and their modification by environmental stress. Journal of Internal Medicine, 259, 7-23. doi:10.1111/j.1365-2796.2005.01592.x
- Li, J., Huo, Y., Zhang, Y., Fang, Z., Yang, J., Zang, T., Xu, X. and Xu, X. (2009). Familial aggregation and heritability of electrocardiographic intervals and heart rate in a rural chinese population. Annals of Noninvasive Electrocardiology, 14, 147-152. doi:10.1111/j.1542-474X.2009.00289.x
NOTES
*Corresponding author.