 Vol.1, No.2, 49-61 (2011) doi:10.4236/ojpm.2011.12008 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ Open Journal of Preventive Medicine Anoikis induction by glycoprotein from Laminaria japonica in HT-29 cells Hiroe Go, Hye-Jung Hwang, In-Hye Kim, Taek-Jeong Nam* Department of Food Science and Nutrition, Pukyong National University, Busan, Korea; *Corresponding author: namtj@pknu.ac.kr Received 10 May 2011; revised 1 July 2011; accepted 20 July 2011. ABSTRACT We extracted a glycoprotein from the brown alga Laminaria japonica (LJGP). We previously demonstrated that LJGP induced apop- tosis in HT-29 colon cancer cells via the Fas- and the mitochondrial signaling pathway, and cell-cycle arrest. However, its effect on the cell membrane remained unknown. In this study, we identified the involvement of matrix metalloproteinase (MMP), integrin, and Epi- thelial (E)-cadherin in LJGP-induced apoptosis in HT-29 cells. LJGP treatment increased the expression and activity of MMP-2 and MMP-9. Furthermore, LJGP de- creased the expression of integrin αν, β3, β5, β6 and E- cadherin. Consistent with a decreased expression of E-cadherin, LJGP inhibited the Wnt signaling pathway. Moreover, activation of downstream molecules of integrin, including focal adhesion kinase (FAK), the Src family of protooncogenic tyrosine kinases, extracellular signal-related kinase (ERK), and phosphatidyl inositol 3 kinase (PI-3K) were also decreased. These findings suggest that LJGP-induced apoptosis of HT-29 cells involves possible ECM disruption and cell detachment, which are exe- cuted principally through the activation of MMPs and by a decrease of adhesion molecules, contributing to a down-regulation of the PI-3K, MAPK, and Wnt signaling pathways. Apoptosis induced by ECM disruption or cell detachment is also known as anoikis. We can say that LJGP induces anoikis in HT-29 cells. Keywords: Anoikis; Glycoprotein; Integrin; Laminaria Japonica; MMP; Seaweed 1. INTRODUCTION Loss of cell anchorage, including cell-matrix anchor- age and cell-cell anchorage, induces apoptosis. The growth of most cells is anchorage-dependent. To main- tain the survival of anchorage-dependent cells, contact with components of the extracellular matrix (ECM) in vivo or contact with hydrophilic, negatively charged sur- faces in vitro are required. Therefore, in the absence of ECM attachment, cell death is induced. This mode of cell-death induction is known as detachment-induced anoikis [1,2]. The term anoikis, a Greek word meaning “homeless- ness,” is a specific type of apoptosis induced by detach- ment from the ECM, loss of cell adhesion, or inappro- priate cell adhesion [3-5]. When epithelial and endothe- lial cells detach from the ECM, these cells normally un- dergo apoptosis; thus, anoikis can suppress the expan- sion of oncogenically transformed cells and allow the maintenance of tissue-cell homeostasis [3-6]. Anoikis has been demonstrated and studied in numerous genu- inely adherent cell types [7]. In contrast to non-trans- formed cells, cancer cells are notoriously able to resist anoikis, and this property has been proposed to contrib- ute to metastasis and new tumor growth beyond the original environment of the cancer cells [8]. Unsurpris- ingly, obtaining resistance to anoikis is a critical step in the metastatic process [2]. Conversely, it may be said that anoikis functions as a physiological barrier to me- tastasis. In this study, we focused on the action induced by LJGP on the cell surface. On the cell surface, there are cell-ECM and/or cell-cell anchorages, and these an- chorages are dependent on the cell-surface receptors. Integrins are cell-surface adhesion receptors composed of heterodimeric complexes of noncovalently linked and subunits, which work alongside other proteins such as cadherins, cell-adhesion molecules, and selectins to mediate cell-cell and cell-matrix adhesive interactions and communications [9]. The functional contribution of integrins is essential for cell proliferation, survival, and migration, as integrins recognize the major adhesive ECM components, fibronectin and laminin [6]. Integrins can sense mechanical forces arising from the ECM,  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 50 converting these stimuli to chemical signals capable of modulating intracellular signal transduction to allow rapid and flexible responses to changes in the environ- ment [5]. Integrin-mediated extracellular signals stimulate a va- riety of intracellular signaling events, including adaptor proteins, such as focal adhesion kinase (FAK), integrin- linked kinase (ILK), the Src family of protooncogenic tyrosine kinases, and Shc, as well as mitogen-activated protein kinase (MAPK) pathways, leading to extracellu- lar signal-regulated kinase (ERK) activation [10]. Acti- vated ERK translocates to the nucleus, where it activates several transcription factors [11]. Binding of integrin to ECM components also stimulates structural proteins, such as phosphatidylinositol-3 kinase (PI-3K), leading to activation of Akt and serves as a link between the ECM and the cytoskeleton [11]. In several studies, degradation of the ECM by MMPs has been shown to be a signal that can induce apoptosis [12]. MMPs constitute a family of extracellular proteases that degrade ECM components important under a variety of normal and pathological conditions [13]. E-cadherin is a type I cadherin transmembrane glyco- protein adhesion molecule that mediates the formation of cell–cell adhesion junctions. In epithelial cells, E-cad- herin is known to play an important role in the develop- ment and maintenance of morphological integrity. Therefore, loss of E-cadherin could have profound ef- fects on epithelial-cell homeostasis [14]. E-cadherins form intracellular complexes with members of the cat- enin family, which link the E-cadherin to the actin cy- toskeleton [15]. Besides playing an important role in cell-cell adhesion, E-cadherin also plays a role in the Wnt signaling pathway through association with β-cat- enin. The Wnt signaling pathway plays important roles during the development of many organ systems and tu- morigenesis [15]. Deregulated Wnt signaling is a key factor for the development of several human cancers, including breast tumorigenesis and colorectal carcinoma [14]. In the present study, we identified a novel action of LJGP on the cell surface, especially in regulation of cell anchorage. In addition, integrin-signaling pathways and the Wnt-signaling pathway may be involved in LJGP- induced apoptosis, that is, detachment-induced anoikis. 2. MATERIALS AND METHODS 2.1. Preparation of LJGP LJGP was prepared as described previously [16]. Briefly, L. japonica was purchased in the traditional market of Gijang-gun, washed, and stored at –20˚C until used. The sample (160 g) was cut into small pieces and steeped in 1 L distilled water for 6 h at room temperature. The aqueous extract was clarified by centrifugation to remove insoluble materials. The extract was then filtered, and three volumes of ethanol were added. The ethanol extract was filtered, and ammonium sulfate was added to precipitate the proteins. The filtrates were combined, pelleted with ammonium sulfate, dissolved in distilled water, dialyzed in distilled water overnight, and concen- trated by rotary evaporation at 40˚C. A concentrated so- lution was distributed into 1.5-ml tubes, lyophilized, and stored at –70˚C until use. Proteins in the lyophilized samples were separated by sodium dodecyl sulfate– polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to periodic acid-Schiff (PAS) staining (Gel- Code®Glycoprotein Staining Kit; Pierce Co., Ltd., Rockford, IL) to determine the presence of glycoprotein. The extracts were solubilized in double-distilled H2O for use in the assays. 2.2. Cell Culture HT-29 colon cancer cells (ATCC HTB-38) were ob- tained from the American Type Culture Collection (Rockville, MD). HT-29 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine se- rum (Hyclone, Inc., South Logan, UT), 100 U/ml peni- cillin, and 100 mg/ml streptomycin and were maintained in a humidified incubator at 37˚C under an atmosphere of 5% CO2. The medium was replaced every two days. 2.3. Western Blot Analysis Cells were cultured to 80% confluence at 37˚C. Cells were then maintained in serum-free medium (SFM) for 12 h. After 12 h, cells were incubated in SFM containing various concentrations (0 - 100 μg/ml) of LJGP for 24 h and then harvested. To prepare whole-cell extracts, the cells were washed with phosphate-buffered saline (PBS) and suspended in a cell-signaling lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophos- phate, 1 mM β-glycerophosphate) containing protease inhibitor (1 mg/ml aprotinin, 1 mg/ml leupeptin, 1 mg/ml pepstatin A, 200 mM Na3VO4, 500 mM NaF, 100 mM PMSF). Lysates were incubated at 4˚C for 30 min and then clarified at 15,000 g for 10 min at 4˚C to remove detergent-insoluble material. The soluble lysates were recovered and used as whole-cell extracts. For the preparation of cytosolic extracts, the cells were washed with PBS, and then 1.0 ml trypsin-EDTA solution was added until the cell layer dispersed. Cells were harvested in PBS and resuspended in lysis buffer (20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl2) containing protease inhibitors. The solutions were incubated for 30 min at 4˚C. During this period, solutions were vortexed five times. The solutions were  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 51 51 centrifuged at 3500 g for 10 min at 4˚C. The super- natants were transferred to new tubes and centrifuged at 1400 g for 15 min at 4˚C. The resulting supernatants were the cytosolic fractions. To prepare nuclear extracts, the cells were suspended in 500 μl buffer A (10 mM Tris (pH 7.5), 5 mM MgCl2, 0.05% TritonX-100) containing protease inhibitors. After incubation at 4˚C for 15 min, the mixtures were centri- fuged at 12,000 g for 15 min. Released nuclei were re- suspended in buffer B (10 mM Tris (pH 7.4), 5 mM MgCl2) and buffer C (10 mM Tris (pH 7.4), 4 mM MgCl2, 1 M NaCl) containing protease inhibitors and incubated for 30 min at 4˚C. After centrifugation at 12,000 g for 15 min, 80% glycerol was added to the su- pernatants to a final concentration of 20%. To prepare cell membranes, cells were suspended in extraction buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100) containing protease inhibitors and stored overnight at –70˚C. The cells were then de- frosted and placed on ice. To lyse the cells, the suspen- sion was repeatedly aspirated and expelled. Finally, the lysates were incubated at 4˚C for 30 min and then clari- fied by centrifugation at 30,000 g for 30 min at 4˚C. The resulting supernatants were the cell membrane fractions. Protein concentration was determined using a bicin- chonic acid protein assay kit (Pierce Biotechnology). The proteins in the extracts (100 g) were separated us- ing 7.5% - 15% SDS-PAGE and transferred to a polyvi- nylidene fluoride membrane (Millipore, Billerica, MA). The membranes were blocked with 1% bovine serum albumin in TBS-T (10 mM Tris–HCl, 150 mM NaCl [pH 7.5], 0.1% Tween 20) and then incubated with the indi- cated antibodies: anti-MMP-9 (H-129; 1:1000), anti- MMP-2 (H-76; 1:1000), anti-TIMP-1 (H-150; 1:1000), anti-GAPDH (FL-335; 1:1000), anti-integrin αν (H-75; 1:1000), anti-integrin β3 (H-96; 1:500), anti-integrin β5 (H-96; 1:1000), anti-integrin β6 (H-110; 1:1000), anti-E- cadherin (H-108; 1:1000), anti- -catenin (C-18; 1:1000), anti-c-myc (C-19; 1:1000), anti-cyclin D1 (H-295; 1: 1000), anti-phospho-ERK1/2 (E-4; 1:1000), anti-phos- pho-p38 (D-8; 1:500), anti-JNK (D-2; 1:1000), anti- phospho-JNK (G-7; 1:1000), anti-p38α/β (H-147; 1: 1000), anti- -actin (C-2; 1:500), anti-PI 3-kinase p85α (B-9; 1:1000), anti-phospho-Akt1/2/3 (Ser473-R; 1:1000), anti-Akt1 (C-20; 1:1000), anti-c-Src (SRC2; 1:1000), and anti-FAK (C-20; 1:1000) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-ERK1/2 (1: 1000), anti-phospho-FAK (Tyr397; 1:1000), and anti- phospho-Src Family (Tyr416; 1:1000) antibodies from Cell Signaling Technology (Beverly, MA). The mem- branes were then incubated overnight at 4˚C with gentle shaking. The secondary antibody was a peroxidase- conjugated goat anti-mouse or -rabbit antibody (1:10,000; GE Healthcare Bioscience, Piscataway, NJ) and horse- radish peroxidase-conjugated goat ExactaCruzTM A (1:10,000) (Santa Cruz). The antibody-bound proteins were visualized using an enhanced chemiluminescence Western blotting kit (Amersham Biosciences, Piscataway, NJ). 2.4. Immunoprecipitation Analysis Formation of E-cadherin and β-catenin was analyzed in cytosolic extracts prepared as follows. Cells were cultured to 80% confluence at 37˚C and then maintained in SFM for 12 h. After 12 h, the cells were incubated with SFM containing various concentrations of LJGP (0 - 100 μg/ml) for 24 h. Cytosolic extracts (1000 μg) were incubated overnight with primary antibodies (anti-E- cadherin) with gentle agitation at 4˚C. Protein A-agarose beads (Sigma, St. Louis, MO, USA) were then added, and the mixture was incubated for 2 h at 4˚C with gentle agitation to capture the immunocomplex. The beads were collected by centrifugation (2 min at 10,000 g) and washed four times with buffer A. The beads were resus- pended in 1 sample buffer and boiled to elute the im- munocomplex. The eluted proteins were analyzed by SDS-PAGE followed by Western blotting with anti-E- cadherin (H-108; 1:1000) or anti- -catenin (C-18; 1: 1000) antibodies (Santa Cruz, CA). 2.5. Gelatin Zymography MMP-2 and MMP-9 activities were analyzed using SDS-substrate gels by adding gelatin (1 mg/ml) to the 10% acrylamide separating gel. Cells in 6-well plates were treated with SFM containing various concentra- tions (0 - 100 μg/ml) of LJGP for 24 h. Tumor-condi- tioned medium (TCM) was mixed with substrate gel sample buffer (13.3% SDS, 40% glycerol, 42 mM Tris- HCl, pH 6.5, 0.013% bromophenol blue) and loaded onto the gel without prior boiling. Following electro- phoresis, the gels were washed three times in 2.5% Tri- tonX-100 for 10 min at room temperature to remove SDS. The gels were then incubated at 37˚C overnight in incubation buffer (50 mM Tris-HCl (pH 8.0), 5 mM CaCl2). The gels were stained with 0.5% Coomassie brilliant blue G-250 in 40% methanol and 7% acetic acid for 1 h at room temperature, and de-stained in the same solution without Coomassie blue. Gelatin-degrading en- zymes were identified as clear bands against the blue background of the stained gel. 3. RESULTS 3.1. Effects of LJGP on Cell Morphology We previously revealed that L. japonica extracts con- tain a glycoprotein of approximately 10-kDa mass by PAS staining, and we named it LJGP [16]. LJGP inhib- 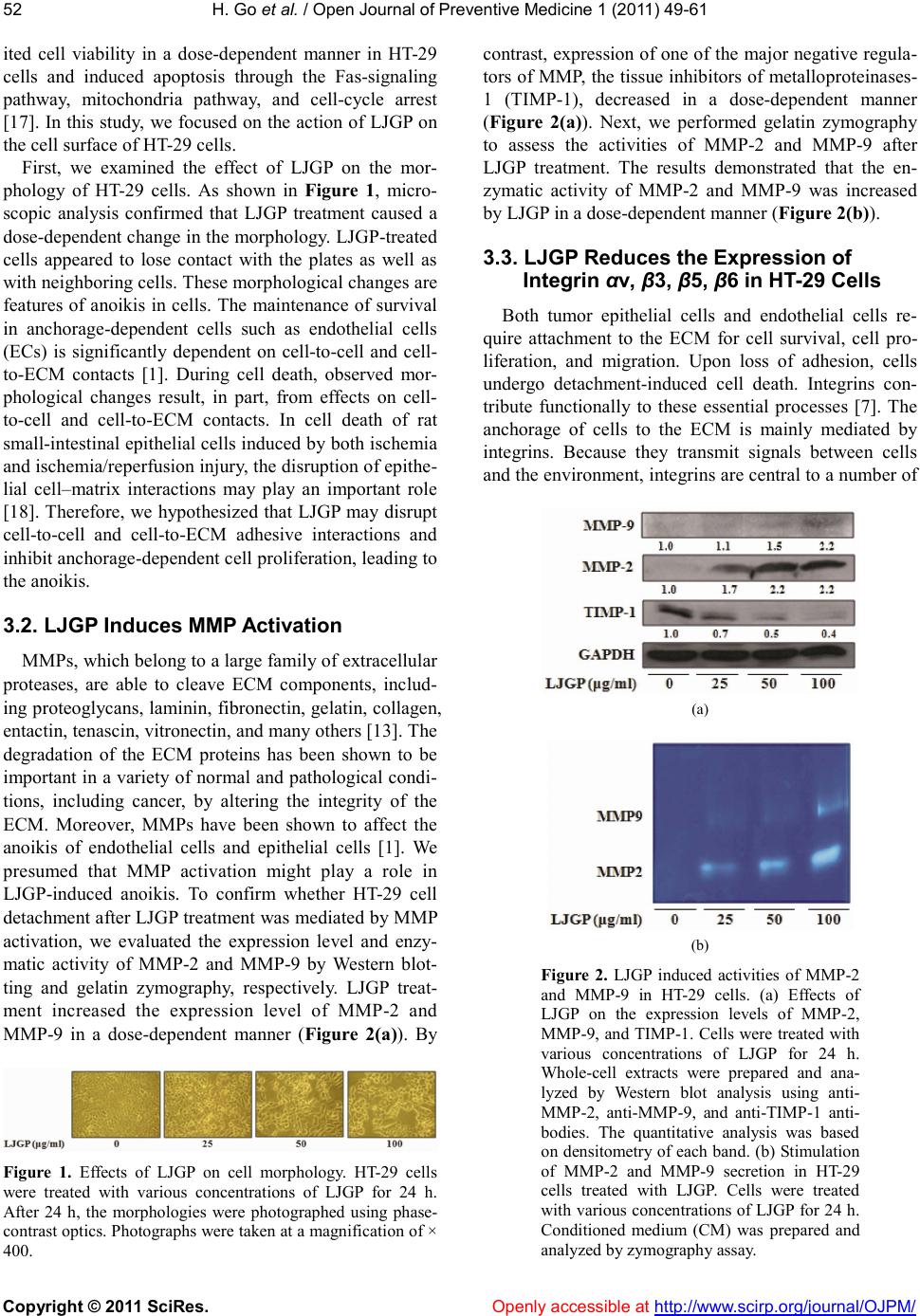 H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 52 ited cell viability in a dose-dependent manner in HT-29 cells and induced apoptosis through the Fas-signaling pathway, mitochondria pathway, and cell-cycle arrest [17]. In this study, we focused on the action of LJGP on the cell surface of HT-29 cells. First, we examined the effect of LJGP on the mor- phology of HT-29 cells. As shown in Figure 1, micro- scopic analysis confirmed that LJGP treatment caused a dose-dependent change in the morphology. LJGP-treated cells appeared to lose contact with the plates as well as with neighboring cells. These morphological changes are features of anoikis in cells. The maintenance of survival in anchorage-dependent cells such as endothelial cells (ECs) is significantly dependent on cell-to-cell and cell- to-ECM contacts [1]. During cell death, observed mor- phological changes result, in part, from effects on cell- to-cell and cell-to-ECM contacts. In cell death of rat small-intestinal epithelial cells induced by both ischemia and ischemia/reperfusion injury, the disruption of epithe- lial cell–matrix interactions may play an important role [18]. Therefore, we hypothesized that LJGP may disrupt cell-to-cell and cell-to-ECM adhesive interactions and inhibit anchorage-dependent cell proliferation, leading to the anoikis. 3.2. LJGP Induces MMP Activation MMPs, which belong to a large family of extracellular proteases, are able to cleave ECM components, includ- ing proteoglycans, laminin, fibronectin, gelatin, collagen, entactin, tenascin, vitronectin, and many others [13]. The degradation of the ECM proteins has been shown to be important in a variety of normal and pathological condi- tions, including cancer, by altering the integrity of the ECM. Moreover, MMPs have been shown to affect the anoikis of endothelial cells and epithelial cells [1]. We presumed that MMP activation might play a role in LJGP-induced anoikis. To confirm whether HT-29 cell detachment after LJGP treatment was mediated by MMP activation, we evaluated the expression level and enzy- matic activity of MMP-2 and MMP-9 by Western blot- ting and gelatin zymography, respectively. LJGP treat- ment increased the expression level of MMP-2 and MMP-9 in a dose-dependent manner (Figure 2(a)). By Figure 1. Effects of LJGP on cell morphology. HT-29 cells were treated with various concentrations of LJGP for 24 h. After 24 h, the morphologies were photographed using phase- contrast optics. Photographs were taken at a magnification of × 400. contrast, expression of one of the major negative regula- tors of MMP, the tissue inhibitors of metalloproteinases- 1 (TIMP-1), decreased in a dose-dependent manner (Figure 2(a)). Next, we performed gelatin zymography to assess the activities of MMP-2 and MMP-9 after LJGP treatment. The results demonstrated that the en- zymatic activity of MMP-2 and MMP-9 was increased by LJGP in a dose-dependent manner (Figure 2(b)). 3.3. LJGP Reduces the Expression of Integrin αν, β3, β5, β6 in HT-29 Cells Both tumor epithelial cells and endothelial cells re- quire attachment to the ECM for cell survival, cell pro- liferation, and migration. Upon loss of adhesion, cells undergo detachment-induced cell death. Integrins con- tribute functionally to these essential processes [7]. The anchorage of cells to the ECM is mainly mediated by integrins. Because they transmit signals between cells and the environment, integrins are central to a number of (a) (b) Figure 2. LJGP induced activities of MMP-2 and MMP-9 in HT-29 cells. (a) Effects of LJGP on the expression levels of MMP-2, MMP-9, and TIMP-1. Cells were treated with various concentrations of LJGP for 24 h. Whole-cell extracts were prepared and ana- lyzed by Western blot analysis using anti- MMP-2, anti-MMP-9, and anti-TIMP-1 anti- bodies. The quantitative analysis was based on densitometry of each band. (b) Stimulation of MMP-2 and MMP-9 secretion in HT-29 cells treated with LJGP. Cells were treated with various concentrations of LJGP for 24 h. Conditioned medium (CM) was prepared and analyzed by zymography assay.  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 53 53 biological processes, including intercellular adhesion, maintenance of cell morphology, cell migration, regula- tion of cell growth and differentiation, and progression through the cell cycle [7,13,19]. Integrins consist of two transmembrane glycoprotein subunits, and , that are non-covalently bound. The receptors always contain one chain and one chain. In this study, we determined the expression level of four types of integrin, integrin αν, integrin β3, integrin β5, and integrin β6, after treatment of HT-29 cells with LJGP. The αν integrins pair with multiple β subunits (β1, β3, β5, β6, and β8). Western blot analyses confirmed that the amount of these proteins decreased in a dose-dependent manner after treatment with LJGP (Figure 3). Especially, in- tegrins αν and β3 significantly decreased, and integrin β6 slightly decreased. 3.4. LJGP Reduces the Phosphorylation of FAK and Src in HT-29 Cells Integrins do not themselves possess a kinase domain or enzymatic activity, but rely on association with other signaling molecules to transmit signals. There are some important key signaling molecules in integrin-mediated signal transduction. One of them is FAK, which is a non-receptor protein tyrosine kinase that colocalizes with integrins in focal adhesions. Upon integrin ligation, FAK is phosphorylated on five different tyrosine resi- dues (Y397, Y407, Y576, Y577, and Y925), with tyro- sine 397 being the major autophosphorylation site [20- 21]. We determined whether activation of FAK occurs after treatment of HT-29 cells with LJGP. The phos- phorylation of tyrosine 397 of FAK has been used as a marker of FAK activity. As shown in Figure 4, phos- Figure 3. LJGP decreased αν, β3, β5, and β6 integrin expression in HT-29 cells. Cells were treated with various concentrations of LJGP for 24 h. Cell membranes were prepared and analyzed by Western blot analysis using anti- integrin αν, -integrin β3, -integrin β5, and -in- tegrin β6 antibodies. The quantitative analysis was based on densitometry of each band and is reported as a fold increase over the control. phorylation of FAK decreased in a dose-dependent man- ner. However, there were no changes in total FAK pro- tein levels. Besides integrins, many down-stream molecules are known to interact with the Y397 autophosphorylation site of FAK, such as c-Src, Shc, Csk, and PI-3K [19]. It has also been reported that abolishing FAK activity alone is not sufficient to abolish apoptosis resistance in colon cancer cells; a concomitant inhibition of Src is also required to do this [22]. Src is a member of the SRC family of kinases (SFK), which consists of nine mem- bers: Blk, Fgr, Fyn, Hck, Lck, Lyn, Src, and Yrk [6]. Within the SFK, Src is the best-known protein and plays a significant role during cancer progression [23]. We determined the levels of phosphorylation of Src follow- ing exposure of cells to LJGP. As shown in Figure 4, LJGP inhibited Src phosphorylation in a dose-dependent manner. 3.5. LJGP Affects the PI3K Pathway Integrin-induced phosphorylation of FAK tyrosine 397 creates a high-affinity binding site for the SFK via their SH2 domains and also for the p85 regulatory sub- unit of PI-3K [24]. PI-3K is a heterodimer lipid kinase composed of a 110-kDa catalytic subunit and an 85-kDa regulatory subunit, which contains two SH2 domains and one SH3 domain. PI-3K phosphorylates phosphoti- dylinositol at the D3 position of the inositol ring in a variety of phosphoinositide substrates, forming 3-phos- phorylated phosphoinositides. The PI-3K signal trans- duction pathway regulates cancer survival, apoptosis, and a number of diverse cellular processes such as growth, proliferation, cell transformation, metastasis, membrane trafficking, and cell survival [25]. Increased Figure 4. Effects of LJGP on the activities of FAK and Src. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole- cell extracts were prepared and analyzed by Western blot analysis using anti-phospho-FAK (Tyr397), -FAK, -phospho-Src family, and -C- Src antibodies. The quantitative analysis was based on densitometry of each band and is re- ported as a fold increase over the control. 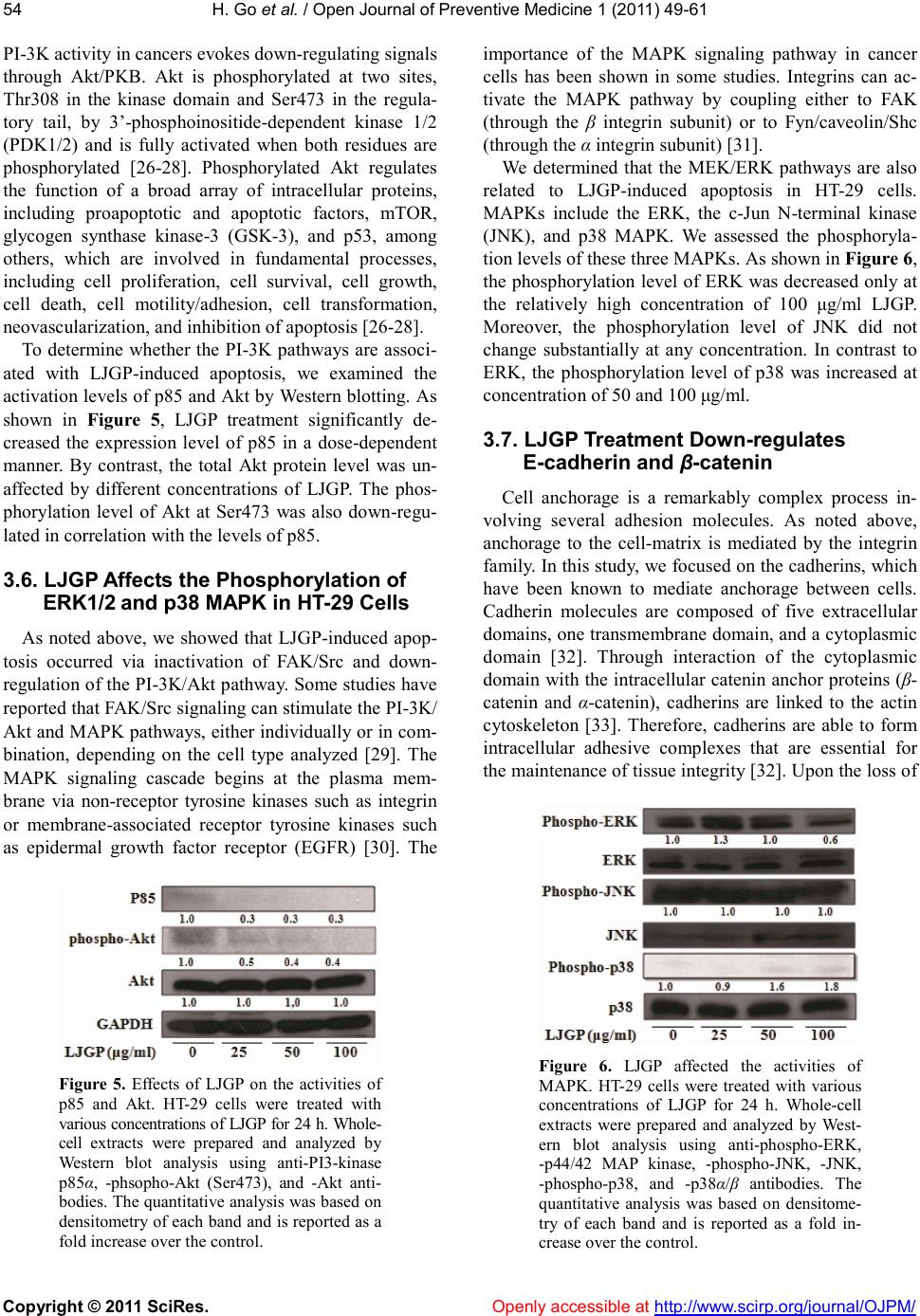 H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 54 PI-3K activity in cancers evokes down-regulating signals through Akt/PKB. Akt is phosphorylated at two sites, Thr308 in the kinase domain and Ser473 in the regula- tory tail, by 3’-phosphoinositide-dependent kinase 1/2 (PDK1/2) and is fully activated when both residues are phosphorylated [26-28]. Phosphorylated Akt regulates the function of a broad array of intracellular proteins, including proapoptotic and apoptotic factors, mTOR, glycogen synthase kinase-3 (GSK-3), and p53, among others, which are involved in fundamental processes, including cell proliferation, cell survival, cell growth, cell death, cell motility/adhesion, cell transformation, neovascularization, and inhibition of apoptosis [26-28]. To determine whether the PI-3K pathways are associ- ated with LJGP-induced apoptosis, we examined the activation levels of p85 and Akt by Western blotting. As shown in Figure 5, LJGP treatment significantly de- creased the expression level of p85 in a dose-dependent manner. By contrast, the total Akt protein level was un- affected by different concentrations of LJGP. The phos- phorylation level of Akt at Ser473 was also down-regu- lated in correlation with the levels of p85. 3.6. LJGP Affects the Phosphorylation of ERK1/2 and p38 MAPK in HT-29 Cells As noted above, we showed that LJGP-induced apop- tosis occurred via inactivation of FAK/Src and down- regulation of the PI-3K/Akt pathway. Some studies have reported that FAK/Src signaling can stimulate the PI-3K/ Akt and MAPK pathways, either individually or in com- bination, depending on the cell type analyzed [29]. The MAPK signaling cascade begins at the plasma mem- brane via non-receptor tyrosine kinases such as integrin or membrane-associated receptor tyrosine kinases such as epidermal growth factor receptor (EGFR) [30]. The Figure 5. Effects of LJGP on the activities of p85 and Akt. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole- cell extracts were prepared and analyzed by Western blot analysis using anti-PI3-kinase p85α, -phsopho-Akt (Ser473), and -Akt anti- bodies. The quantitative analysis was based on densitometry of each band and is reported as a fold increase over the control. importance of the MAPK signaling pathway in cancer cells has been shown in some studies. Integrins can ac- tivate the MAPK pathway by coupling either to FAK (through the β integrin subunit) or to Fyn/caveolin/Shc (through the α integrin subunit) [31]. We determined that the MEK/ERK pathways are also related to LJGP-induced apoptosis in HT-29 cells. MAPKs include the ERK, the c-Jun N-terminal kinase (JNK), and p38 MAPK. We assessed the phosphoryla- tion levels of these three MAPKs. As shown in Figure 6, the phosphorylation level of ERK was decreased only at the relatively high concentration of 100 μg/ml LJGP. Moreover, the phosphorylation level of JNK did not change substantially at any concentration. In contrast to ERK, the phosphorylation level of p38 was increased at concentration of 50 and 100 μg/ml. 3.7. LJGP Treatment Down-regulates E-cadherin and β-catenin Cell anchorage is a remarkably complex process in- volving several adhesion molecules. As noted above, anchorage to the cell-matrix is mediated by the integrin family. In this study, we focused on the cadherins, which have been known to mediate anchorage between cells. Cadherin molecules are composed of five extracellular domains, one transmembrane domain, and a cytoplasmic domain [32]. Through interaction of the cytoplasmic domain with the intracellular catenin anchor proteins (β- catenin and α-catenin), cadherins are linked to the actin cytoskeleton [33]. Therefore, cadherins are able to form intracellular adhesive complexes that are essential for the maintenance of tissue integrity [32]. Upon the loss of Figure 6. LJGP affected the activities of MAPK. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole-cell extracts were prepared and analyzed by West- ern blot analysis using anti-phospho-ERK, -p44/42 MAP kinase, -phospho-JNK, -JNK, -phospho-p38, and -p38α/β antibodies. The quantitative analysis was based on densitome- try of each band and is reported as a fold in- crease over the control.  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 55 55 anchorage, modulation of E-cadherin-cytoskeleton com- plexes might be involved in the apoptosis-signaling cas- cade. Three major catenins are known: α-catenin, β-catenin, and γ-catenin (plakoglobin). β-catenin and γ-catenin di- rectly bind to cadherin in a mutually exclusive manner, whereas α-catenin binds to β-catenin or γ-catenin and makes an additional direct or indirect contact to the actin cytoskeleton, leading to strong cell-cell adhesion [34]. First, we determined the expression levels of E-cad- herin and β-catenin after LJGP treatment of HT-29 cells. As shown in Figure 7(a), LJGP decreased the expres- sion level of E-cadherin and β-catenin in a dose-de- pendent manner. β-catenin associates with E-cadherin at cell–cell junc- tions. Therefore, we next examined the binding level of E-cadherin and β-catenin after LJGP treatment of HT-29 cells. To test this, E-cadherin was immunoprecipitated, followed by immunoblotting with anti-β-catenin and E- (a) (b) Figure 7. LJGP suppressed cell-cell anchorage. (a) E-cadherin and β-catenin levels during LJGP-induced anoikis. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole-cell extracts were prepared and ana- lyzed by Western blot analysis using anti-E- cadherin and anti-β-catenin antibodies. The quantitative analysis was based on densitome- try of each band and is reported as a fold in- crease over the control. (b) LJGP interferes with E-cadherin-β-catenin linkage formation. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole-cell extracts were prepared and immunoprecipitated with E-cad- herin. Immunocomplexes were analyzed by Western blot analysis using anti-E-cadherin and β-catenin antibodies. The quantitative ana- lysis was based on densitometry of each band and is reported as a fold increase over the con- trol. cadherin antibodies. The level of E-cadherin-associated β-catenin was reduced after 24 h of LJGP treatment in a dose-dependent manner (Figure 7(b)). Furthermore, de- creased amounts of E-cadherin protein were also de- tected in the immunoprecipitates. Thus, β-catenin disso- ciated from E-cadherin in response to the LJGP treat- ment. 3.8. LJGP Down-Regulates Wnt Signaling Cadherins not only provide anchorage between neigh- boring cells, but they are also elements in the complex network of the Wnt/Wingless survival-signaling pathway. E-cadherin and β-catenin act as downstream components of this pathway and play a crucial role [33]. Accumula- tion of β-catenin stimulates activation of the Wnt/Wing- less signaling pathway and its interaction with members of the lymphoid enhancer factor (LEF)/T-cell factor (TCF) family of transcription factors, leading to cellular proliferation and possibly neoplastic transformation [7]. Therefore, an understanding of the mechanism that regulates the function and expression of E-cadherin and β-catenin is critical for the understanding of invasion, metastasis, and proliferation of cancer cells. The devel- opment of colon cancer might begin by aberrant active- tion of Wnt signaling. β-catenin is a multi-functional protein that plays essential roles both at adherens junc- tions and in Wnt signaling. In the Wnt signaling pathway, β-catenin plays a central role as a cotranscriptional acti- vator of genes in the nucleus, together with LEF/TCF [33]. In Figure 7, we determined the participation of β- catenin at adherens junctions during LJGP-induced apoptosis in HT-29 cells. Next, we investigated its in- volvement as a component of the Wnt signaling pathway. To investigate whether LJGP affected the Wnt signaling pathway, we investigated the translocation of β-catenin from the cytosol to the nucleus, and the expression levels of Wnt signal target proteins, such as c-myc and cyclin D1. After treatment of the cells with LJGP, we measured the amount of β-catenin in the cytosol and nucleus. As shown in Figure 8, LJGP slightly increased the amount of cytosolic β-catenin. By contrast, nuclear β-catenin decreased in a dose-dependent manner after treatment of cells with LJGP. These results suggest that LJGP blocked the translocation of β-catenin from the cytosol to the nucleus. We therefore hypothesized that the ex- pression level of the β-catenin target gene might also be decreased. To confirm this hypothesis, we investigated the expression level of c-myc and cyclin D1 by Western blotting. As shown in Figure 7, both c-myc and cyclin D1 significantly decreased after treatment of cells with LJGP. These results suggest that Wnt signaling is related to LJGP-induced anoikis in HT-29 cells.  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 56 Figure 8. LJGP reduced translocation of β-cat- enin and expression of target proteins of Wnt signaling. HT-29 cells were treated with various concentrations of LJGP for 24 h. Whole-cell ex- tracts, cytosol cell extracts, and nuclear cell ex- tracts were prepared and analyzed by Western blot analysis using anti-β-catenin, -c-myc, and -cyclinD1 antibodies. The quantitative analysis was based on densitometry of each band and is reported as a fold increase over the control. 4. DISCUSSION Natural marine products, like seaweeds, have recently become the focus of increased research interest due to their potential pharmacological activities [35]. Marine organisms have evolved various functions to adapt to a wide variety of environmental changes, including salt density, water pressure, drying at low tide, and ultravio- let radiation. As a result, many of them contain various physiologically active substances that are known to benefit health. The brown alga L. japonica is commonly consumed in Korea, Japan, and China, and less so in Europe and the United States [36]. It is known to have many biological activities, including an antioxidant effect, anti-mutagenic activity, and antibacterial activity [12,37]. We assumed that its medicinal effects were mainly due to the carbo- hydrate or protein components, as L. japonica contains 60.9% carbohydrate and 10.3% protein [38]. Previously, we extracted a glycoprotein from L. japonica (named LJGP), which we demonstrated had apoptotic effects on HT-29 cells [16,17]. LJGP inhibited the growth of HT-29 cells by arresting the cell cycle and activating apop- tosis-related pathways, including the Fas-signaling path- way and mitochondrial pathway [17]. In the present study, we examined the actions of LJGP on the cell-surface membrane during LJGP-induced apoptosis. As shown in Figure 1, LJGP treatment caused a dose-dependent increase in morphological changes that are characteristic of anoikis in cells. Anoikis is a form of apoptosis involving disruption of cell-to-cell or cell-to- ECM adhesive interactions at the cell-surface membrane. We hypothesized that the LJGP-induced apoptosis of HT-29 cells was due to cell detachment-induced death; that is anoikis. To examine the mechanism of anoikis after treatment with LJGP, we studied the degradation of the ECM and cell-anchorage-related signaling pathway. There are some reports that cell detachment is related to anoikis. Doxazosin induced anoikis and affected cell morphology in prostate cancer cells [39]. In DU-145 prostate cancer cells, cell detachment was induced by methyl selenium as a prerequisite for anoikis [40]. In human osteosarcoma cells, receptor activator of nuclear factor κβ ligand (RANKL) inhibited malignancy and evoked caspase-3-mediated anoikis [41]. It was reported that 15-deoxy-Delta (12,14)-prostaglandin J (2) decreased the adherence rate of BEL-7402 hepatocellular carci- noma cells, leading to anoikis [42]. MMP is associated with degradation of the ECM and is known to induce cell death [43]. It was reported that MMP induced apoptosis of human endothelial cells [44]. Moreover, FOXO3a induced apoptosis of endothelial cells through activation of MMP [1]. Furthermore, some studies have indicated that TIMP-1 functions as a potent inhibitor of apoptosis in mammalian cells [45]. It was reported that caspase-mediated apoptotic pathways in- duced by a variety of stimuli, including anoikis, stauro- sporine exposure, hydrogen peroxide, x-ray irradiation, growth factor withdrawal, and adriamycin treatment, were down-regulated by TIMP-1-activated cell survival signaling [44]. Moreover, downregulation of TIMP-1 expression enhanced apoptotic cell death. In the present study, we observed that LJGP increased the activities of MMP-2 and MMP-9 and decreased the activity of TIMP-1 (Figure 2). Down regulation of TIMP-1 may be the cause of the increased MMP-2 and MMP-9 activities. It may be that activation of MMP and suppression of TIMP-1 induces anoikis in LJGP-treated HT-29 cells. Signal transduction from the extracellular environ- ment to the intracellular network is mediated by integrin- activated signaling molecules, such as FAK, PI3-K, ILK, SHC, ERK, MAPK, EGFR, and caveolin. These many helper molecules are necessary for the cell to react spe- cifically to varying cues from the environment and from within the cell, and to precisely modulate the cell’s be- havior [19]. Loss of integrin-mediated epithelial cell– ECM interactions causes loss of phosphorylation of these downstream effectors, which are known to mediate cell susceptibility to anoikis and a variety of biological processes [6]. In the present study, we showed that LJGP treatment caused a dose-dependent decrease in the ex- pression levels of integrin αν, integrin β3, integrin β5, and integrin β6 (Figure 3). Because integrins are a kind of cell-surface adhesion receptor, their decreased expression after treatment with LJGP would weaken the adhesion of cells to the ECM, thus inducing cell death. These results are expected from  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 57 57 the fact that both MMP-2 and MMP-9 are known to de- grade a wide variety of proteins, including chemotactic molecules, adhesion molecules, proteinase inhibitors, cell-surface receptors, blood clotting factors, latent growth factors, and growth factor-binding proteins [46]. The decreased expression of integrin could be the result of a degradation of the ECM due to an increase in MMP activity (Figure 2). FAK controls a variety of biological processes, in- cluding cell survival, spreading, invasion, proliferation, motility, migration, and cell-cycle progression [19]. Disruption of FAK signaling in cancer cells induces cell death after the induction of cell rounding and detach- ment from the basal stratum. FAK has also been reported to become over-expressed in tumor cells, indicating that FAK activity is critical for resistance to apoptosis [47]. It also been reported that over-expression of FAK occurs in a number of human malignancies and that the degree of over expression is correlated with greater aggressiveness of the cancer [48,49]. In spite of the loss of cell anchor- age, over-expression of active FAK blocks anoikis in cells, thereby supporting the role of this kinase in anoikis regulation [5]. In the present study, LJGP decreased the phosphorylation levels of FAK without changing the total FAK level (Figure 4). LJGP may have the potential to reduce the phosphorylation of FAK, thereby inducing anoikis in HT-29 cells. As shown in Figure 4, LJGP also decreased the phos- phorylation level of Src. Upon cell adhesion, ECM-in- tegrin-mediated signals activate Src through FAK, and the loss of adhesion results in inactivation of Src, leading to anoikis. Several studies have reported the inactivation of Src through FAK. Garcinol and disruption of caveolae decreased not only the phosphorylation of FAK, but also that of Src during anoikis [47,50]. We hypothesize that LJGP treatment, by reducing FAK phosphorylation, may cause down-regulation of Src in HT-29 cells, thereby inducing anoikis. Alteration of the PI-3K/Akt pathway has been de- tected in various human cancers. Many studies have re- ported that the PI-3K/Akt pathway is constitutively acti- vated and plays an important role in tumor formation in several cancers, including gastric, renal cell, ovarian, and lung cancers. Therefore, it may be effective to de- velop potential treatment agents that target this pathway. In fact, some small molecule inhibitors, including in- hibitors of PI-3K, Akt, and mTOR, have been developed and/or approved as treatment agents for various human cancers [28]. As shown in Figure 5, LJGP decreased the PI-3K/Akt pathway. It is known that integrin-mediated signals activate the PI-3K/Akt pathway and its subse- quent signaling functions. Furthermore, it has been re- ported that integrins induce Akt phosphorylation through PI-3K [25,51]. The suppression of integrin expression by LJGP treatment may, therefore, induce the down-regula- tion of FAK, Src, and PI-3K/Akt survival signaling in HT-29 cells. LJGP also affected the MAPK pathway (Figure 6). The MAPK pathway is highly activated during the late progression of colorectal cancer and in primary tumors of diverse origins [30]. In intestinal epithelial cancer cells, anoikis resistance is mediated by the activation of MEK/ERK [29]. In colorectal carcinoma, the MAPK pathway is aberrantly activated [52]. Because MAPKs are believed to be responsible for the proliferation, sur- vival, cell-cycle progression, and metastasis of cancer cells, inhibition of this pathway is a potential therapeutic approach. Indeed, highly potent inhibitors of MAPK activation have been shown to be capable of inhibiting human cancer growth in immune-deficient mice [30]. In the present study, we found that integrin/FAK/Src sig- naling down-regulated both the PI3K signaling pathway and the MAPK signaling pathway in LJGP-treated HT- 29 cells. LJGP might, therefore, be a promising new therapeutic agent for the treatment of colon cancer. In embryonic tissue and primary mouse mammary gland epithelial cells, anoikis induced by blockage of cadherin after treatment with blocking antibodies was inhibited when cell-to-cell anchorage was preserved, despite loss of the cell-to-ECM anchorage [7]. Also, inactivation of E-cadherin through mutations caused cell death in mice embryo. These reports demonstrate the importance of cadherins in cell survival. In epithelial cells, physical integrity and polarity are maintained through intercellular junctions consisting of adherens junctions, tight junctions, desmosomes, hemidesmo- somes, and gap junctions [53]. E-cadherin is an impor- tant component of adherens junctions, where E-cadherin interacts with catenin that mediates binding to the actin cytoskeleton. E-cadherin indirectly interacts with the actin cytoskeleton, and this interaction is essential for the stabilization and strength of cell-cell junctions. Cad- herin engagement has been shown to protect squamous carcinoma cells and normal proximal tubular cells from anoikis [54]. Disruption of cell-cell anchorage may also play a role in anoikis, because expression of a dominant negative N-cadherin mutant in the intestinal epithelium was shown to increase the frequency of apoptosis in cells, while perturbing migration and differentiation [54]. Upon the loss of anchorage, normal enterocytes executed a strikingly rapid apoptosis program [54]. During induc- tion of apoptosis in endothelial cells, degradation of VE-cadherin and β-catenin occurred [12]. NIH3T3 cells grown in serum-free media exhibited a reduction of cell-cell contacts during apoptosis [54]. In the present study, disruption of E-cadherin and β-catenin appeared  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 58 to reduce cell-cell contact during LJGP-induced anoikis in HT-29 cells (Figure 7). Interaction between β-catenin and E-cadherin is a prerequisite for cell adhesion due to the role of β-catenin in protecting the cytoplasmic do- main of cadherin from rapid degradation, enhancing the efficiency of transport from the endplas- mic reticulum to the cell surface, and recruiting α-catenin to the sites of cell-cell contact [55]. Although anoikis has been known to be induced by the disruption of integrin-mediated signaling, the disruption of E-cadherin signaling also affects anoikis. There appears to be a connection be- tween these two pathways. One report suggests the pos- sibility that β-catenin may lie downstream from the in- tegrins, and several integrin-stimulated signaling path- ways might lead to the induction of β-catenin signaling. Because the anti-apoptotic kinase Akt is known to inhibit the activity of glycogen synthase kinase 3-β, a serine kinase that functions directly to reduce β-catenin signal- ing, it is expected that Akt is a connection between in- tegrins and β-catenin [56]. A report that activated Akt could be localized at cell–cell contacts also supports the existence of this connection. E-cadherin-blocking anti- bodies, as well as the calcium chelator EGTA, induced apoptosis and loss of Akt activity in granulose cells [57]. E-cadherin bound to renal epithelial cells and promoted survival in a PI-3K-dependent fashion [58]. Down- regulation of E-cadherin resulted in changes of ανβ5 and ανβ1 integrin expression and migration toward tenascin [59]. We showed here that LJGP induced anoikis via both integrin-mediated and E-cadherin-mediated signals. Based on these results, we conclude that LJGP intri- cately affected both cell-cell and cell-matrix contacts, leading to anoikis in HT-29 cells. There are some data to support the notion that cad- herin-associated β-catenin promotes the carcinogenesis process. First, β-catenin associates with and is down- regulated by the tumor suppressor APC. Second, β-cat- enin transduces, at least partly, the oncogenic Wnt growth factor signal to the nucleus. Third, β-catenin is mutated in a significant number of human cancers, and overexpression of an NH2 terminally truncated form of β-catenin in the epidermis of transgenic mice produced well-differentiated hair tumors [56]. A dynamic pool of β-catenin in the cytosol and nucleus is responsible for the transduction of Wnt signals. Wnt proteins play criti- cal regulatory roles in many biological processes, in- cluding the development, maintenance, differentiation, and regulation of embryonic and adult stem cells. Fur- thermore, deregulation of Wnt signaling is associated with multiple diseases, including various cancers. In the absence of a Wnt signal, cytosolic β-catenin is captured and phosphorylated by the cytoplasmic protein complex called the β-catenin-destruction complex; this complex includes APC, axin, GSK-3β, and CKI, which catalyzes the phosphorylation of N-terminal serines and threonine in β-catenin, thus labeling it for ubiquitin-dependent degradation by the proteasome. In the presence of a Wnt signal, the disruption of the complex containing APC, axin, GSK-3β, and β-catenin is triggered, which prevents the phosphorylation-dependent degradation of β-catenin, causing β-catenin to accumulate in the cytosol. Accu- mulated β-catenin is recruited to the promoters of Wnt- response genes through its interaction with members of the Tcf/LEF family of DNA-binding proteins, and to- gether, they turn on the transcription of a plethora of genes [60]. β-catenin has been known to significantly protect cells from anoikis and is known as a potent in- hibitor of apoptosis. Similarly, it may be said that LJGP is a potent anti-cancer treatment. In the nucleus, β-catenin stimulates the expression of target genes of Wnt signaling, such as cyclin D1 and c- myc, which have been implicated as having key roles in oncogenesis. Both the cyclin D1 and c-myc proteins contain TCF binding sites and appear to be constitutively active in several colon cancer cell lines. In Figure 8, we show that LJGP inhibited the translocation of β-catenin from the cytosol to the nucleus and, consequently, inhib- ited the expression of cyclin D1 and c-myc. LJGP-in- duced anoikis might be associated with Wnt signaling. Some reports have indicated that Wnt signaling is asso- ciated with apoptosis in cancer cells. Resveratrol sup- presses human colon cancer cell proliferation and ele- vates apoptosis via suppression of the Wnt signaling pathway [61]. Lignans inhibit cell growth via inhibition of Wnt/β-catenin signaling [62]. Quercetin inhibits hu- man SW480 colon cancer growth through Wnt/β-catenin signaling [52]. In acute myeloid leukemia cells, apop- tosis is induced by small molecule inhibitors of Wnt signaling [63]. In conclusion, LJGP could qualify as a promising, in- Figure 9. Signal transduction following loss of cell anchorage, leading to anoikis.  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 59 59 novative treatment for colon cancer. LJGP-induced anoikis of HT-29 cells involved cell detachment and possibly ECM disruption, which were executed principally through the activation of MMPs and a decrease of adhe- sion molecules, contributing to down-regulation of the PI-3K, MAPK, and Wnt pathways (Figure 9). 5. ACKNOWLEDGMENTS This research was a part of the project titled “Development for novel biofunctional protein source from marine algae produced in the coastal area of Busan,” funded by the Ministry of Land, Transport and Mari- time Affairs, Korea. REFERENCES [1] Lee, H.-Y., You, H.-J., Won, J.-Y., Youn, S.-W., Cho, H.-J., Park, K.-W., Park, W.-Y., Seo, J.-S., Park, Y.-B., Walsh, K., Oh, B.-H. and Kim, H.-S. (2008) Forkhead factor, FOXO3a, induces apoptosis of endothelial cells through activation of matrix metalloproteinases. Ameri- can Heart Journal, 28, 302-308. [2] Kim, J.-B., Yu, J.-H., Ko, E., Lee, K.-W., Song, A.-K., Park, S.-Y., Shin, I., Han, E. and Noh, D.-Y. (2010) The alkaloid berberine inhibits the growth of anoikis-resistant MCF-7 and MDA-MB-231 breast cancer cell lines by in- cluding cell cycle arrest. Phytomedicine, 17, 436-440. doi:10.1016/j.phymed.2009.08.012 [3] Michel, J.B. (2003) Anoikis in the cardiovascular system known and unknown extracellular mediators. American Heart Journal, 23, 2146-2154. [4] Coates, J.M., Galante, J.M. and Bold, R.J. (2010) Cancer therapy beyond apoptosis: Autophagy and anoikis as mechanisms of cell death. Journal of Surgical Research, 164, 301-308. doi:10.1016/j.jss.2009.07.011 [5] Chiarugi, P. and Giannoni, E. (2008) Anoikis: A neces- sary death program for anchorage-dependent cells. Bio- chemical Pharmacology, 76, 1352-1364. doi:10.1016/j.bcp.2008.07.023 [6] Sakamoto, S. and Kyprianou, N. (2010) Targeting anoikis resistance in prostate cancer metastasis. Molecular As- pects of Medicine, 31, 205-214. doi:10.1016/j.mam.2010.02.001 [7] Grossmann, J. (2002) Molecular mechanisms of “de- tachment-induced apoptosis-Anoikis”. Apoptosis, 7, 247- 260. doi:10.1023/A:1015312119693 [8] Duxbury, M.S., Ito, H., Zinner, M.J., Ashley, S.W. and Whang, E.E. (2004) Focal adhesion kinase gene silen- ceing promotes anoikis and suppresses metastasis of hu- man pancreatic adenocarcinoma cells. Surgery, 135, 555-562. doi:10.1016/j.surg.2003.10.017 [9] Aplin, A.E., Howe, A., Alahari, S.K. and Juliano, R.L. (1998) Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, im- munoglobulin-cell adhesion molecules, and selectins. Pharmacological Reviews, 50, 197-263. [10] Peng, C., Liu, X., Liu, E., Xu, K., Niu, W., Chen, R., Wang, J., Zhang, Z., Lin, P., Wang, J., Agrez, M. and Niu, J. (2009) Norcantharidin induces HT-29 colon cancer cell apoptosis through the ανβ6-extracellular signal-related kinase signaling pathway. Cancer Science, 100, 2302- 2308. doi:10.1111/j.1349-7006.2009.01320.x [11] Maity, G., Fahreen, S., Banerji, A., Choudhury, P.R., Sen, T., Dutta, A. and Chatterjee, A. (2010) Fbronectin–in- tegrin mediated signaling in human cervical cancer cells (SiHa). Molecular and Cellular Biochemistry, 336, 65- 74. doi:10.1007/s11010-009-0256-5 [12] Lee, B.-J., Senevirathne, M., Kim, J.-S., Kim, Y.-M., Lee, M.-S., Jeong, M.-H., Kang, Y.-M., Kim, J.-I., Nam, B.-H., Ahn, C.-B. and Je, J.-Y. (2010) Protective effect of fer- mented sea tangle against ethanol and carbon tetrachlo- ride-induced hepatic damage in Sprague-Dawley rats. Food and Chemical Toxicology, 48, 1123-1128. doi:10.1016/j.fct.2010.02.006 [13] Wagenaar-Miller, R.A., Gorden, L. and Matrisian, L.M. (2004) Matrix metalloproteinases in colorectal cancer: Is it worth talking about? Cancer and Metastasis Reviews, 23, 119-135. doi:10.1023/A:1025819214508 [14] Li, L.N., Zhang, H.D., Yuan, S.J., Yang, D.X., Wang, L. and Sun, Z.X. (2008) Differential sensitivity of colorectal cancer cell lines to artesunate is associated with expres- sion of beta-catenin and E-cadherin. European Journal of Pharmacology, 588, 1-8. doi:10.1016/j.ejphar.2008.03.041 [15] Oloumi, A., McPhee, T. and Dedhar, S. (2004) Regula- tion of E-cadherin expression and β-catenin/Tcf tran- scriptional activity by the integrin-linked kinase. Bio- chimica et Biophysica Acta, 1691, 1-15. doi:10.1016/j.bbamcr.2003.12.002 [16] Go, H., Hwang, H.-J. and Nam, T.-J. (2009) Glycoprotein extraction from Laminaria japonica promotes IEC-6 cell proliferation. International Journal of Molecular Medi- cine, 24 , 819-824. [17] Go, H., Hwang, H.-J. and Nam, T.-J. (2010) A glycopro- tein from Laminaria japonica induces apoptosis in HT-29 colon cancer cells. Toxicology in Vitro, 24, 1546-1553. doi:10.1016/j.tiv.2010.06.018 [18] Ikeda, H., Suzuki, Y., Suzuki, M., Koike, M., Tamura, J., Nomura, M. and Itoh, G. (1998) Apoptosis is a major mode of cell death caused by ischaemia and ischaemia/ reperfusion injury to the rat intestinal epithelium. Gut, 42, 530-537. doi:10.1136/gut.42.4.530 [19] Kuphal, S., Bauer, R. and Bosserhoff, A.K. (2005) In- tegrin signaling in malignant melanoma. Cancer Metas- tasis Reviews, 24, 195-222. do i:10. 1007/s10555-005-1572-1 [20] Schlaepfer, D.D., Hanks, S.K., Hunter, T. and Van Der Geer, P. (1994) Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhe- sion kinase. Nature, 372, 786-791. [21] Calalb, M.B., Polte, T.R. and Hanks, S.K. (1995) Tyro- sine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity. Molecular and Cellular Biology, 15, 954-963. [22] Golubovskaya, V.M., Gross, S., Kaur, A.S., Wilson, R.I., Xu, L.H., Yang, X.H. and Cance, W.G. (2003) Simulta- neous inhibition of focal adhesion kinase and SRC en- hances detachment and apoptosis in colon cancer cell lines. Molecular Cancer Research, 1, 755-764. [23] Fizazi, K. (2007) The role of Src in prostate cancer. An- nals of Oncology, 18, 1765-1774. doi:10.1093/annonc/mdm086  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. Openly accessible at http://www.scirp.org/journal/OJPM/ 60 [24] Jones, R.J., Brunton, V.G. and Frame, M.C. (2000) Adhe- sion-linked kinases in cancer; emphasis on Src, focal ad- hesion kinase and PI 3-kinase. European Journal of Can- cer, 36, 1595-1606. doi:10.1016/S0959-8049(00)00153-2 [25] Murillo, C.A., Rychahou, P.G. and Evers, B.M. (2004) Inhibition of alpha5 integrin decreases PI3K activation and cell adhesion of human colon cancers. Surgery, 136, 143-149. doi:10.1016/j.surg.2004.04.006 [26] Dai, R.Y., Chen, S.K., Yan, D.M., Chen, R., Lui, Y.P., Duan, C.Y., Li, J., He, T. and Li, H. (2010) PI3K/Akt promotes GRP78 accumulation and inhibits endoplasmic reticulum stress-induced apoptosis in HEK293 cells. Folia Biologica, 56, 37-46. [27] Jiang, H., Fan, D., Zhou, G., Li, X. and Deng, H. (2010) Phosphatidylinositol 3-kinase inhibitor (LY294002) in- duces apoptosis of human nasopharyngeal carcinoma in vitro and in vivo. Journal of Experimental & Clinical Cancer Research, 29, 34. doi:10.1186/1756-9966-29-34 [28] Li, Q., Wu, J., Zheng, H., Liu, K., Guo, T.L., Liu, T., Eblen, S.T., Grant, S. and Zhang, S. (2010) Discovery of 3-(2-aminoethyl)-5-(3-phenyl-propylidene)-thiazolidine- 2,4-dione as a dual inhibitor of the Raf/MEK/ERK and the PI3K/Akt signaling pathways. Bioorganic & Medici- nal Chemistry Letters, 20, 4526-4530. doi:10.1016/j.bmcl.2010.06.030 [29] Demers, M.J., Thibodeau, S., Noel, D., Fujita, N., Tsuruo, T., Gauthier, R., Arguin, M. and Vachon, P.H. (2009) In- testinal epithelial cancer cell anoikis resistance: EGFR- mediated sustained activaton of Src overrides Fak- deoendent signaling to MEK/Erk and/or PI3-K/Akt-1. Journal of Cellular Biochemistry, 107, 639-654. doi:10.1002/jcb.22131 [30] Ahmed, N., Niu, J., Dorahy, D.J., Gu, X., Andrews, S., Meldrum, C.J., Scott, R.J., Baker, M.S., Macreadie, I.G. and Agrez, M.V. (2002) Direct integrin ανβ6-ERK bind- ing: Implications for tumor growth. Oncogene, 21, 1370- 1380. doi:10.1038/sj.onc.1205286 [31] Walker, J.L. and Assoian, R.K. (2005) Integrin-dependent signal transduction regulating cyclinD1 expression and G1 phase cell cycle progression. Cancer Metastasis Re- views, 24, 383-393. doi:10.1007/s10555-005-5130-7 [32] Blaschuk, O.W. and Devemy, E. (2009) Cadherins as novel targets for anti-cancer therapy. European Journal of Pharmacology, 625, 195-198. doi:10.1016/j.ejphar.2009.05.033 [33] Gavert, N. and Ben-Ze`ev, A. (2007) β-catenin signaling in biological control and cancer. Journal of Cellular Bio- chemistry, 102, 820-828. doi:10.1002/jcb.21505 [34] Steinhusen, U., Badock, V., Bauer, A., Behrens, J., Witt- man-Liebold, B., Dorken, B. and Bommert, K. (2000) Apoptosis-induced cleavage of β-catenin by caspase -3 results in proteolytic fragments with reduced transactiva- tion potential. The Journal of Biological Chemistry, 275, 16345-16353. doi:10.1074/jbc.M001458200 [35] Yasuhara-Bell, J. and Lu, Y. (2010) Marine compounds and their antiviral activities. Antiviral Research, 86, 231-240. doi:10.1016/j.antiviral.2010.03.009 [36] Yuan, Y.V. and Walsh, N.A. (2006) Antioxidant and anti- proliferative activities of extracts from a variety of edible seaweeds. Food and Chemical Toxicology, 44, 1144- 1150. doi:10.1016/j.fct.2006.02.002 [37] Zhu, Z., Zhang, Q., Chen, L., Ren, S., Xu, P., Tang, Y. and Luo, D. (2010) Higher specificity of the activity of low molecular weight fucoidan for thrombin-induced platelet aggregation. Thrombosis Research, 125, 419- 426. doi:10.1016/j.thromres.2010.02.011 [38] Cho, D.-M., Kim, D.-S., Lee, D.-S., Kim, H.-R. and Pyeun, J.-H. (1995) Trace components and functional saccharides in seaweed-1. Changes in proximate compo- sition and trace elements according to the harvest season and places. Bulletin of the Korean Fisheries, 28, 49-59. [39] Walden, P.D., Globina, Y. and Nieder, A. (2004) Induc- tion of anoikis by doxazosin in prostate cancer cells is associated with activation of caspase-3 and a reduction of focal adhesion kinase. Urological Research, 32, 261-265. do i:10. 1007/s00240-003-0365-7 [40] Jiang, C., Wang, Z., Ganther, H. and Lu, J. (2001) Cas- pases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Research, 61, 3062-3070. [41] Akiyama, T., Choong, P.F. and Dass, C.R. (2010) RANK- Fc inhibits malignancy via inhibiting ERK activation and evoking caspase-3-mediated anoikis in human osteosar- coma cells. Clinical and Experimental Metastasis, 27, 207-215. doi:10.1007/s10585-010-9319-y [42] Cao, L.Q., Wang, Q., Chen, X.L., Zhen, M.C., Fu, X.H., and Huang, X.H. (2007) 15-Deoxy-delta (12,14)-pros- taglandin J (2) induces anoikis of hepatocellular carci- noma cells: An in vitro experiment. National Medical Journal of China, 87, 3001-3005. [43] Kochtebane, N., Choqueux, C., Passefort, S., Nataf, P., Messika-Zeitoun, D., Bartagi, A., Michel, J.B., An- gles-Cano, E. and Jacob, M.P. (2010) Plasmin induces apoptosis of aortic valvular myofibroblasts. The Journal of Pathology, 221, 37-48. doi:10.1002/path.2681 [44] Wu, W.B., Chang, S.C., Liau, M.Y. and Huang, T.F. (2001) Purification, molecular cloning and mechanism of action of graminelysin I, a snake-venom-derived metal- loproteinase that induces apoptosis of human endothelial cells. Biochemical Journal, 35 7, 719-728. doi:10.1042/0264-6021:3570719 [45] Liu, X.W., Bernardo, M.M., Fridman, R. and Kim, H.R. (2003) Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidy- linositol 3-kinase and MAPK signaling pathway. The Journal of Biological Chemistry, 278, 40364-40372. doi:10.1074/jbc.M302999200 [46] Cierniewski, C.S., Papiewska-Pajak, I., Malinowsi, M., Sacewicz-Hofman, I., Wiktorska, M., Kryczka, J., Wy- socki, T., Niewiarowska, J. and Bednarek, R. (2010) Thymosin beta4 regulates migration of colon cancer cells by a pathway involving interaction with Ku80. Annals of the New York Academy of Sciences, 1194, 60-71. doi:10.1111/j.1749-6632.2010.05480.x [47] Park, E.K., Park, M.J., Lee, S.H., Li, Y.C., Kim, J., Lee, J.S., Lee, J.W., Ye, S.K., Park, J.W., Kim, C.W., Park, B.K. and Kim, Y.N. (2009) Cholesterol depletion induces anoiki-like apoiptosis via FAK down-regulation and caveolae internalization. The Journal of Pathology, 218, 337-349. doi:10.1002/path.2531 [48] Owens, L.V., Xu, L., Dent, G.A., Yang, X., Sturge, G.C., Craven, R.J. and Cance, W.G. (1996) Focal adhesion kinase as a marker of invasive potential in differentiated  H. Go et al. / Open Journal of Preventive Medicine 1 (2011) 49-61 Copyright © 2011 SciRes. http://www.scirp.org/journal/OJPM/Openly accessible at 61 61 human thyroid cancer. Annals of Surgical Oncology, 3, 100-105. doi:10.1007/BF02409059 [49] Agochiya, M., Brunton, V.G., Owens, D.W., Parkinson, E.K., Paraskeva, C., Keith, W.N. and Frame, M.C. (1999) Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene, 18, 5646-5653. doi:10.1038/sj.onc.1202957 [50] Liao, C.H., Sang, S., Ho, C.T. and Lin, J.K. (2005) Gar- cinol modulates tyrosine phosphorylation of FAK and subsequently induces apoptosis through down-regulation of Src, ERK, and Akt survival signaling in human colon cancer cells. Journal of Cellular Biochemistry, 96, 155- 169. doi:10.1002/jcb.20540 [51] Zheng, D.Q., Woodard, A.S., Tallini, G. and Languino, L.R. (2000) Substrate specificity of alpha(v)beta(3) in- tegrin-mediated cell migration and phosphatidylinositol 3-kinase/ AKT pathway activation. The Journal of Bio- logical Chemistry, 275, 24565-24574. doi:10.1074/jbc.M002646200 [52] Shan, J.Z., Xuan, Y.Y., Zheng, S., Dong, Q. and Zhang, S.Z. (2009) Ursolic acid inhibits proliferation and in- duces apoptosis of HT-29 colon cancer cells by inhibiting the EGFR/MAPK pathway. Journal of Zhejiang Univer- sity (Science B), 10, 668-674. doi:10.1631/jzus.B0920149 [53] Sullivan, R. and Graham, C.H. (2007) Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis Reviews, 26, 319-331. doi:10.1007/s10555-007-9062-2 [54] Fouquet, S., Lugo-Martinez, V.H., Faussat, A.M., Renaud, F., Cardot, P., Chambaz, J., Pincon-Raymond and M., Thenet, S. (2004) Early loss of E-cadherin from cell-cell contacts is involved in the onset of anoikis in enterocytes. The Journal of Biological Chemistry, 279, 43061-43069. doi:10.1074/jbc.M405095200 [55] Daugherty, R.L. and Gottardi, C.J. (2007) Phospho- regulation of β-catenin adhesion and signaling functions. Physiology, 22, 303-309. doi:10.1152/physiol.00020.2007 [56] Orford, K., Orford, C.C. and Byers, S.W. (1999) Exoge- nous expression of β-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation- induced cell cycle arrest. The Journal of Cell Biology, 146, 855-868. doi:10.1083/jcb.146.4.855 [57] Peluso, J.J., Pappalardo, A. and Fernandez, G. (2001) E-cadherin-mediated cell contact prevents apoptosis of spontaneously immortalized granulose cells by regulating Akt kinase activity. Biology of Reproduction, 64, 1183- 1190. doi:10.1095/biolreprod64.4.1183 [58] Bergin, E., Levine, J.S., Koh, J.S. and Lieberthal, W. (2000) Mouse proximal tubular cell-cell adhesion inhib- its apoptosis by a cadherin-dependent mechanism. Ame- rican Journal of Physiology, 278, F758-F768. [59] Von Schlippe, M., M., Marshall, J.F., Perry, P., Stone, M., Zhu, A.J. and Hart, I.R. (2000) Functional interaction between E-cadherin and alphav-containing integrins in carcinoma cells. Journal of Cell Science, 11 3, 425-437. [60] Xu, W. and Kimelman, D. (2007) Mechanistic insights from structural studies of β-catenin and its binding part- ners. Journal of Cell Science, 120, 3337-3344. doi:10. 1242/ jcs.013771 [61] Vanamala, J., Reddivari, L., Radhakrishnan, S. and Tarver, C. (2010) Resveratrol suppresses IGF-1 induced human colon cacner cell proliferation and elevates apop- tosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer, 10, 238. doi:10.1186/1471-2407-10-238 [62] Yoo, J.-H., Lee, H.-J., Kang, K., Jho, E.-H., Kim, C.-Y., Baturen, D., Tunsag, J. and Nho, C.-W. (2010) Lignans inhibit cell growth via regulation of Wnt/beta-catenin signaling. Food and Chemical Toxicology, 48, 2247- 2252. doi:10.1016/j.fct.2010.05.056 [63] Minke, K.S., Staib, M., Puetter, A., Gehrke, I., Gan- dhirajan, R.K., Schlosser, A., Schmitt, E.K., Hallek, M. and Kreuzer, K.A. (2009) Small molecule inhibitors of WNT signaling effectively induce apoptosis in acute myeloid leukemia cells. European Journal of Haematol- ogy, 82, 165-175. doi:10.1111/j.1600-0609.2008.01188.x
|