International Journal of Organic Chemistry
Vol.05 No.04(2015), Article ID:62156,11 pages
10.4236/ijoc.2015.54027
Chemoenzymatic Synthesis of an Enantiomerically Enriched Bicyclic Carbocycle Using Candida parapsilosis ATCC 7330 Mediated Enantioselective Hydrolysis
Thangavelu Saravanan1, Anju Chadha1,2*, Tarur Konikkaledom Dinesh3,4, Namasivayam Palani4*, Sengottuvelan Balasubramanian3*
1Laboratory of Bioorganic Chemistry, Department of Biotechnology, Indian Institute of Technology Madras, Chennai, India
2National Center for Catalysis Research, Indian Institute of Technology Madras, Chennai, India
3Department of Inorganic Chemistry, University of Madras, Guindy Campus, Chennai, India
4ITC Life Sciences and Technology Centre, ITC Limited, Peenya Industrial Area, Phase I, Bangalore, India

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 November 2015; accepted 21 December 2015; published 24 December 2015
ABSTRACT
Enantiomerically enriched (R)-1-(2-bromocycloalkenyl)-3-buten-1-ol and its derivatives were obtained via enantioselective hydrolysis [resolution] with good enantioselectivities (E = 31 to >500) using Candida parapsilosis ATCC 7330. The various reaction parameters were optimized for enantioselective hydrolysis to achieve high enantiomeric excess (ee) and conversions. Among the substrates tested, (RS)-1-(2-bromocyclohex-1-en-1-yl) but-3-yn-1-yl acetate was hydrolysed by the biocatalyst in 12 h to the corresponding (R)-alcohol in 49% conversion and >99 ee. The optically pure allylic alcohol thus obtained was used as a chiral starting material for the synthesis of an enantiomerically enriched bicyclic alcohol effectively establishing achemoenzymatic route.
Keywords:
Chemoenzymatic Synthesis, Enantioselective Hydrolysis, Candida parapsilosis ATCC 7330, Hydrolases, Bicyclic Carbocycle

1. Introduction
Enantiomerically enriched allylic and propargylic alcohols are very useful intermediates in the asymmetric synthesis of more complex molecules [1] [2] and are also resourceful synthons for the synthesis of various biologically active compounds [3] [4] . Several methods are available for the synthesis of such systems, e.g. enantioselective addition of chiral allylboranes [5] and allyltitanation [6] to aldehydes, asymmetric organozinc additions to aldehydes [7] [8] and the elimination of chiral vinyl sulfoxides [9] . The widely used methods for the preparation of these intermediates are enantioselective acylation of secondary allylic alcohols and enantioselective hydrolysis of the corresponding esters catalysed by enzymes and nonenzymatic chiral catalysts [1] [10] - [13] . In particular, enantiomerically pure 1-(2’-bromocycloalkenyl)-3-buten-1-ol and its derivatives can be cyclised and utilized for the synthesis of optically active condensed carbocycles (Scheme 1). These bicyclic allylic alcohols can be oxidized to bicyclicenones which are important chiral synthons for the development of biologically active compounds [14] [15] and is an important structural feature of many natural products [16] - [19] . Synthesis of various bicyclicenones by intramolecular cyclization has been described in literature [20] - [22] . Intramolecular tri butyl tin hydride mediated cyclization of optically active 1-(2’-bromocycloalkenyl)-3-buten-1-ols is a potential route for the synthesis of optically pure condensed carbocyles [23] - [25] . However, synthesis of optically pure carbocycles from the corresponding chiral intermediates i.e., enantiopure allylic alcohols has not been reported.
Various chemical [1] [22] and biocatalytic [26] - [29] methods are available for the synthesis of enantiomerically pure aryl/alkyl-allyl alcohols. Lipases, generally obtained from microorganisms, are the most widely used enzymes in organic synthesis, mainly due to their high enantioselectivity [30] - [32] , the possibility of using non- conventional solvents like organic solvents and ionic liquids [33] and are economical, stable, and easy to reprocess. Among the methods reported, only few specific examples are available for the lipase mediated resolution of aliphatic cyclic allylic alcohols. In this context, Mc Cubbin et al. resolved a series of aryl and aliphatic vinyl chlorohydrins by enzymatic kinetic resolution using Amano lipase AK. The resolution of alkyl and cyclic aliphatic vinyl chlorohydrins was a fast reaction but the enantioselectivity was lower as compared to that of aryl substrates [34] . Takabe et al. reported the one-pot acid promoted catalyzed cyclisation of geranyl amine N-oxide to synthesise 1-acetyl-4,4-dimethyl-1-cyclohexene which in turn was reduced and resolved by Amano lipase AK with good enantioselectivity [35] . Similarly Kuriata et al. synthesized chiral bicycle [3.1.0] hexane derivatives via chemoenzymatic synthesis in which the synthesized (+)-1-[(1S,5R)-6,6-dimethylbicyclo[3.1.0] hex-2-en-2- yl)]ethanol was resolved by a transesterification strategy using Amano PS lipase from B. cepacia [36] . However, the enantioselective hydrolysis of the corresponding acetate using various lipases from B. cepacia and C. rugosa was unsuccessful. The versatility of the biocatalyst Candida parapsilosis ATCC 7330 was established in our lab. This yeast is used for the preparation of a variety of enantiopure compounds by deracemization [36] - [39] , asymmetric reduction [40] [41] and oxidative kinetic resolution [42] .
This biocatalyst is also reported for resolution of N-protected amino acid esters with excellent enantioselectivity (E = 40 to >500) [43] . In continued efforts to expand the scope of the biocatalyst, herein we report the enantioselective hydrolysis of 1-(2-bromocycloalkenyl) but-3-en-1-yl acetates using fermenting cells of C. parapsilosis ATCC 7330. These cyclic acetates with different functionalities (bromo and allyl/propargyl), in addition to yielding important chiral synthons, provide an interesting set of substrates to study hydrolysis using C. parapsilosis ATCC 7330. To the best of our knowledge, this is the first report for the synthesis of enantiomerically enriched1-(2-bromocycloalkenyl) but-3-en-1-ol and its derivatives by biocatalyst mediated enantioselective hydrolysis. The enantiopure enriched 1-(2-bromocycloalkenyl) but-3-en-1-ol thus obtained was further cyclized to 3-methyl-2,3,4,5,6,7-hexahydro-1-H-inden-1-ol which can be used for the synthesis of optically pure condensed carbocycles.
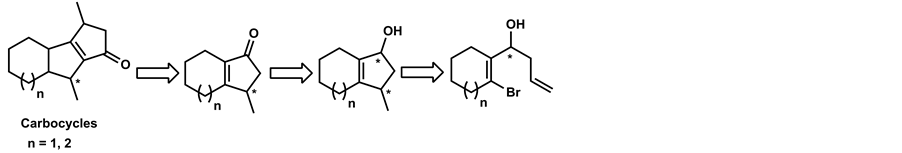
Scheme 1. Retrosynthetic route for carbocycle from bicyclic allylic alcohol.
2. Result and Discussion
Initially, the model substrate (RS)-1-(2-bromocyclohex-1-en-1-yl) but-3-en-1-yl acetate 1a was subjected to enantioselective hydrolysis with commercially available lipases (Lipase PS, Amano lipase AK and CAL-B) under reported conditions [44] . All the lipases tested gave <5% conversion even after 3 days. Further, enantioselective hydrolysis of (RS)-1-(2-bromocyclohex-1-en-1-yl) but-3-en-1-yl acetate 1a was carried out with the resting cells (14 h culture) of Candida parapsilosis ATCC 7330 using our earlier reported conditions [43] . In this reaction, the substrate 1a (0.2 mmol) was incubated with the resting cells of C. parapsilosis ATCC 7330 suspended in water (20 mL; 6 g wet cell mass from 300 mL culture) using ethanol as a cosolvent (0.5% v/v). The product 2a was obtained with moderate conversion (26%) and only 20% ee as analysed by gas chromatography (GC) using a chiral column (Scheme 2). The poor enantioselectivity could be due to the presence of multiple hydrolases with opposite enantioselectivities for the same substrate. Next, the substrate 1a (0.08 mmol) was dissolved in cosolvent ethanol (0.5% v/v) and directly added in to a 12 h fermenting culture (50 mL) at 25˚C and incubation was continued. After 48 h the product was obtained with an improvement in ee (89%) but the conversion was very poor (8%).
To further improve the conversion and enantiomeric excess, the reaction conditions were optimized with 1a as follows.
2.1. Optimization of Cosolvents
Reportedly, conversion and enantioselectivity in lipase catalysed hydrolysis can be improved by the addition of a suitable cosolvent [45] . First, the reaction was carried out in the absence of a cosolvent and the product was obtained with 82% ee and 19% conversion. When the reaction was carried out with ethanol, isopropyl ether or acetone as a cosolvent, the conversion dropped from 19 to 8%. Similar observations have been reported by Szymanski et al. for the enzymatic hydrolysis of α-acetoxyamides when acetone was used as a cosolvent [46] . Dioxane and DMSO showed moderate conversion (25% - 37%). All long chain hydrophopic cosolvents (n-octane, isooctane and hexane) gave good conversion (up to 56%). This result is consistent with the literature reports that hydrophobic solvents favor the binding of hydrophobic substrates to lipase [27] . Among the cosolvents tested in this study, only hexane showed reasonable conversion (40%) and good ee (90%) (Figure 1).

Scheme 2. Enantioselective hydrolysis of (RS)-1a.

Figure 1. Optimization of cosolvents.
2.2. Optimization of Reaction Time
Further, the reaction time was varied from 12 to 60 h using hexane as cosolvent. It was found that 48 h showed improvement in both conversion (40%) and enantiomeric excess (89.6%) (Table 1). Further increase in the reaction time resulted in decrease in ee (82.2%). Hence 48 h was chosen as the optimum incubation time for further experiments.
2.3. Effect of Temperature
To study the effect of temperature on conversion and ee of enantioselective hydrolysis, the reaction was performed at various temperatures from 25˚C - 35˚C. The product 2a was obtained with good conversion (40%) and ee (89%) at 25˚C (Table 2). With increasing temperature (30˚C & 35˚C), the conversion also increased (66 & 68% respectively) but the enantiomeric excess reduced sharply (61 & 57% respectively) (Table 2). This Candida grows best at 25˚C [36] , which supports the observation that maximum conversion is obtained at 25˚C. All further experiments were carried out at 25˚C.
2.4. Optimization of Substrate Concentration
In whole cell biocatalysis, conversion and enantioselectivity can be controlled by optimizing the substrate concentration due to the presence of multiple enzymes with opposite stereo preferences and different Km values for the same substrate [45] . The substrate 1a was added in varied concentrations (1 - 2.5 mM) keeping all the conditions identical (Figure 2). Notably, higher conversion (53%) was observed with low substrate concentration (1 mM), while the enantiomeric excess was moderate (75%). At a substrate concentration of 1.5 mM, the enanti-

Figure 2. Optimization of substrate concentration (1a).

Table 1. Optimization of reaction time for improved conversion and enantiomeric excess.
Table 2. Effect of temperature.
omeric excess increased to 89% with good conversion (39%). Further increase in the substrate concentration showed a drop in conversion however the ee was maintained. Hence, 1.5 mM of substrate was chosen as the optimum substrate concentration.
2.5. Substrate Scope
Under the optimized reaction conditions, three substrates were used (Scheme 3). The experiment with substrate 1a yielded product (R)-1-(2-bromocyclohex-1-en-1-yl)but-3-en-1-ol 2a in 33% isolated yield and 89% ee (E = 31) (Table 3, entry 2a). To further explore the substrate scope of this biocatalyst, substrate 1b (Scheme 3) was subjected to hydrolysis using fermenting cells of C. parapsilosis ATCC 7330. Initially, under the optimized reaction time of 48 h, a moderate conversion (20%) was obtained with excellent ee (>99%). The conversion was further improved to 32% by extending the reaction time to 60 h and the product (R)-1-(2-bromocyclohept-1-en-1-yl) but-3-en-1-ol 2b was isolated with moderate yield (29%) and excellent ee (> 99%) (E => 200) (Table 3, entry 2b). The obtained high enantioselectivity suggests that the substrate with the 7-membered ring is seemingly better than the one with the 6-membered ring for the biocatalyst. Another substrate 1c (Scheme 3) with an alkyne chain was subjected for enantioselective hydrolysis and the product (R)-1-(2-bromocyclohex-1-en-1-yl) but-3- yn-1-ol 2c was obtained with excellent ee (>99%) in lesser reaction time (12h) compared to substrates 1a & 1b. Further increase in the reaction time resulted in decrease in ee. The product 2c was isolated in 42% yield and 99% ee (E => 500) (Table 3, entry 2c).
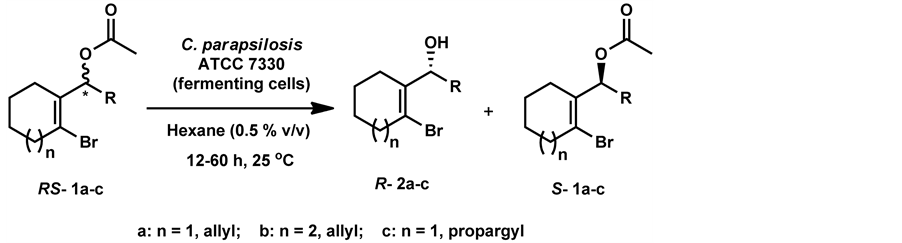
Scheme 3. Enantioselective hydrolysis of (RS)-1a-c.
Table 3. Enantioselective hydrolysis of (RS)-1a-1c.
aAbsolute configuration was assigned by comparing specific rotation value with the analogue substrate [47] ; bConversion and ee was determined by GC using Chirasil Dex CB chiral column; cEnantiomeric excess of the unreacted acetates were determined after hydrolysis using 5% methanolic KOH; dEnantioselectivity ratios were calculated according to: E = ln[(eep(1 − ees)/( eep+ees)]/ln[(eep(1 + ees)/(eep + ees) [48] ; E values in the range 200 - 500 are mentioned as >200, more than 500 as >500.
1H NMR (500 MHz,CDCl3) δ:5.86 (dd, J = 8,5.5 Hz, 1H), 5.74 - 5.82 (m, 1H), 5.10 - 5.15 (ddd, J = 17, 3.5, 1.5 Hz, 1H), 5.06 - 5.09 (ddd, J = 9.5, 1.5, 0.5 Hz, 1H), 2.51 - 2.55 (m, 2H), 2.44 - 2.50 (m, 1H), 2.36 - 2.41 (m, 1H), 2.08 - 2.17 (m, 2H), 2.06 (brs, 3H), 1.66 - 1.72 (m, 3H), 1.61 - 1.64 (m, 1H); 13C NMR (125 MHz, CDCl3) δ: 169.9, 133.7, 133.3, 121.3, 117.6, 75.7, 36.7, 36.6, 25.9, 24.5, 21.9, 20.9; HRMS for C12H17O2Br (Cal: 295.0310 [M + Na]+, Found: 295.0310); ee = 59%, retention times 15.0 (S, major), 15.5 min (R, minor); [α] D25 + 1.1 (c 1, CHCl3).
1H NMR (500 MHz, CDCl3) δ: 5.74 - 5.79 (m, 2H), 5.06 - 5.11 (m, 1H), 5.03 - 5.04 (m, 1H), 2.72 - 2.79 (m, 2H), 2.37 - 2.43 (m, 1H), 2.27 - 2.35(m, 1H), 2.19 - 2.26 (dd, J = 6.5, 5 Hz, 2H), 2.02 (brs, 3H), 1.70 - 1.74 (m, 2H), 1.53 - 1.56 (m, 3H), 1.38 - 1.52 (m, 1H); 13C NMR (125 MHz, CDCl3) δ: 169.9, 138.7, 133.2, 125.0, 117.7, 76.7, 41.5, 36.7, 31.6, 27.9, 25.9, 25.1, 20.9; HRMS forC13H19O2Br (Cal: 309.0466 [M + Na]+, Found: 309.0466); ee = 46%, retention times 20.3 (S, major), 21.5 min (R, minor); [α] D25 + 1.7 (c 1, CHCl3).
1H NMR (500 MHz, CDCl3) δ: 5.87 - 5.89 (m, 1H), 2.56 - 2.66 (m, 2H), 2.52 - 2.54 (m, 2H), 2.18 - 2.28 (m, 1H), 2.08 (s, 3H), 1.97 - 1.98 (t, J = 2.5 Hz, 1H), 1.62 - 1.75 (m, 4H), 1.59 (brs, 1H); 13CNMR (125 MHz, CDCl3) δ: 169.7, 133.1, 122.1, 79.4, 74.0, 70.2, 36.7, 26.0, 24.4, 22.4, 21.8, 20.8; HRMS for C12H15O2Br (Cal: 293.0153 [M + Na]+, Found: 293.0154); ee => 99%, retention times19.4 (R, minor), 19.8 min (S, major); [α] D25 + 3.2 (c 0.5, CHCl3).
1H NMR (500 MHz,CDCl3) δ: 5.78 - 5.86 (m, 1H), 5.12 - 5.18 (m, 2H), 4.78 - 4.81 (dd = t, J = 6.5 Hz, 1H), 2.50 - 2.52 (m, 2H), 2.28 - 2.34 (m, 3H), 2.04 - 2.10 (m, 1H), 1.63 - 1.74 (m, 5H); 13C NMR (125 MHz, CDCl3) δ:136.9, 134.3, 120.3, 118.0, 73.2, 38.9, 36.8, 25.4, 24.7, 22.1; ee = 89%; retention times 15.0 (S, minor), 15.5 min (R, major); [α]D25 + 25.4 (c 0.5, CHCl3).
Lipase PS (from Burkholderia cepacia) and Amano lipase AK (from Pseudomonas fluorescence) were obtained from Amano Pharmaceuticals Co., Nagoya, Japan. CAL B (Candida antarctica lipase B, Novozym 435) was purchased from Sigma. Candida parapsilosis ATCC 7330 was obtained from ATCC (Manassas, VA) and maintained at 4˚C in yeast malt agar medium that contained 5 g/L peptic digest of animal tissue, 3 g/L malt extract, 3 g/L yeast extract, 10 g/L dextrose and 20 g/L agar. All chemicals used were of analytical grade and distilled prior to use. TLC was carried out on Kieselgel 60 F254 aluminium sheets (Merck1.05554). 1H and 13C NMR spectra were recorded in CDCl3 solution on a Bruker AVANCE III 500 MHz spectrometer. Chemical shifts are expressed in ppm values using TMS as an internal standard. The enantiomeric excess was determined by gas chromatography using Perkin Elmer Clarus 600 gas chromatograph and Shimadzu GC 2014 gas chromatograph fitted with flame ionisation detector using VARIAN Chirasil Dex CB chiral column (0.25 μm × 25 mm × 30 m). The injector and detector were set at 220˚C, respectively. Nitrogen was used as the carrier gas. Optical rotations were determined using Autopol-IV automatic polarimeter.
3.2. General Procedure for the Synthesis of (RS)-1a-1c [23]
Tin powder (1.5 mmol) was suspended in a mixture of water (2 mL) and diethyl ether (2mL). To this mixture allyl bromide (3 mmol) was added and treated with few drops of 48% hydrobromic acid and the mixture was stirred well for 5 min at RT. To this reaction mixture bromoaldehyde (1 mmol) was added and the resultant mixture was stirred for 6 h at RT (reaction was monitored by TLC). After the complete conversion, the reaction was quenched with water (5 ml) and extracted with 20% dichloromethane/hexane mixture (10 mL × 3). The organic layers were combined and dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude reaction mixture was purified by silica gel column chromatography using hexane/ethyl acetate (9:1) as a mobile phase (Yield: 60% - 80%). The respective bromo alcohol (1 mmol) was stirred with (2 mL) of acetic anhydride containing catalytic amount of sodium acetate for 24 h at room temperature. The progress of the reaction was monitored by TLC. After the completion of reaction, the reaction mixture was diluted with dichloromethane and washed with water. The organic layer was dried over sodiumsulphate and concentrated under reduced pressure. The crude reaction mixture was purified by column chromatography over silica gel (100 - 200 mesh) using hexane/ethyl acetate mixture (9:1) as a mobile phase to obtain the pure product (Yield: 70% - 80%).

Scheme 4. Radical cyclization of 2a.
3.3. Growth Conditions for Candida parapsilosis ATCC 7330
Candida parapsilosis ATCC 7330 was pre-cultured for 12 h at 25˚C with shaking at 200rpm in yeast malt broth medium that contained 5 g/L peptic digest of animal tissue, 3 g/L malt extract, 3 g/L yeast extract and 10 g/L dextrose. The pre-cultured broth, 4 mL [4% (v/v)] was transferred to a 500 mL Erlenmeyer flask that contained 96 mL of yeast malt broth. The culture was grown on rotatory shaker at 25˚C and 200 rpm for 12 h.
3.4. Typical Procedure for Enantioselective Hydrolysis Using Candida parapsilosis ATCC 7330
To the 500 mL Erlenmeyer flask that contained 12 h cultivated cells (100 mL culture), the substrate (racemic acetate, 1.5 mM) in 500 μL of hexane as co-solvent was added and the incubation was continued at 25˚C and 200 rpm. After getting the maximum conversion and enantiomeric excess of the product, the cells were centrifuged at 6000 rpm for 10 min. The formed product and the remaining substrate were extracted from the supernatant using ethylacetate. The organic layer was dried over anhydrous sodium sulphate. The solvent was removed by evaporation and the crude extract was purified by silica gel column chromatography using hexane/ ethyl acetate (95:05) as a mobile phase eluent.
3.5. General Procedure for the Synthesis of 3a and 3b [23]
The respective enantiomerically enriched (R)-1-(2-bromocyclohex-1-en-1-yl)but-3-en-1-ol 2a (1 mmol) was dis- solved in ethanol (25 mL). To this bis-tri-n-butyltinoxide (5 mol%), sodium borohydride (1 mol) and catalytic amount of azobisisobutyro nitrile (AIBN) were added. The reaction mixture was refluxed for 3 h at 80˚C under N2 atmosphere and the reaction was monitored by TLC. After the completion of reaction, the reaction mixture was concentrated under reduced pressure. The products were purified by column chromatography over silica gel (100 - 200 mesh) using hexane/ethyl acetate mixture (9:1) as a mobile phase to obtain the cyclized product 3a and 3b (Yield: 70% - 80%).
3.6. Spectroscopic Characterization
3.6.1. (S)-1-(2-bromocyclohex-1-enyl) but-3-enyl acetate 1a
1H NMR (500 MHz,CDCl3) δ:5.86 (dd, J = 8,5.5 Hz, 1H), 5.74 - 5.82 (m, 1H), 5.10 - 5.15 (ddd, J = 17, 3.5, 1.5 Hz, 1H), 5.06 - 5.09 (ddd, J = 9.5, 1.5, 0.5 Hz, 1H), 2.51 - 2.55 (m, 2H), 2.44 - 2.50 (m, 1H), 2.36 - 2.41 (m, 1H), 2.08 - 2.17 (m, 2H), 2.06 (brs, 3H), 1.66 - 1.72 (m, 3H), 1.61 - 1.64 (m, 1H); 13C NMR (125 MHz, CDCl3) δ: 169.9, 133.7, 133.3, 121.3, 117.6, 75.7, 36.7, 36.6, 25.9, 24.5, 21.9, 20.9; HRMS for C12H17O2Br (Cal: 295.0310 [M + Na]+, Found: 295.0310); ee = 59%, retention times 15.0 (S, major), 15.5 min (R, minor); [α] D25 + 1.1 (c 1, CHCl3).
GC conditions: Perkin Elmer Clarus 600 gas chromatograph. The initial column temperature of 100˚C was held for 1 min, then raised to150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 180˚C at a rate of 1˚C/min and finally held at 170˚C for 3 min.
3.6.2. (S)-1-(2-bromocyclohept-1-enyl) but-3-enyl acetate 1b
1H NMR (500 MHz, CDCl3) δ: 5.74 - 5.79 (m, 2H), 5.06 - 5.11 (m, 1H), 5.03 - 5.04 (m, 1H), 2.72 - 2.79 (m, 2H), 2.37 - 2.43 (m, 1H), 2.27 - 2.35(m, 1H), 2.19 - 2.26 (dd, J = 6.5, 5 Hz, 2H), 2.02 (brs, 3H), 1.70 - 1.74 (m, 2H), 1.53 - 1.56 (m, 3H), 1.38 - 1.52 (m, 1H); 13C NMR (125 MHz, CDCl3) δ: 169.9, 138.7, 133.2, 125.0, 117.7, 76.7, 41.5, 36.7, 31.6, 27.9, 25.9, 25.1, 20.9; HRMS forC13H19O2Br (Cal: 309.0466 [M + Na]+, Found: 309.0466); ee = 46%, retention times 20.3 (S, major), 21.5 min (R, minor); [α] D25 + 1.7 (c 1, CHCl3).
GC conditions: Shimadzu GC 2014 gas chromatograph. The initial column temperature of 120˚C was held for 1 min, then raised to 150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 4 min.
3.6.3. (S)-1-(2-bromocyclohex-1-enyl) but-3-ynyl acetate 1c
1H NMR (500 MHz, CDCl3) δ: 5.87 - 5.89 (m, 1H), 2.56 - 2.66 (m, 2H), 2.52 - 2.54 (m, 2H), 2.18 - 2.28 (m, 1H), 2.08 (s, 3H), 1.97 - 1.98 (t, J = 2.5 Hz, 1H), 1.62 - 1.75 (m, 4H), 1.59 (brs, 1H); 13CNMR (125 MHz, CDCl3) δ: 169.7, 133.1, 122.1, 79.4, 74.0, 70.2, 36.7, 26.0, 24.4, 22.4, 21.8, 20.8; HRMS for C12H15O2Br (Cal: 293.0153 [M + Na]+, Found: 293.0154); ee => 99%, retention times19.4 (R, minor), 19.8 min (S, major); [α] D25 + 3.2 (c 0.5, CHCl3).
GC conditions: Shimadzu GC 2014 gas chromatograph. The initial column temperature of 120˚C was held for 1 min, then raised to 150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 4 min.
3.6.4. (R)-1-(2-bromocyclohex-1-enyl) but-3-en-1-ol 2a [20]
1H NMR (500 MHz,CDCl3) δ: 5.78 - 5.86 (m, 1H), 5.12 - 5.18 (m, 2H), 4.78 - 4.81 (dd = t, J = 6.5 Hz, 1H), 2.50 - 2.52 (m, 2H), 2.28 - 2.34 (m, 3H), 2.04 - 2.10 (m, 1H), 1.63 - 1.74 (m, 5H); 13C NMR (125 MHz, CDCl3) δ:136.9, 134.3, 120.3, 118.0, 73.2, 38.9, 36.8, 25.4, 24.7, 22.1; ee = 89%; retention times 15.0 (S, minor), 15.5 min (R, major); [α]D25 + 25.4 (c 0.5, CHCl3).
GC conditions: Perkin Elmer Clarus 600 gas chromatograph. The initial column temperature of 100˚C was held for 1 min, then raised to 150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 3 min. Split Ratio = 1:10. Flow rate = 1.5 ml/min.
3.6.5. (R)-1-(2-bromocyclohept-1-enyl) but-3-en-1-ol 2b [20]
1H NMR (500 MHz, CDCl3) δ: 5.79 - 5.88 (m, 1H), 5.13 - 5.19 (m, 2H), 4.78 - 4.81 (dd = t, J = 7 Hz, 1H), 2.74 - 2.84 (ddd, J = 11.5, 7, 3.5 Hz, 2H), 2.28 - 2.34 (m, 4H), 1.75 - 1.80 (m, 3H), 1.55 - 1.62 (m, 2H), 1.50 - 1.55 (m, 2H); 13C NMR (125 MHz, CDCl3) δ:142.2, 134.3, 123.4, 117.9, 74.7, 41.6, 39.0, 31.6, 27.3, 26.3, 25.3; ee => 99%; retention times 20.3 (S, minor), 21.5 min (R, major); [α] D25 + 15.8 (c 0.6, CHCl3).
GC conditions: Shimadzu GC 2014 gas chromatograph. The initial column temperature of 120˚C was held for 1 min, then raised to 150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 4 min.
3.6.6. (R)-1-(2-bromocyclohex-1-enyl) but-3-yn-1-ol 2c [21]
1H NMR (500MHz,CDCl3) δ: 4.89 - 4.91 (dd = t, J = 6.5 Hz 1H), 2.49 - 2.54 (m, 2H), 2.47 - 2.49 (m, 2H), 2.46 - 2.47 (m, 1H), 2.05 - 2.06 (dd = t, J = 3 Hz, 1H), 1.63 - 1.74 (m, 5H), 1.24 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 136.1, 121.0, 80.5, 72.3, 70.7, 36.8, 25.3, 24.7, 24.6, 22.0; ee = > 99%, retention times 19.4 (R, major), 19.8 min (S, minor); [α] D25 + 12.2 (c 0.5, CHCl3).
GC conditions: Shimadzu GC 2014 gas chromatograph. The initial column temperature of120˚C was held for 1 min, then raised to150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 4 min.
3.6.7. (1R, 3R)-2,3,4,5,6,7-hexahydro-3-methyl-1-H-inden-1-ol 3a
1H NMR (400 MHz, C6D6) δ:4.46 - 4.47 (d, J = 6.4 Hz 1H); 2.56 (brs, 1H); 2.15 - 2.19 (m, 1H); 1.85 - 1.90 (m, 3H); 1.50 - 1.66 (m, 7H); 0.86 - 0.88 (d, J = 6.8 Hz 3H); 13C NMR (100 MHz, CDCl3) δ: 143.9, 135.6, 78.6, 42.6, 39.6, 23.5, 22.8, 22.7, 22.7, 19.5; HRMS for C10H16O (Cal: 151.1123 [M]+, Found: 151.1121); (Diasteromeric ratio 1:1) ee = > 88%; retention times 8.52 min (R, major), 8.60 min (S, minor); [α] D25 + 8.3 (c 0.5, CHCl3).
GC conditions: Shimadzu GC 2014 gas chromatograph. The initial column temperature of 120˚C was held for 1 min, then raised to 150˚C at a rate of 8˚C/min and held at 150˚C for 2 min. Further the temperature raised to 170˚C at a rate of 1˚C/min and finally held at 170˚C for 4 min.
4. Conclusion
Enantiomerically enriched 1-bromohexa-1,5-dien-3-ols 2a-2c were synthesized with good ee (up to >99%) by enantioselective hydrolysis from the corresponding racemic acetates using fermenting cells of C. parapsilosis and are reported here for the first time. The reaction parameters like cosolvent (hexane), incubation time (12 - 60 h), temperature (25˚C) and substrate concentration (1.5 mM) were optimized to obtain maximum conversion and enantiomeric excess. Notably, the product 1-(2-bromocyclohex-1-en-1-yl) but-3-yn-1-ol 2c was obtained with good yield and excellent ee in less reaction time than the other two substrates (E => 500). (R)-1-(2-bromocyclo- hex-1-en-1-yl)but-3-en-1-ol was used as chiral starting material for the first time to prepare enantiomerically enriched (1R, 3R)-3-methyl-2,3,4,5,6,7-hexahydro-1-H-inden-1-ol and (1R, 3S) 3-methyl 2,3,4,5,6,7-hexahydro- 1-H-inden-1-ol with good ee (>85% ) via a chemoenzymatic approach.
Acknowledgements
One of the authors [TS] thanks UGC, New Delhi for Senior Research Fellowship, SAIF, IIT Madras for instrumentation facilities and Mr. T. Rathnavel Pandian for assistance in optimization study. TKD and NP thank ITC Life Sciences Technology Centre, Bangalore for providing the necessary infrastructure to carry out the experiments. Professor K. K. Balasubramanian, INSA Senior Scientist, Department of Biotechnology, IIT Madras is thanked for reviewing the paper and providing valuable input. Dr. Sowmyalakshmi Venkataraman, IIT Madras for helping with the analysis.
Disclaimer
All views expressed herein are authors’ views and in no way, expressed or implied, are that of or necessarily represent the positions of ITC Limited, my current employer.
Cite this paper
ThangaveluSaravanan,AnjuChadha,Tarur KonikkaledomDinesh,NamasivayamPalani,SengottuvelanBalasubramanian, (2015) Chemoenzymatic Synthesis of an Enantiomerically Enriched Bicyclic Carbocycle Using Candida parapsilosis ATCC 7330 Mediated Enantioselective Hydrolysis. International Journal of Organic Chemistry,05,271-281. doi: 10.4236/ijoc.2015.54027
References
- 1. Bellemin-Laponnaz, S., Tweddell, J., Ruble, J.C., Breitling, F.M. and Fu, G.C. (2000) The Kinetic Resolution of Allylic Alcohols by a Non-Enzymatic Acylation Catalyst; Application to Natural Product Synthesis. Chemical Communications, 1009-1010.
http://dx.doi.org/10.1039/b002041i - 2. Hoveyda, A.H., Evans, D.A. and Fu, G.C. (1993) Substrate-Directable Chemical Reactions. Chemical Reviews (Washington DC), 93, 1307-1370.
http://dx.doi.org/10.1021/cr00020a002 - 3. Fontana, A., d’Ippolito, G., D’Souza, L., Mollo, E. and Parameswaram, P.S. (2001) New Acetogenin Peroxides from the Indian Sponge Acarnus bicladotylota. Journal of Natural Products, 64, 131-133.
http://dx.doi.org/10.1021/np0002435 - 4. Roush, W.R. and Sciotti, R.J. (1998) Enantioselective Total Synthesis of (-)-Chlorothricolide via the Tandem Inter- and Intramolecular Diels-Alder Reaction of a Hexaenoate Intermediate. Journal of the American Chemical Society, 120, 7411-7419.
http://dx.doi.org/10.1021/ja980611f - 5. Marino, J.P., McClure, M.S., Holub, D.P., Comasseto, J.V and Tucci, F.C. (2002) Stereocontrolled Synthesis of (-)- Macrolactin A. Journal of the American Chemical Society, 124, 1664-1668.
http://dx.doi.org/10.1021/ja017177t - 6. BouzBouz, S., Pradaux, F., Cossy, J., Ferroud, C. and Falguieres, A. (2000) Enantioselective Synthesis of Propargylic Alcohols by Addition of Enantiopure Cyclopentadienyldialkoxyallyltitanium Complexes to Acetylenic Aldehydes. Tetrahedron Letters, 41, 8877-8880.
http://dx.doi.org/10.1016/S0040-4039(00)01597-5 - 7. Frantz, D.E., Faessler, R. and Carreira, E.M. (2000) Facile Enantioselective Synthesis of Propargylic Alcohols by Direct Addition of Terminal Alkynes to Aldehydes. Journal of the American Chemical Society, 122, 1806-1807.
http://dx.doi.org/10.1021/ja993838z - 8. Pu, L. and Yu, H.-B. (2001) Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds. Chemical Reviews (Washington DC), 101, 757-824.
http://dx.doi.org/10.1021/cr000411y - 9. Nakamura, S., Kusuda, S., Kawamura, K. and Toru, T. (2002) Preparation of Optically Pure Propargylic and Allylic Alcohols from 2-(Trimethylsilyl)vinyl Sulfoxides as a Chiral Ethynyl Anion Synthon: Computational Studies on Elimination Reaction of 2-(Trimethylsilyl)vinyl Sulfoxides. Journal of Organic Chemistry, 67, 640-647.
http://dx.doi.org/10.1021/jo0157223 - 10. Birman, V.B. and Jiang, H. (2005) Kinetic Resolution of Alcohols Using a 1,2-Dihydroimidazo[1,2-a]quinoline Enantioselective Acylation Catalyst. Organic Letters, 7, 3445-3447.
http://dx.doi.org/10.1021/ol051063v - 11. Rotticci, D., Norin, T. and Hult, K. (2000) Mass Transport Limitations Reduce the Effective Stereospecificity in Enzyme-Catalyzed Kinetic Resolution. Organic Letters, 2, 1373-1376.
http://dx.doi.org/10.1021/ol005639m - 12. Vedejs, E. and Daugulis, O. (1999) 2-Aryl-4,4,8-trimethyl-2-phosphabicyclo[3.3.0]octanes: Reactive Chiral Phosphine Catalysts for Enantioselective Acylation. Journal of the American Chemical Society, 121, 5813-5814.
http://dx.doi.org/10.1021/ja990133o - 13. Onaran, M.B. and Seto, C.T. (2003) Using a Lipase as a High-Throughput Screening Method for Measuring the Enantiomeric Excess of Allylic Acetates. Journal of Organic Chemistry, 68, 8136-8141.
http://dx.doi.org/10.1021/jo035067u - 14. Therasse, P., Arbuck, S.G., Eisenhauer, E.A., Wanders, J. and Kaplan, R.S. (2000) New Guidelines to Evaluate the Response to Treatment in Solid Tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. Journal of the National Cancer Institute, 92, 205-216.
http://dx.doi.org/10.1093/jnci/92.3.205 - 15. Trost, B.M. and Pinkerton, A.B. (2000) A New Strategy for Cyclopente-none Synthesis. Organic Letters, 2, 1601-1603.
http://dx.doi.org/10.1021/ol005853a - 16. Herrmann, J.L., Richman, J.E. and Schlessinger, R.H. (1973) Novel Linch-Pin Construction of Dihydrojasmone. High Yield Synthesis of Cis-Jasmone. Tetrahedron Letters, 14, 3275-3278.
http://dx.doi.org/10.1016/S0040-4039(01)86893-3 - 17. Mathew, J. and Alink, B. (1990) A Novel Route to Substituted Cyclopent-2-en-1-one; Application to the Synthesis of Cis-Jasmone and Dihydrojasmone. Journal of the Chemical Society, Chemical Communications, No. 9, 684-686.
http://dx.doi.org/10.1039/c39900000684 - 18. Mikolajczyk, M. and Balczewski, P. (1987) Methylenomycin B: A New Synthesis from a β-Keto Phosphonate. Synthesis, 1987, 659-661.
http://dx.doi.org/10.1055/s-1987-28041 - 19. Smith III, A.B., and Boschelli, D. (1983) Stereocontrolled Total Synthesis of (±)-Xanthocidin, Two Diastereomers, (±)-Epixanthocidin and (±)-β-Isoxanthocidin, and (±)-Dedihydroxy-4,5-didehydroxanthocidin, a Likely Biosynthetic Precursor. The Journal of Organic Chemistry, 48, 1217-1226.
http://dx.doi.org/10.1021/jo00156a015 - 20. Ray, D., Mal, S.K. and Ray, J.K. (2005) Palladium-Catalyzed Novel Cycloisomerization: An Unprecedented Domino Oxidative Cyclization towards Substituted Carbocycles. Synlett, 2005, 2135-2140.
http://dx.doi.org/10.1055/s-2005-872241 - 21. Ray, D., Nasima, Y., Sajal, M.K., Ray, P. and Urinda, S. (2013) Palladium-Catalyzed Intramolecular Oxidative Heck Cyclization and Its Application toward a Synthesis of (±)-β-Cuparenone Derivatives Supported by Computational Studies. Synthesis, 45, 1261-1269.
http://dx.doi.org/10.1055/s-0032-1316884 - 22. Vedejs, E. and MacKay, J.A. (2001) Kinetic Resolution of Allylic Alcohols Using a Chiral Phosphine Catalyst. Organic Letters, 3, 535-536.
http://dx.doi.org/10.1021/ol006923g - 23. Dinesh, T.K., Palani, N. and Balasubramanian, S. (2015) Intramolecular Radical Cyclization of Vinyl, Aryl and Alkyl Halides Using Catalytic Amount of Bis-tri-n-butyltin Oxide/Sodium Borohydride: A Novel Entry to Functionalized Carbocycles. Synlett, 26, 1055-1058.
http://dx.doi.org/10.1055/s-0034-1380325 - 24. Hart, D.J. (1984) Free-Radical Carbon-Carbon Bond Formation in Organic Synthesis. Science, 223, 883-887.
http://dx.doi.org/10.1126/science.223.4639.883 - 25. Stork, G. and Mook Jr., R., (1983) Vinyl Radical Cyclization. 2. Dicyclization via Selective Formation of Unsaturated Vinyl Radicals by Intramolecular Addition to Triple Bonds. Applications to the Synthesis of Butenolides and Furans. Journal of the American Chemical Society, 105, 3720-3722.
http://dx.doi.org/10.1021/ja00349a067 - 26. Lee, D., Huh, E.A., Kim, M.-J., Jung, H.M. and Koh, J.H. (2000) Dynamic Kinetic Resolution of Allylic Alcohols Mediated by Ruthenium- and Lipase-Based Catalysts. Organic Letters, 2, 2377-2379.
http://dx.doi.org/10.1021/ol006159y - 27. Kadnikova, E.N. and Thakor, V.A. (2008) Enantioselective Hydrolysis of 1-Arylallyl Acetates Catalyzed by Candida antarctica Lipase. Tetrahedron: Asymmetry, 19, 1053-1058.
http://dx.doi.org/10.1016/j.tetasy.2008.04.018 - 28. Marques, FA., Oliveira, M.A., Frensch, G., Sales Maia, B.H.L.N. and Barison, A. (2011) Highly Efficient Kinetic Resolution of Allylic Alcohols with Terminal Double Bond. Letters in Organic Chemistry, 8, 696-700.
http://dx.doi.org/10.2174/157017811799304151 - 29. Chen, P. and Xiang, P. (2011) Kinetic Resolution of Allylic Alcohols via Stereoselective Acylation Catalyzed by Lipase PS-30. Tetrahedron Letters, 52, 5758-5760.
http://dx.doi.org/10.1016/j.tetlet.2011.08.093 - 30. Lau, R.M., van Rantwijk, F., Seddon, K.R. and Sheldon, R.A. (2000) Lipase-Catalyzed Reactions in Ionic Liquids. Organic Letters, 2, 4189-4191.
http://dx.doi.org/10.1021/ol006732d - 31. Reetz, M.T. (2002) Lipases as Practical Biocatalysts. Current Opinion in Chemical Biology, 6, 145-150.
http://dx.doi.org/10.1016/S1367-5931(02)00297-1 - 32. Palani, N., Chadha, A. and Balasubramanian, K.K. (1998) Mechanism of Lithium Perchlorate/Diethyl Ether-Catalyzed Rearrangement of α- and β-Endo- and -Exo-Dicyclopentadienyl Vinyl Ethers: Use of Deuterium Labeling and a Chiral Probe. The Journal of Organic Chemistry, 63, 5318-5323.
http://dx.doi.org/10.1021/jo970715t - 33. Vidya, P. and Chadha, A (2010) Pseudomonas cepacia Lipase Catalyzed Esterification and Transesterification of 3-(Furan-2-yl) Propanoic Acid/Ethyl Ester: A Comparison in Ionic Liquids vs Hexane. Journal of Molecular Catalysis B: Enzymatic, 65, 68-72.
http://dx.doi.org/10.1016/j.molcatb.2010.01.032 - 34. McCubbin, J.A., Maddess, M.L. and Lautens, M. (2008) Enzymatic Resolution of Chlorohydrins for the Synthesis of Enantiomerically Enriched 2-Vinyloxiranes. Synlett, 2008, 289-293.
- 35. Takabe, K., Yamada, T., Miyamoto, T. and Mase, N. (2008) Cyclization of N,N-Diethylgeranylamine N-Oxide in One-Pot Operation: Preparation of Cyclic Terpenoid-Aroma Chemicals. Tetrahedron Letters, 49, 6016-6018.
http://dx.doi.org/10.1016/j.tetlet.2008.08.001 - 36. Chadha, A. and Baskar, B. (2002) Biocatalytic Deracemization of α-Hydroxy Esters: High Yield Preparation of (S)-Ethyl 2-Hydroxy-4-phenylbutanoate from the Racemate. Tetrahedron: Asymmetry, 13, 1461-1464.
http://dx.doi.org/10.1016/S0957-4166(02)00403-2 - 37. Baskar, B., Pandian, N.G., Priya, K. and Chadha, A. (2005) Deracemization of Aryl Substituted α-Hydroxy Esters Using Candida parapsilosis ATCC 7330: Effect of Substrate Structure and Mechanism. Tetrahedron, 61, 12296-12306.
http://dx.doi.org/10.1016/j.tet.2005.09.104 - 38. Saravanan, T., Jana, S. and Chadha, A. (2014) Utilization of Whole Cell Mediated Deracemization in a Chemoenzymatic Synthesis of Enantiomerically Enriched Polycyclic Chromeno[4,3-b] Pyrrolidines. Organic & Biomolecular Chemistry, 12, 4682-4690.
http://dx.doi.org/10.1039/c4ob00615a - 39. Titu, D. and Chadha, A. (2008) Enantiomerically Pure Allylic Alcohols: Preparation by Candida parapsilosis ATCC 7330 Mediated Deracemisation. Tetrahedron: Asymmetry, 19, 1698-1701.
http://dx.doi.org/10.1016/j.tetasy.2008.07.012 - 40. Mahajabeen, P. and Chadha, A. (2011) One-Pot Synthesis of Enantiomerically Pure 1,2-Diols: Asymmetric Reduction of Aromatic α-Oxo Aldehydes Catalyzed by Candida parapsilosis ATCC 7330. Tetrahedron: Asymmetry, 22, 2156- 2160.
http://dx.doi.org/10.1016/j.tetasy.2011.12.008 - 41. Venkataraman, S. and Chadha, A. (2015) Preparation of Enantiomerically Enriched (S)-ethyl 3-Hydroxy 4,4,4-Trifluorobutanoate Using Whole Cells of Candida parapsilosis ATCC 7330. Journal of Fluorine Chemistry, 169, 66-71.
http://dx.doi.org/10.1016/j.jfluchem.2014.11.004 - 42. Sivakumari, T. and Chadha, A. (2014) Regio- and Enantio-Selective Oxidation of Diols by Candida parapsilosis ATCC 7330. RSC Advances, 4, 60526-60533.
http://dx.doi.org/10.1039/C4RA08146C - 43. Stella, S. and Chadha, A. (2010) Resolution of N-Protected Amino Acid Esters Using Whole Cells of Candida parapsilosis ATCC 7330. Tetrahedron: Asymmetry, 21, 457-460.
http://dx.doi.org/10.1016/j.tetasy.2010.02.011 - 44. Waldinger, C., Schneider, M., Botta, M., Corelli, F. and Summa, V. (1996) Aryl Propargylic Alcohols of High Enantiomeric Purity via Lipase Catalyzed Resolutions. Tetrahedron: Asymmetry, 7, 1485-1488.
http://dx.doi.org/10.1016/0957-4166(96)00166-8 - 45. Kinoshita, M. and Ohno, A. (1996) Factors Influencing Enantioselectivity of Lipase-Catalyzed Hydrolysis. Tetrahedron, 52, 5397-5406.
http://dx.doi.org/10.1016/0040-4020(96)00179-2 - 46. Szymanski, W. and Ostaszewski, R. (2007) Chemoenzymatic Synthesis of Enantiomerically Enriched α-Hydroxyamides. Journal of Molecular Catalysis B: Enzymatic, 47, 125-128.
http://dx.doi.org/10.1016/j.molcatb.2007.04.007 - 47. Malkov, A.V., Barlog, M., Jewkes, Y., Mikusek, J. and Kocovsky, P. (2011) Enantioselective Allylation of α,β-Unsaturated Aldehydes with Allyltrichlorosilane Catalyzed by METHOX. The Journal of Organic Chemistry, 76, 4800-4804.
http://dx.doi.org/10.1021/jo200712p - 48. Rakels, J.L.L., Straathof, A.J.J. and Heijnen, J.J. (1993) A Simple Method to Determine the Enantiomeric Ratio in Enantioselective Biocatalysis. Enzyme and Microbial Technology, 15, 1051-1056.
http://dx.doi.org/10.1016/0141-0229(93)90053-5
NOTES

*Corresponding authors.