 J. Biomedical Science and Engineering, 2009, 2, 345-358 doi: 10.4236/jbise.2009.25050 Published Online September 2009 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online September 2009 in SciRes. http://www.scirp.org/journal/jbise Review: structure of amyloid fibril in diseases Arezou Ghahghaei1*, Nasim Faridi1 1Department of Biology, Faculty of Science, University of Sistan and Baluchestan, Zahedan, Iran. Email: Arezou@chem.usb.ac.ir Received 8 May 2009; revised 13 July 2009; accepted 21 July 2009. ABSTRACT Tissue deposition of normally soluble proteins, or their fragments, as insoluble amyloid fibrils causes both acquired and hereditary systemic amyloidoses, which is usually fatal. Amyloid is associated with serious diseases such as Alz- heimer’s disease, type 2 diabetes, Parkinson’s Disease, Huntington’s Disease, cancer and the transmissible spongiform encephalopathies. In- formation concerning the structure and mecha- nism of formation of fibrils in these diseases is critical for understanding the process of pathol- ogy of the amyloidoses and to the development of more effective therapeutic agents that target the underlying disease mechanisms. Structural models have been made using information from a wide variety of techniques, including electron microscopy, X-ray diffraction, solid state NMR, and Congo red and CD spectroscopy. Although each type of amyloidosis is characterised by a specific amyloid fibril protein, the deposits share pathognomonic histochemical properties and the structural morphology of all amyloid fibrils is very similar. In fact, the structural similarity that defines amyloid fibres exists principally at the level of β-sheet folding of the polypeptides within the protofilament, while the different types vary in the supramolecular assembly of their proto- filaments. Keywords: Amyloid; Protofilaments; Aggregation; Neurodegenerative 1. INTRODUCTION Amyloid fibril formation arises from the slow aggrega- tion of intermediately folded peptide or protein mole- cules. During this process the protein goes from its na- tive soluble form to insoluble fibril, which is highly β- sheet in character [1,2,3,4]. The aggregation of the amy- loid β peptide has been examined in various solvents and conditions and this has led to a model by which a con- formational switching occurs from an α-helix or random coil to a β-sheet structure early on the amyloid-forming pathway [5], prior to a nucleation-dependent process leading to the elongation of the fibril. Along this path- way, small oligomeric intermediates and short fibrillar structures (protofibrils) have been observed. In cross- section, the fibril appears to be composed of several sub- fibrils or protofilaments. Each of these protofilaments is rich in stacked β-sheet structures in which hydrogen bonded β-strands run perpendicular to the fibril’s axis, and the backbone hydrogen bonds are parallel to it [6]. The basic process of amyloid formation is known to in- volve the construction of fibrils from individual polypep- tide monomer units held together by noncovalent interac- tions generated by the formation of intermolecular β-sheets or the correct stacking of intramolecular β-sheet structures [7]. Small polypeptide fragments (as small as five or six residues long) are able to form amyloid fibrils from fully denatured conformations, larger sequences, typically be- tween 80 and 150 residues, appear to require the popula- tion of more compact or partially folded states to form amyloids [7]. In a number of systems it has been demon- strated that amyloid fibril formation by proteins in vitro is preceded by the formation of metastable, nonfibrillar forms often referred to as protofibrils. These species often have the appearance of spherical beads 2–5 nm in diameter, beaded chains, where the individual beads again have a diameter of 2–5 nm, or annular structures, formed appar- ently by the circularisation of the beaded chains. Figure 1 suggests that the various fates awaiting a polypeptide chain, once it has been synthesized in the cell, will depend on the kinetics and thermodynamics of the various equilibria between various possible states [5,8]. In its monomeric state, the protein is believed to fold from its highly disordered unfolded state (3) through a partially structured intermediate state (2) to a globular native state (1) [8,9]. The unfolded and partially folded states can form aggregated species that are fre- quently disordered. Highly ordered amyloid fibrils can form through a mechanism of nucleation and elongation [8,9,10] showed that the β-domain is the destabilized region of the human lysozyme. This implies that the ag- gregation process may be initiated by intermolecular rather than intramolecular association [8,10,11]. 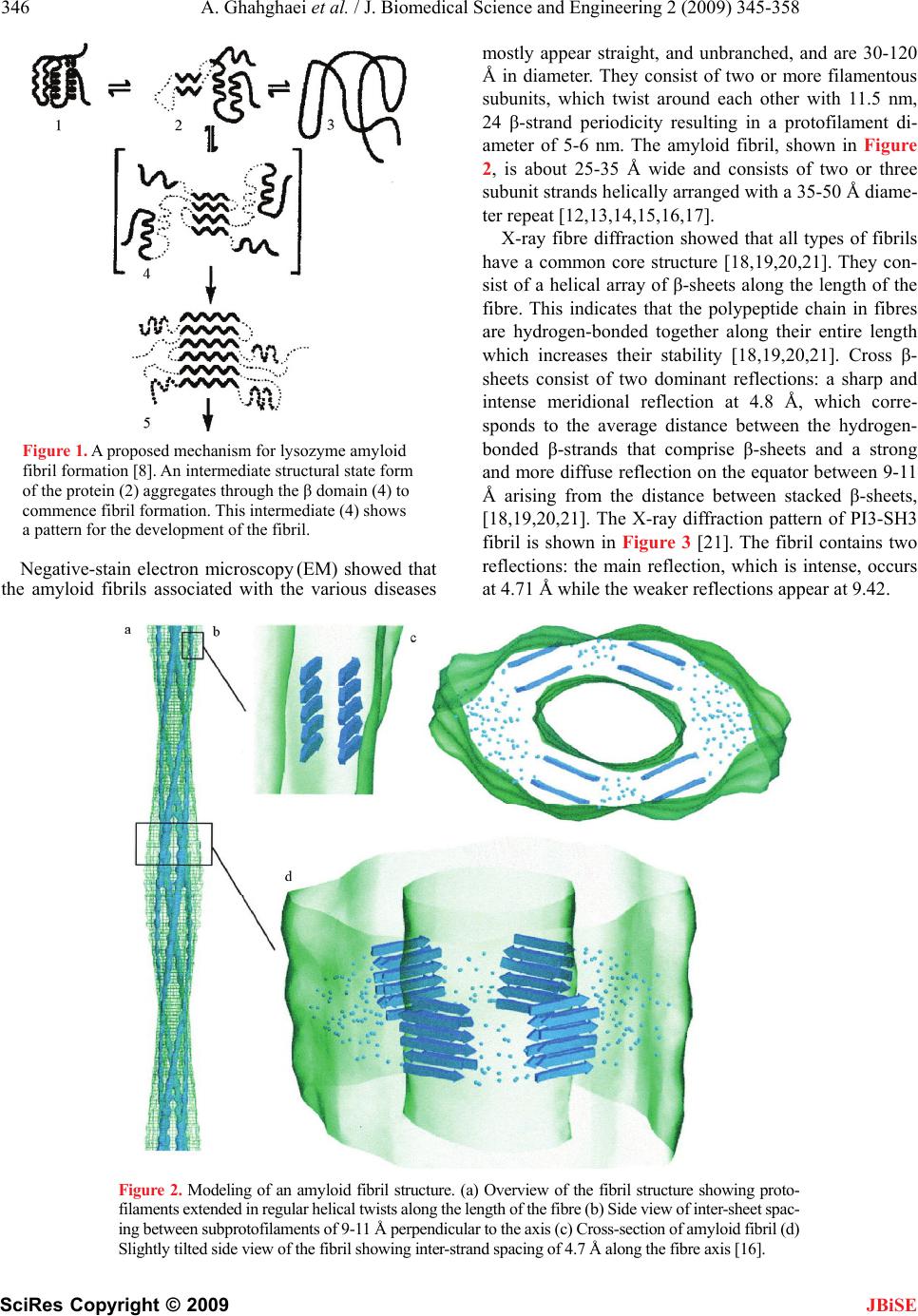 346 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE Figure 1. A proposed mechanism for lysozyme amyloid fibril formation [8]. An intermediate structural state form of the protein (2) aggregates through the β domain (4) to commence fibril formation. This intermediate (4) shows a pattern for the development of the fibril. Negative-stain electron microscopy (EM) showed that the amyloid fibrils associated with the various diseases mostly appear straight, and unbranched, and are 30-120 Å in diameter. They consist of two or more filamentous subunits, which twist around each other with 11.5 nm, 24 β-strand periodicity resulting in a protofilament di- ameter of 5-6 nm. The amyloid fibril, shown in Figure 2, is about 25-35 Å wide and consists of two or three subunit strands helically arranged with a 35-50 Å diame- ter repeat [12,13,14,15,16,17]. X-ray fibre diffraction showed that all types of fibrils have a common core structure [18,19,20,21]. They con- sist of a helical array of β-sheets along the length of the fibre. This indicates that the polypeptide chain in fibres are hydrogen-bonded together along their entire length which increases their stability [18,19,20,21]. Cross β- sheets consist of two dominant reflections: a sharp and intense meridional reflection at 4.8 Å, which corre- sponds to the average distance between the hydrogen- bonded β-strands that comprise β-sheets and a strong and more diffuse reflection on the equator between 9-11 Å arising from the distance between stacked β-sheets, [18,19,20,21]. The X-ray diffraction pattern of PI3-SH3 fibril is shown in Figure 3 [21]. The fibril contains two reflections: the main reflection, which is intense, occurs at 4.71 Å while the weaker reflections appear at 9.42. Figure 2. Modeling of an amyloid fibril structure. (a) Overview of the fibril structure showing proto- filaments extended in regular helical twists along the length of the fibre (b) Side view of inter-sheet spac- ing between subprotofilaments of 9-11 Å perpendicular to the axis (c) Cross-section of amyloid fibril (d) Slightly tilted side view of the fibril showing inter-strand spacing of 4.7 Å along the fibre axis [16]. 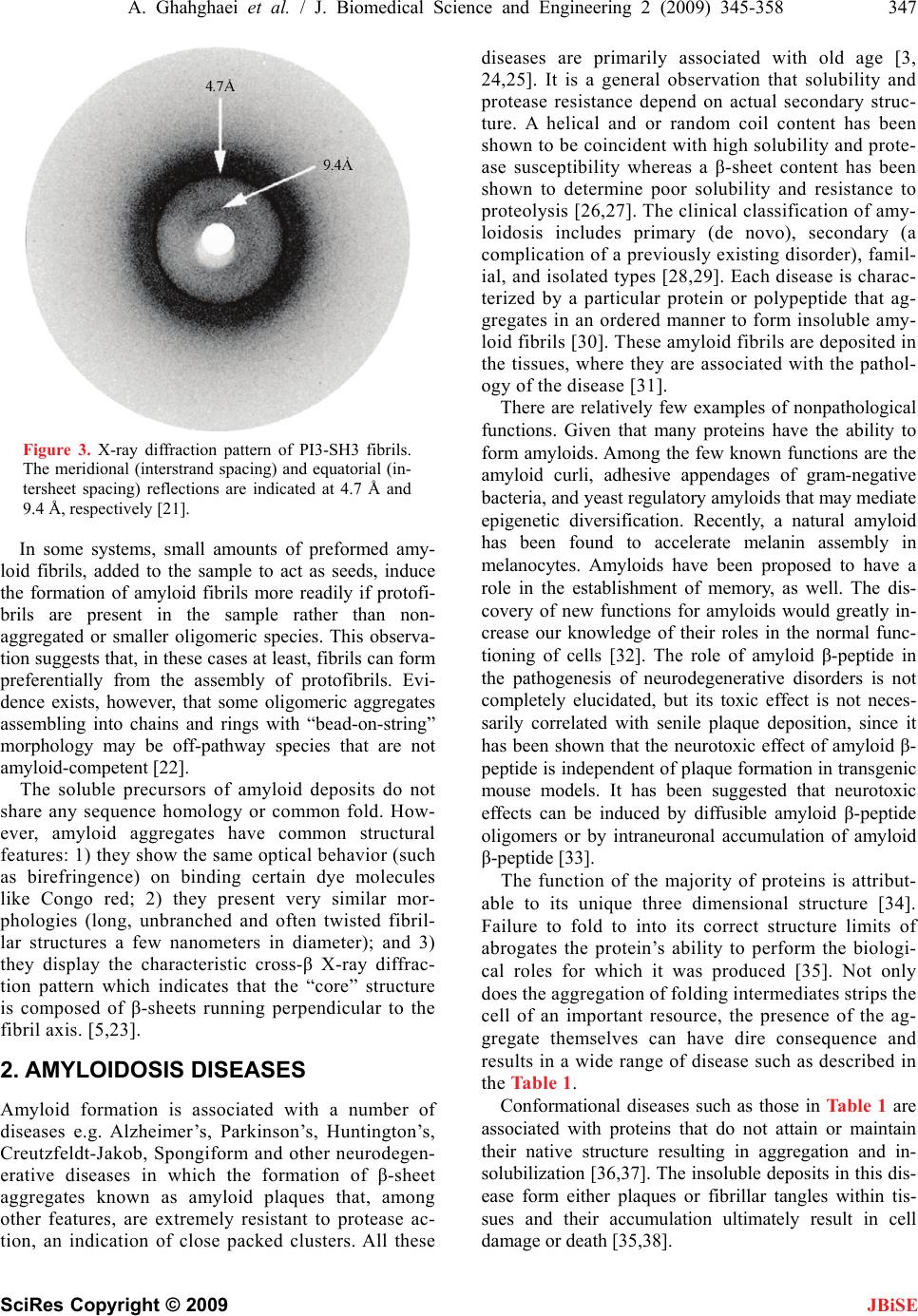 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 347 SciRes Copyright © 2009 JBiSE Figure 3. X-ray diffraction pattern of PI3-SH3 fibrils. The meridional (interstrand spacing) and equatorial (in- tersheet spacing) reflections are indicated at 4.7 Å and 9.4 Å, respectively [21]. In some systems, small amounts of preformed amy- loid fibrils, added to the sample to act as seeds, induce the formation of amyloid fibrils more readily if protofi- brils are present in the sample rather than non- aggregated or smaller oligomeric species. This observa- tion suggests that, in these cases at least, fibrils can form preferentially from the assembly of protofibrils. Evi- dence exists, however, that some oligomeric aggregates assembling into chains and rings with “bead-on-string” morphology may be off-pathway species that are not amyloid-competent [22]. The soluble precursors of amyloid deposits do not share any sequence homology or common fold. How- ever, amyloid aggregates have common structural features: 1) they show the same optical behavior (such as birefringence) on binding certain dye molecules like Congo red; 2) they present very similar mor- phologies (long, unbranched and often twisted fibril- lar structures a few nanometers in diameter); and 3) they display the characteristic cross-β X-ray diffrac- tion pattern which indicates that the “core” structure is composed of β-sheets running perpendicular to the fibril axis. [5,23]. 2. AMYLOIDOSIS DISEASES Amyloid formation is associated with a number of diseases e.g. Alzheimer’s, Parkinson’s, Huntington’s, Creutzfeldt-Jakob, Spongiform and other neurodegen- erative diseases in which the formation of β-sheet aggregates known as amyloid plaques that, among other features, are extremely resistant to protease ac- tion, an indication of close packed clusters. All these diseases are primarily associated with old age [3, 24,25]. It is a general observation that solubility and protease resistance depend on actual secondary struc- ture. A helical and or random coil content has been shown to be coincident with high solubility and prote- ase susceptibility whereas a β-sheet content has been shown to determine poor solubility and resistance to proteolysis [26,27]. The clinical classification of amy- loidosis includes primary (de novo), secondary (a complication of a previously existing disorder), famil- ial, and isolated types [28,29]. Each disease is charac- terized by a particular protein or polypeptide that ag- gregates in an ordered manner to form insoluble amy- loid fibrils [30]. These amyloid fibrils are deposited in the tissues, where they are associated with the pathol- ogy of the disease [31]. There are relatively few examples of nonpathological functions. Given that many proteins have the ability to form amyloids. Among the few known functions are the amyloid curli, adhesive appendages of gram-negative bacteria, and yeast regulatory amyloids that may mediate epigenetic diversification. Recently, a natural amyloid has been found to accelerate melanin assembly in melanocytes. Amyloids have been proposed to have a role in the establishment of memory, as well. The dis- covery of new functions for amyloids would greatly in- crease our knowledge of their roles in the normal func- tioning of cells [32]. The role of amyloid β-peptide in the pathogenesis of neurodegenerative disorders is not completely elucidated, but its toxic effect is not neces- sarily correlated with senile plaque deposition, since it has been shown that the neurotoxic effect of amyloid β- peptide is independent of plaque formation in transgenic mouse models. It has been suggested that neurotoxic effects can be induced by diffusible amyloid β-peptide oligomers or by intraneuronal accumulation of amyloid β-peptide [33]. The function of the majority of proteins is attribut- able to its unique three dimensional structure [34]. Failure to fold to into its correct structure limits of abrogates the protein’s ability to perform the biologi- cal roles for which it was produced [35]. Not only does the aggregation of folding intermediates strips the cell of an important resource, the presence of the ag- gregate themselves can have dire consequence and results in a wide range of disease such as described in the Table 1. Conformational diseases such as those in Table 1 are associated with proteins that do not attain or maintain their native structure resulting in aggregation and in- solubilization [36,37]. The insoluble deposits in this dis- ease form either plaques or fibrillar tangles within tis- sues and their accumulation ultimately result in cell damage or death [35,38].  348 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE Table 1. Representative human disease associated with defective protein folding [113,114,115]. Disease Protein Phenotype Alzheimer’s disease β-Amyloid Aggregation Tay-Sachs disease β-Hexasominidase Improper trafficking Cancer PS3 Misfolding/improper trafficking Spongiform Prion Protein Aggregation Creutz feldt-jakob diseases Prion protein Aggregation Maifan syndrom Fibrillin Misfolding Parkinson diseases α-Synuclein Aggregation Gaucher’s disease β-Glucosidase Improper trafficking Scurvy Collagen Misfolding Huntington’s diseases Huntington Aggregation Familial amyloiosis Transthyretin Aggregation 3. THE ALZHEIMER’S DISEASE Alzheimer’s disease (AD) is one of the most common neurodegenerative disorders associated with aging, and is characterized by fibrillar deposits of amyloid β (Aβ) peptides in the brain parenchyma and cortical blood ves- sels [5,37,39]. Patients in Alzheimer’s disease often have psychiatric manifestations of disease, such as psychosis (e.g., delusions and hallucinations) and disruptive behav- iors (e.g., psychomotor agitation and physical aggres- sion), especially in the later stages of the disease (1). One of the characteristics of brains afflicted with AD is the presence of extracellular structural elements referred to as amyloid plaques. Plaques are, in part, composed of masses of filaments, which are in turn composed of the insoluble form of the Aβ peptide [5,39,40]. These Aβ aggregates may cause neuronal injury directly by acting on synapses, or indirectly by activating microglia and astrocytes and therefore pharmacological interventions have been developed to target the sequential events originating from Aβ synthesis [40,41]. Usually neuritic plaques composed of amyloid β-protein (Aβ) are an early and invariant neuropathological feature of Alz- heimer's disease (AD), [41]. Genetic and neuropatho logical studies suggest that the processing of amyloid precursor protein (APP) to yield amyloid β-protein (Aβ), and its subsequent aggregation, play important roles in the pathogenesis of Alzheimer’s disease (AD) as the numbers of A plaques and A burden increased over time in the brain with AD and A deposition precedes clinical symptom of AD. The other pathological feature of this disease is intraneural neurofibrillary tangles [42, 43,44]. The Aβ family of peptides is derived by the enzymatic breakdown of the amyloid precursor protein (APP), a 563-770 residue membrane protein expressed in neu- ronal and non-neuronal tissue [45]. Patients with Down syndrome develop AD because of an extra copy of the amyloid precursor protein (APP) gene on chromosome 21 [46]. Amyloid precursor protein (APP) is a ubiquitous membrane glycoprotein encoded by a single gene on chromosome 21 and formed as a cleavage byproduct by three proteases, β-, γ- nd α-secretase [47]. It’s cleavege via the α-secretase or the β-secretase pathway, often re- ferred to as the amyloidogenic pathway. When APP is cleaved by α-secretase, it produces a large amino- terminal fragment APPα destined for secretion and a smaller carboxyl-terminal fragment. Further processing of the carboxyl-terminal fragment by γ-secretase pro- duces a 22- to 24-residue fragment termed P3, which may or may not be amyloidogenic. Alternatively, when APP is cleaved by β-secretase it produces a soluble amino-terminal fragment, APPβ, and a carboxyl-terminal fragment containing the Aβ peptide. Cleavage of the carboxyl-terminal fragment by γ-secretase results in the formation of multiple Aβ variants of 40-43 amino acids, which are prone to aggregate. The most abundant forms are 40 and 42 amino acids in length, Aβ 40 and Aβ 42. Both forms are capable of assembling into 60-100 Å diameter β-sheet fibrils that exhibit the characteristic cross-β X-ray fiber diffraction pattern, and yield a red- green birefringence when stained with Congo red [45]. The ratio of Aβ 42 to Aβ 40 is about 1:10. Aβ 42 plays a critical role in the pathogenesis of AD since its aggrega- tive ability and neurotoxicity are much greater than those of Aβ 40. Aβ 42 oligomers initially formed as a seed accelerate the aggregation of Aβ 40 to form the amyloid plaques that eventually lead to the neurodegeneration (amyloid cascade hypothesis) [47]. 4. THE STRUCTURE OF AMYLOID IN ALZHEIMER DISEASE The formation of insoluble Aβ deposits in the brain is a pathological hallmark of AD. If the hypothesis that the neurotoxicity of Aβ is mediated by amyloid fibril forma- tion is correct, inhibition of Aβ fibril formation might  A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 349 SciRes Copyright © 2009 slow progression or prevent the disease. However, more recent studies have shown that fibrils are not the only neurotoxic structures and that Aβ also assembles into soluble forms like small oligomers and protofibrils, which could be responsible for neurotoxicity [48]. Aβ (1-40) and Aβ (1-42) differ structurally by the absence or presence of two C-terminal amino acids [49]. The amino acid sequence of the Aβ 1–42 peptide is D1AEFRHDSG9 YEVHHQKLVFFAED23VGSNK28GAIIGLMVGGVV4 0I42, where subscripts indicate residue numbers. JBiSE The solid state NMR data are consistent with an Aβ1– 40 monomer secondary structure composed of a struc- turally disordered N-terminal region followed by two β- strand segments connected by a “loop” or “bend” seg- ment. Structural model resulted by NMR data support a parallel β-sheet structure in fibrils formed by both the 40 and 42-residue β amyloid peptides (Aβ 1–40,42). Paral- lel β-sheets have been found by solid state NMR origi- nally in fibrils formed by residues 10–35 of β-amyloid (Aβ 10–35) [23,31,50]. However, antiparallel β-sheets have also been found by solid state NMR in fibrils formed by shorter β-amyloid fragment. In agreement with the NMR experiment MD analysis of various struc- tural models of short β-amyloid fragments, including parallel and antiparallel β-sheet structures suggested a strand-loop-strand structure with parallel β-sheets for Aβ10–35 fibrils [51]. Recent solid state NMR and elec- tron microscopy experiments have shown that Aβ 1–40 can form at least two distinct amyloid fibril structures, with distinct and self-propagating morphologies and molecular-level structural features, dependent on subtle variations in fibril growth conditions. Although all Aβ1– 40 fibril structures studied to date contain parallel β- sheets, distinct structures differ in the specific details of side-chain-side-chain contacts and in mass-per length (MPL) values [52]. Thin sections of amyloid fibrils formed from fragments of amyloid β (Aβ), the amyloid fibril protein of Alz- heimer's disease, show cross-sections containing five or six protofilaments [23,53,54,55,56]. The protofilaments were over 40Å in diameter and appeared as “beaded” structures with a 200 Å periodicity [53,54,57,58,59]. Electron microscopy studies have shown that amyloid fibrils consist of three to six protofilaments. It is possible that the exposed hydrophobic residues could be involved in helical packing among the protofilaments. It is known, for instance, that side-chain packing plays an important role in stabilizing helical coiled-coil bundles. While the center of pleated β-sheet structure is hydrophobic, the ends of the sheets are somewhat hydrophilic. Ionic inter- actions and/or hydrogen bond interactions as additional forces stabilizing the cross-β structure have been pro- posed [60]. Oligomerization of Aβ42 induced by the intermolecu- lar β-sheet at positions 15–21 and 24–32 would confine this radical species in the oligomer, making it possible to damage the cells continuously. However, this oligomer would not lead to the fibrils since the C-terminal β-sheet is intramolecular (Figure 4(b)). As shown in Figure 4(a) and (b), the C-terminal residues in Aβ42 play a critical role in its aggregative and neurotoxicity [61] and the intermolecular β-sheet in the C-terminal region of Aβ42 seems to be preferable to long fibrils [47,62]. Aβ (1-42) did not adopt a unique fold, but rather a mixture of rap- idly interconverting conformations that were classified into three distinct families. The secondary structure analysis revealed that these conformations were domi- nated by loops and turns but that some helical structure formed in the C-terminal hydrophobic tail. Experimental studies of full-length Aβ monomers in water (organic solvent mixtures) showed that the monomer structure consists of two α-helical regions connected through an exible turn- or bend-like kink. The model of one helical turn of the twisted pleated β-sheet is composed of 48 monomers of Aβ. Each monomer contains an antipar- allel β-sheet. Overall, there are 96 β-strands. Four strands form a unit and 24 units stack together along the fibril axis. If each unit were twisted by 15° relative to its immediate neighbors (above and below) in the same way, 24 units would make a complete helical turn, with the Figure 4. (a) A representation of the hydrophobic residue at the C-terminus of A42 [61]. (b) The in- tramolecular anti-parallel -sheet at the C-terminus of A42 [112].  350 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 helical axis parallel to the fibril axis. Given that the angle between the two immediate strands from two different monomers was set at 15°, the helical twist formed by the 24 units in the model was actually nearly a complete turn (~350°). Four strands in each unit layer are in an identi- cal orientation, and those between two adjacent (above, below) unit layers are in an antiparallel arrangement (Figure 5(a) and (b)). In other words, the unit layers stack in an antiparallel fashion. The distances between the two adjacent strands in different unit layers and that between two strands within a unit layer are ~4.7 and ~9.7 Å, respectively (Figure 5(c) and (d)). These dis- tances are consistent with results from x-ray fibril dif- fraction studies on amyloid fibrils. NMR experiments on Aβ (10-35) monomer structure in an aqueous solution show a collapsed structure with loops, strands, and turns without any significant amount of α-helical or β-strand content. These studies suggest that Aβ monomer struc- ture is very sensitive to external conditions, such as tem- perature, pH, and solvent. An Aβ (1-40) monomer struc- ture considered with little α-helix or β-strand at low temperatures. As the temperature was increased to physiological, substantial β-sheet content developed. A folded Aβ (1-42) monomer, but not an Aβ (1-40) mono- mer, possesses a turn at G37-G38 stabilized by a hydro- phobic interaction between V36 and V39 [63]. Prelimi- nary cryo-electron microscopy of the amyloid fibrils formed from Aβ (11-25) and from Aβ (1-42) revealed that although they resembled one another closely in di- ameter and morphology, Aβ (11-25) appeared to form more consistently homogeneous, straight, uniform fibrils with clearly definededges (Figure 6(a). The Aβ (1-42) fibril showed less contrast, poorly defined edges and did not appear as straight or rigid (Figure 6(b). Close in- spection of Aβ (1-42) fibril images did not reveal any additional features (Figure 6(b). The Aβ (1-42) fibrils showed the spacing between the bands measured 4.7-4.8 Å, which corresponds to the hydrogen bonding distance between β-strands. These results appear to directly re- veal the β-sheet structure a single amyloid fibril [5,56]. A model has been suggested for Aβ amyloid protofila- ments in which, Aβ (12-42) folds into a β-hairpin and associates into four β-sheets, which twist around a cen- tral axis. In these models the β-strands are in register be- tween the sheets. Aβ (11-25) fibrils were more ordered, uniform structures. Aβ (1-42) fibrils appeared to have less defined edges and this meant that selecting fibril images was more difficult. It is possible that this is because Aβ (1-42) fibril structure is complicated by loops and disor- dered protein around the periphery of the fibrils whereas Aβ (11-25) fibrils constitute the core β-sheet structure. The β-strands clearly run perpendicular to the fibre axis. The strong, obvious striations visible across the fibre in these projections indicate that the β-sheets within the JBiSE Figure 5. (a) A four-strand antiparallel β-sheet unit from the dimer interface. (b) A dimer of Aβ 12-42 in an antiparallel β-sheet conformation. The distance between any two adjacent strands is ~4.7 Å. (c) The dimer is translated three times in a direction perpendicular to the plane defined by the C atoms of the dimer, each by ~9.7 Å. An 8-mer results. (d) A perfectly stacked 16-mer. It was constructed by stacking (parallel) two 8-mers together in the direction of the fibril axis [61]. 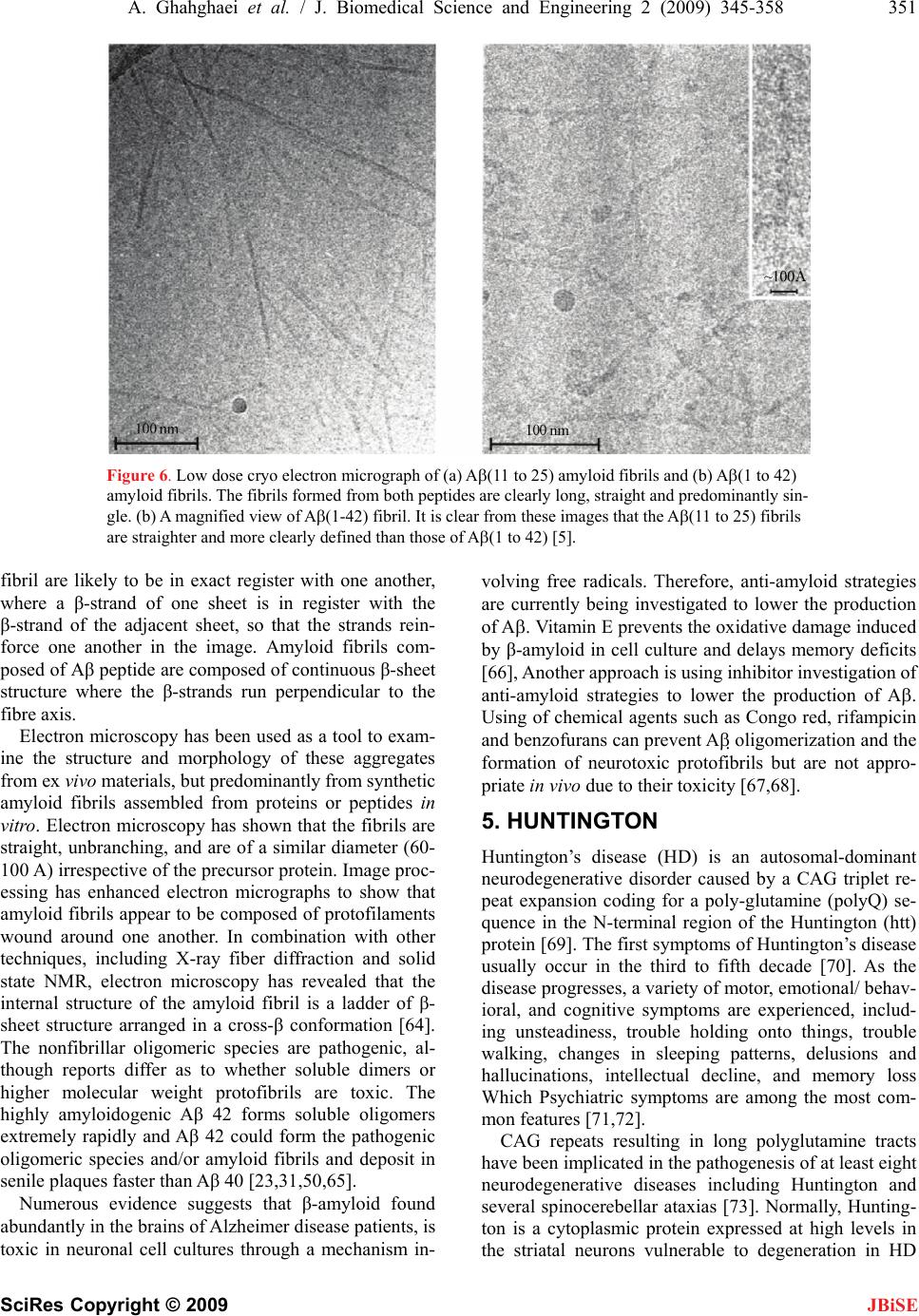 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 351 SciRes Copyright © 2009 JBiSE Figure 6. Low dose cryo electron micrograph of (a) A(11 to 25) amyloid fibrils and (b) A(1 to 42) amyloid fibrils. The fibrils formed from both peptides are clearly long, straight and predominantly sin- gle. (b) A magnified view of A(1-42) fibril. It is clear from these images that the A(11 to 25) fibrils are straighter and more clearly defined than those of A(1 to 42) [5]. fibril are likely to be in exact register with one another, where a β-strand of one sheet is in register with the β-strand of the adjacent sheet, so that the strands rein- force one another in the image. Amyloid fibrils com- posed of Aβ peptide are composed of continuous β-sheet structure where the β-strands run perpendicular to the fibre axis. Electron microscopy has been used as a tool to exam- ine the structure and morphology of these aggregates from ex vivo materials, but predominantly from synthetic amyloid fibrils assembled from proteins or peptides in vitro. Electron microscopy has shown that the fibrils are straight, unbranching, and are of a similar diameter (60- 100 A) irrespective of the precursor protein. Image proc- essing has enhanced electron micrographs to show that amyloid fibrils appear to be composed of protofilaments wound around one another. In combination with other techniques, including X-ray fiber diffraction and solid state NMR, electron microscopy has revealed that the internal structure of the amyloid fibril is a ladder of β- sheet structure arranged in a cross-β conformation [64]. The nonfibrillar oligomeric species are pathogenic, al- though reports differ as to whether soluble dimers or higher molecular weight protofibrils are toxic. The highly amyloidogenic Aβ 42 forms soluble oligomers extremely rapidly and Aβ 42 could form the pathogenic oligomeric species and/or amyloid fibrils and deposit in senile plaques faster than Aβ 40 [23,31,50,65]. Numerous evidence suggests that β-amyloid found abundantly in the brains of Alzheimer disease patients, is toxic in neuronal cell cultures through a mechanism in- volving free radicals. Therefore, anti-amyloid strategies are currently being investigated to lower the production of A. Vitamin E prevents the oxidative damage induced by β-amyloid in cell culture and delays memory deficits [66], Another approach is using inhibitor investigation of anti-amyloid strategies to lower the production of A. Using of chemical agents such as Congo red, rifampicin and benzofurans can prevent A oligomerization and the formation of neurotoxic protofibrils but are not appro- priate in vivo due to their toxicity [67,68]. 5. HUNTINGTON Huntington’s disease (HD) is an autosomal-dominant neurodegenerative disorder caused by a CAG triplet re- peat expansion coding for a poly-glutamine (polyQ) se- quence in the N-terminal region of the Huntington (htt) protein [69]. The first symptoms of Huntington’s disease usually occur in the third to fifth decade [70]. As the disease progresses, a variety of motor, emotional/ behav- ioral, and cognitive symptoms are experienced, includ- ing unsteadiness, trouble holding onto things, trouble walking, changes in sleeping patterns, delusions and hallucinations, intellectual decline, and memory loss Which Psychiatric symptoms are among the most com- mon features [71,72]. CAG repeats resulting in long polyglutamine tracts have been implicated in the pathogenesis of at least eight neurodegenerative diseases including Huntington and several spinocerebellar ataxias [73]. Normally, Hunting- ton is a cytoplasmic protein expressed at high levels in the striatal neurons vulnerable to degeneration in HD  352 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE and at low or undetectable levels in the neurons resistant to degeneration. In HD brain, N-terminal fragments of mutant huntington were reported to accumulate and form inclusions in the nucleus and identified as a characteris- tic neuronal intracellular inclusion (NII) [74]. This inclu- sions contained β-pleated sheet stabilized by hydrogen bonds. In several cases, NI is spherical aggregates, sometimes assembled by insoluble amyloid-like fibrils. Aggregates outside the nucleus have also been observed, but it has been shown recently that nuclear localization is required for toxicity [75]. The poly-Q disorders are also associated with the formation of neuronal nuclear inclusions (NI) whose average size is about 5–7 μm, and formed by aggregates of the protein with the expanded polyglutamine sequence [75,76,77]. The hypothesis that amyloid structures are formed in Huntington’s disease (HD) is supported by evidence that polyglutamine forms amyloid like protein aggregates in vitro, which stain with Congo Red (a histological stain for amyloid) and exhibit green birefringence under po- larized light, [78,79]. Ultrastructural studies of brains of HD transgenic mice revealed neuronal intranuclear in- clusion that contained aggregated huntingtin protein with granular or fibrillar morphology. Neuronal intranuclear inclusion with similar structural features also were de- tected in postmortem brains of HD and spinocerebellar ataxia type 3 patients as well as in stable and transiently transfected cell lines. Furthermore, recombinant proteins with an expanded poly(Q) stretch (51–122 glutamines) were found to form insoluble high molecular weight protein aggregates in vitro [80]. Increasing PG length correlates with the number of inclusions, which contributes to earlier onset of illness. In vivo inclusions occur primarily in subpopulations of neurons that are affected in the illnesses. In vitro expres- sion of PG tracts has been shown to lead to aggregation of proteins by either cross linking or by polar-zipper hydrogen bonding [81,82]. Consistent with this, it was found that the rate of aggregate formation in vitro di- rectly correlates with repeat length: the longer the poly(Q) tract, the faster the aggregation rate. Similarly, the protein concentration required for aggregate forma- tion decreased with an increase of the poly(Q) repeat length. In vitro aggregation of N-terminal, poly(Q)- containing huntingtin peptides is self initiated and, like a crystallization or Aβ formation, follows a nucleation- dependent pathway. [83]. Thus the formation of amyloidlike huntingtin aggre- gates in vitro not only depends on poly(Q) repeat length but also critically depends on protein concentration and time [79]. Among these, formation of ordered huntingtin aggregates is highly poly(Q) repeat length-dependent, which correlated the age of onset and the severity of HD. The majority of adult-onset cases have expansions rang- ing from 41 to 55 units, whereas expansions of 70 and above invariably cause the juvenile form of the disease. Interestingly, pathological effects occur in patients only when the length of poly-Q exceeds a rather sharp thresh- old of 35±40 glutamines. Thus length of the poly-Q tract correlates directly with the age of onset and with the severity of the symptoms in the diseases [79,83]. Discovery of the gene underlying HD can be used as a genetic therapy for a logical step towards finding a cure. Animal models that closely mimic the neurobiological and clinical symptoms of the disease can be used to test experimental treatments for HD across different stages of the disease [71,84]. 6. PARKINSON Parkinson’s disease (PD) is an age-related neurodegen- erative disorder. In 80% cases PD is most common Parkinsonism which clinically is a syndrome character- ized by tremor at rest, rigidity, slowness or absence of voluntary movement, postural, instability, and freezing. PD is a progressive disease, which affects both women and men. It has an effect on 1% of people beyond 65 years of age, with a higher prevalence in men [85-88]. The postmortem PD substantia nigra is characterized by sporadic intraneuronal cytoplasmic inclusions known as Lewy bodies (LB). The presence of Lewy bodies are associated with neurodegenerative disorders such as sporadic and familial Parkinson’s disease (PD), dementia with LBs and the LB variant of Alzheimer’s disease. The principal component of LBs is the protein α-synuclein, but they also contain various amounts of other proteins, including the molecular chaperones, αB-crystallin, clus- terin, torsin A, Hsp27 and Hsp70 [89,90,91,92,93]. α-Synuclein protein and a fragment of it, called NAC. α-Synuclein is a presynaptic protein, which was origi- nally identified as the precursor protein for the non-β- amyloid component (NAC) of Alzheimer's disease (AD) senile plaques. NAC is a 35 amino acid peptide compris- ing amino acids 61-95 of the α-syn sequence and has been identified as the second major constituent in the plaques of AD brains [94,95,96]. The formation of NAC peptides in a crossed β-pleated sheet conformation was assessed by thioflavine-S staining. Only the aged sam- ples of NAC(1-35) and NAC(1-18) were thioflavine-S positive, indicative of the presence of amyloid-like fila- ments [97,98]. The involvement of α-synuclein in neurodegenerative diseases was first suspected after the isolation of a α- synuclein fragment (NAC) from amyloid plaques in Alzheimer’s disease (AD). Later, two different α-synu- clein mutations were shown to be associated with auto- somal-dominant Parkinson’s disease (PD), but only in a small number of families. However, the discovery that α-synuclein is a major component of Lewy bodies and Lewy neurites, the pathological hallmarks of PD, con- firmed its role in PD pathogenesis [99].  A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 353 SciRes Copyright © 2009 JBiSE Two autosomal dominant mutations in the α-synuclein gene were linked to familial early onset PD: A53T and A30P. The mutations, A30P and A53T, do not affect the conformational behavior of the monomeric protein; wild-type α-synuclein (WT), A53T, and A30P are all ‘‘natively unfolded’’ at low concentration. At higher concentrations, WT, A53T, and A30P all form amyloid fibrils of similar morphology by what appears to be a nucleation-dependent mechanism [97]. All three proteins also produce nonfibrillar oligomers that may be assem- bly intermediates, analogous to the Aβ protofibril. Fibril- lization of α-synuclein is clearly accelerated by the A53T mutation but the effect of the A30P mutation on fibril formation has not been determined, although A30P ac- celerates the formation of nonfibrillar oligomers and the disappearance of unsedimentable protein from solution [100,101]. Circular dichroism spectroscopy indicated that α-synuclein undergoes a conformational change from random coil to β-sheet structure during assembly. X-ray diffraction and electron diffraction of the α- synuclein assemblies showed a cross-β conformation characteristicof amyloid [90]. In vitro multiple factors have been shown to acceler- ate α-synuclein aggregation. In aqueous solution, α- synuclein is natively unfolded within extended structure composed of random coil without a hydrophobic core. In vivo, it binds to rat brain vesicles via the first four 11- mer N-terminal repeats. In vitro, it binds to monolayer phospholipid membranes, acquiring an α-helical secon- dary structure probably formed by the seven N-terminal 11-residue repeats containing the conserved core se- quence Lys-Thr-Lys-Glu-Gly-Val [102]. Therapeutic stra- tegies aimed to prevent this aggregation are therefore envisaged. Although little has been learned about its normal function, α-synuclein appears to interact with a variety of proteins and membrane phospholipids, and may therefore participate in a number of signaling path- ways. In particular, it may play a role in regulating cell differentiation, synaptic plasticity, cell survival, and dopaminergic neurotransmission. Thus, pathological mechanisms based on disrupted normal function are also possible [102,103]. 7. SPONGIFORM AND FAMILIAL AMYLOIDOSIS DISEASES The transmissible spongiform encephalopathies (TSEs) or prion diseases form a group of fatal neurodegenera- tive diseases including bovine spongiform encephalopa- thy, Creutzfeldt-Jakob Disease (CJD) and Gerstmann- Stra¨ussler-Scheinker syndrome (GSS). TSEs, or prion diseases, are mammalian neurodegenerative disorders characterized by a posttranslational conversion and brain accumulation of an insoluble, protease-resistant isoform (PrPSc) of the host-encoded cellular prion protein (PrPC) [104]. The diseases are rare, but outbreaks of acquired forms of CJD, such as variant CJD and iatrogenic CJD with cadaveric growth hormone or dura grafts, have prompted the development of therapeutic interventions and new diagnostic methods [105]. In humans, prion diseases result from infectious modes of transmission, Gerstmann-Sträussler-Scheinker Syndrome, Fatal Famil- ial Insomnia; and modes of transmission. The clinical symptoms associated with each of the human prion dis- ease forms vary dramatically [106,107]. Cellular prion protein, PrPC, is a predominantly α- helical glycoprotein that remains attached to the outer membrane of the cells through a glycophosphatidyl inositol linkage. A -sheet-rich conformational isoform of PrPC, termed PrPSC, has been considered to be the infectious agent of the fatal neurodegenerative diseases transmissible spongiform encephalopathies (TSE) and its hereditary forms of spongiform encephalopathies (SE). [104]. PrPSC exists as oligomers and amyloid polymers (fibres) and, unlike PrPC, is resistant to digestion by pro- teinase K (PK), which is considered as an indicator of formation of PrPSC. However, PrPC may undergo dis- ease-associated structural modifications that do not lead to a protease-resistant molecule, indicating that prion disease can occur in the absence of PrPSC; further prote- ase-resistant prion protein forms without any infectivity can be generated. [108]. Familial amyloidosis may occur in patients with fa- milial Mediterranean fever or it may arise from transthyretin mutations. Approximately 30 such muta- tions have been described [28]. Transthyretin, previously called prealbumin because it migrates ahead of albumin in standard electrophoretic separations, is a serum carrier of thyroid hormones and vitamin A. The mutant transthyretin is deposited as extracellular twisted β- pleated sheet fibrils in peripheral somatic and autonomic nerves and visceral organs; it causes autonomic and pe- ripheral somatic disorders, and the disease is ultimately fatal [28]. Clinically, patients are susceptible to neuro- pathic, cardiopathic, or nephropathic complications. Type I familial hereditary generalized amyloidosis is inherited as an autosomal dominant gene mutation with a single amino acid substitution of methionine for valine at posi- tion 30 in transthyretin [28]. 8. CONCLUSIONS The topics discussed in this review provide a great deal of evidence for the proteins that have an intrinsic capac- ity of aggregating and forming structures such as amy- loidfibrils. Aggregates are most commonly formed from the interaction of partially folded intermediates contain- ing significant native-like structure. These interactions involve extended chain or -sheet-like conformations. Thus, both ordered and disordered aggregates show in- creased structure relative to the native conformation (in the case of all-proteins, the increased structure is dis-  354 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE tinct from that of the native protein). Misfolded and aggregated species are likely to owe their toxicity to the exposure on their surfaces of regions of proteins that are buried in the interior of the structures of the correctly folded native states. The exposure of large patches of hydrophobic groups is likely to be par- ticularly significant as such patches favour the interac- tion of the misfolded species with cell membranes [109,110,111]. Interactions of this type are likely to lead to the impairment of the function and integrity of the membranes involved, giving rise to a loss of regulation of the intracellular ion balance and redox status and eventually to cell death. The data reported so far strongly suggest that the conversion of normally soluble proteins into amyloid fibrils and the toxicity of small aggregates appearing during the early stages of the formation of the latter are common or generic features of polypeptide chains. Moreover, the molecular basis of this toxicity also appears to display common features between the different systems that have so far been studied. Amyloidosis comprises a group of diseases in which protein tissue deposits have common morphologic struc- tural, and staining properties but variable protein compo- sition [28,37]. In this review we highlighted the rele- vance of amyloid fibril formation of different protein to diseases. The fibrillar morphology, diagnostic staining characteristics and underlying β-sheet structure of the proteins deposits led to them being classified as amyloid fibrils, and the diseases are now regarded as a protein misfolding disease. There are still many outstanding and critical questions regarding protein aggregation. Among these are questions about the detailed mechanism of the aggregation process, factors determining the kinetics of aggregation, the structural nature of the intermolecular interactions, and how aggregation may be effectively and efficiently prevented, especially in vivo. REFERENCES [1] Come, J. H., Fraser, P. E. and Lansbury, J., P. T., (1993) A kinetic model for amyloid formation in the prion dis- eases: Importance of seeding. P. Natl. Acad. Sci. USA. 90, 5959–5963. [2] Chiti, F., Webster, P., Taddei, N., Clark, A., Stefani, M., Ramponi, G. and Dobson, C. M. (1999) Designing condi- tions for in vitro formation of amyloid protofilaments and fibrils. Biochemistry. 96, 3590–3594. [3] Arrigo, P. A. and Muller, G. E. W. (2001) ((W. E. G. Mul- ler (Managing Editor), P. J., I.Kostovic, Y. Kuchino, A. Macieira-coelho, R. E. Rhoads, Ed.). Ed.). [4] van Montfort, L. R., Slingsby, C. and Vierling, E. (2002) Structure and function of the small heat shock protein/ α- crystallin family of molecular chaperones. Protein. Chem. 59, 105–155. [5] Serpell, C. L. a. (2000) Alzheimer's amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta. 1502, 16–30. [6] Pellarin, R. and Caflisch, A. (2006) Interpreting the ag- gregation kinetics of amyloid peptides. J. Mol. Biol 360, 882–892. [7] Hall, D., Hirota, N. and Dobson, M. C. (2005) A toy model for predicting the rate of amyloid formation from unfolded protein. J. Mol. Biol. 195, 195–205. [8] Dobson, M. C. (2001) The structure basis of protein fold- ing and its links with human disease. Phil. Trans. R. Soc. Lond. B.. 356, 133–145. [9] Dobson, M. C. (1999) Protein misfolding, evolution and disease. TIBS. 24, 329–332. [10] Canet, D., Sunde, M., Last, A. M., Miranker, A., Spencer, A., Robinson, C. V. and Dobson, C. M. (1999) Mechanis- tic studies of the folding of human lysozyme and the ori- gin of amyloidogenic behavior in its disease-related vari- ants. Biochemistry. 38, 6419–27. [11] MacPhee, E. C. and Dobson, C. M. (2000) Chemical dissection and reassembly of amyloid fibril formed by a peptide fragment of transthyretin. J. Mol. Biol. 297, 1203–1215. [12] Shirahama, T. and Cohens, A. S. (1967) High-resolution electron microscopic. Analysis of the amyloid fibril. J. Cell. Biol.. 33, 679–708. [13] Chamberlain, K. A., MacPhee, E. C., Zurdo, J., Moro- zova-Roche, A. L., Hill, A. H., Dobson, M. C. and Davis, J. J. (2000) Ultra structural organization of amyloid fi- brils by atomic force microscopy. Biophys. J.. 79, 3282– 3293. [14] Conway, K. A. and Harper, J. D. (2000) Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 39, 2552–63. [15] Cohen, A. S., Shirahama, T. and Skinner, M. (1982) Elec- tron microscopy of amyloid. In electron microscopy of protein. Academic Press Inc, London UK. 3, 165–205. [16] Jiménez, J. L., Guijarro, J. I., Orlova, E., Zurdo, J., Dob- son, C. M., Sunde, M. and Saibil, H. R. (1999) Cryo- electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. EMBO. 18, 815– 821. [17] Serpell, L. C., Sunde, M., E., F. P., Luther, P. K., Morris, E. P., Sangren, O., Lundgren, E. and Blake, C. C. (1995) Examination of the structure of the transthyretin amyloid fibril by image reconstruction from electron micrographs. J. Mol. Biol.. 254, 113–8. [18] Kirschner, A. D., Abraham, C. and Selkoe, J. D. (1986) X-ray diffraction from intraneuronal paired helical fila- ments and extraneuronal amyloid fibers in alzheimer dis- ease indicates cross-β conformation. P. Natl. Acad. Sci. USA. 83, 503–508. [19] Blake, C. and Serpell, L. (1996) Synchrotron X-ray stud- ies suggest that the core of the transthyretin amyloid fi- bril is a continuous β-sheet helix. Structure. 4, 989–98. [20] Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B. and Blake, C. F. (1997) Common core structure of amyloid fibrils by synchrotron X-ray diffrac- tion. J. Mol. Biol. 273, 729–739. [21] Guijarro, I. L., Sunde, M., Jones, A. J., Campbell, D. I. and Dobson, M. C. (1998) Amyloid fibril formation by an SH3 domain. Biochemistry. 95, 4224–4228. [22] Plakoutsi, G., Bemporad, F., Calamai, M., Taddei, N., Dobson, M. C. and Chiti, F. (2005) Evidence for a mechanism of amyloid formation involving molecular  A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 355 SciRes Copyright © 2009 JBiSE reorganisation within native-like precursor aggregates. J. Mol. Biol. 351, 910–922. [23] Cecchini, M., Curcio, R., Pappalardo, M., Melki, R. and Caflisch, A. (2006) A Molecular Dynamics Approach to the Structural Characterization of Amyloid Aggregation. J. Mol. Biol. 357, 1306–1321. [24] Hatters, D. M., Wilson, M. R., Easterbrook-Smith, S. B. and Howlett, G.J. (2002) Suppression of apolipoprotein C-II amyloid formation by the extracellular chaperone, clusterin Eur. J. Biochem. 269, 2789–2794. [25] Haley, D. A., Horwitz, J. and Stewart, P. L. (1998) The small Heat-Shock protein,aB-crystallin, has a variable quaternary structure. J. Mol. Biol. 277, 27–35. [26] Jacchieri, G. S. (1998) Study of a-helix to b-strand to b- sheet transitions in amyloid: the role of segregated hy- drophobic b-strands. Biophysical Chemistry. 74, 23–34. [27] Milner-White, J. E., Watson, D. J., Qi, G. and Hayward, S. (2006) Amyloid formation may involve a- to b-sheet interconversion via peptide plane flipping. Structure. 14, 1369–1376. [28] Horwitz, S., Thomas, C., Gruener, G., Nand, S. and Shea, F. J. (1998) MR of Leptomeningeal Spinal and Posterior Fossa Amyloid. AJNR Am J Neuroradiol. 19, 900–902. [29] Horowitz, S., Thomas, C., Gruener, G., Nand, S. and Shea, F. J. (1998) MR of Leptomeningeal Spinal and Posterior Fossa Amyloid. AJNR Am J Neuroradiol. 19, 900–902. [30] Tycko, R. (2000) Solid-state NMR as a probe of amyloid fibril structure. Curr. Opin. Chem. Biol. 4, 500–506. [31] Makin, S. O., Atkins, E., Sikorski, P., Johansson, J. and Serpell, C. L. (2005) Molecular basis for amyloid fibril formation and stability. PNAS. 102, 315–320. [32] Otoo, N. H., Lee, G.K., Qiu, W. and Lipke, N. P. (2008) Candida albicans Als adhesins have conserved amyloid- forming sequences. Eukaryotic Cell. 7, 776–782. [33] Religa, D., Laudon, H., Styczynska, M., Winblad, B., Näslund, J. and Haroutunian, V. (2003) Amyloid β Pa- thology in Alzheimer’s Disease and Schizophrenia. Am J Psychiatry. 160, 867–872. [34] Barrel, J. M., Broadley, S. A., Schaffar, G. and Hart, F. U. (2004) Role of molecular chaperones in protein misfold- ing diseases. Semin. Cell. Dev. Biol. 15, 17–29. [35] Ellidson and Bottomley (2004) The role of Misfolding in the pathogenesis of human diseases. IUBMB life. 56(3), 119–123. [36] Carrell, R. W. and Lomas, D. A. (1997) Conformational diseas. Lancet. 350, 134–138. [37] Small, W. G., Kepe, V., Ercoli, M. L., Siddarth, P., Book- heimer, Y. S., Miller, J. K., Lavretsky, H., Burggren, C. A., Cole, M. G., Vinters, V. H., Thompson, M. P., Huang, C. S., Satyamurthy, N., Phelps, E. M. and Barrio, R. J. (2006) PET of brain amyloid and Tau in mild cognitive impairment. N. Engl. J. Med. 355, 2652–2663. [38] Selkoe (2003) Folding proteins in fatal ways. Nature. 426(6968), 900–904. [39] Han, H., Weinreb, H. P. and Lansbury, T. P. (1995) The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by p-amyloid: is NAC a com- mon trigger or target in neurodegenerative disease? Chemistry & Biology. 2, 163–l69. [40] Shoghi-Jadid, K., Barrio, R. J., Kepe, V., Wu, M. H., Small, W. G., Phelps, E. M. and Huang, C. S. (2005) Im- aging b-amyloid fibrils in Alzheimer’s disease: a critical analysis through simulation of amyloid fibril polymeriza- tion. Nuclear Medicine and Biology. 32, 337–351. [41] Miura, Y., You, C. and Ohnishi, R. (2008) Inhibition of Alzheimer amyloid aggregation by polyvalent trehalose. SCIENCE AND TECHNOLOGY OF ADVANCEDMA- TERIALS (Sci. Technol. Adv. Mater). 9, 1–6. [42] Sefton, F. C. and Yu, G. (2008) Ab Predictor of Alz- heimer disease symptoms Arch Neurol. 65, 875–876. [43] Malm, T., Ort, M., Tä htivaara, L., Jukarainen, N., Gold- steins, G., Puolivä li, J., Nurmi, A., Pussinen, R., Ah- toniemi, T., Miettinen, K. T., Kanninen, K., Leskinen, S., Vartiainen, N., Yrjä nheikki, J., Laatikainen, R., Harris- White, E. M., Koistinaho, M., Frautschy, A. S., Bures, J. and Koistinaho, J. (2006) b-Amyloid infusion results in delayed and age-dependent learning deficits without role of inflammation or b-amyloid deposits. PNAS 103, 8852–8857. [44] Grundman, M., Petersen, R. C., Ferris, H. S., Thomas, R. G., Aisen, S. P., Bennett, A. D., Foster, L. N., Jack, R. C., Galasko, R. D., Doody, R., Kaye, J., Sano, M., Mohs, R., Gauthier, S., Kim, T. H., Jin, S., Schultz, N. A., Schafer, K., Mulnard, R., Dyck, V. H. C., Mintzer, J., Zamrini, Y. E., Cahn-Weiner, D. and Thal, J. L. (2004) Mild cogni- tiveimpairment can be distinguished from Alzheimer Disease and normal aging for clinical trials. Arch Neurol. 61, 59–66. [45] Huang, J. H. T., Yang, S. D., Plaskos, N. P., Go, S., Yip, M. C., Fraser, E. P. and Chakrabartty, A. (2000) Struc- tural Studies of Soluble Oligomers of the Alzheimer b- Amyloid Peptide. J. Mol. Biol. 297, 73–87. [46] Rosenberg, N. R. (2000) Explaining the cause of the amyloid burden in Alzheimer disease. Arch Neurol. 59, 1367–1368. [47] Irie, K., Murakami, K., Masuda, Y., Morimoto, A., Ohi- gashi, H., Ohashi, R., Takegoshi, K., Nagao, M., Shi- mizu, T. and Shirasawa, T. (2005) Structure of B-amyloid fibrils and its relevance to their neurotoxicity: implica- tions for the pathogenesis of Alzheimer’s disease. Journal of Bioscience and Bioengineering. 99, 437–447. [48] Rivière, C., Richard, T., Quentin, L., Krisa, S., Mérillon, M. J. and Monti, P. J. (2007) Inhibitory activity of stil- benes on Alzheimer’s b-amyloid fibrils in vitro. Bioor- ganic & Medicinal Chemistry. 15, 1160–1167. [49] Hardy, J. and Selkoe, D. J. (2002) The amyloid hypothe- sis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 297, 353–356. [50] Sachse, C., Xu, C., Wieligmann, K., Diekmann, S., Grig- orieff, N. and Fändrich, M. (2006). Quaternary structure of a mature amyloid fibril from Alzheimer’s Aβ(1-40) peptide. J. Mol. Biol. 362, 347–354. [51] Ma, B. Y. and Nussinov, R. (2002) Stabilities and con- formations of Alzheimer’s b-amyloid peptide oligomers (Ab (16–22), Ab (16–35) and Ab (10–35)): Sequence ef- fects. Proc. Natl Acad. Sci. USA. 99, 14126–14131. [52] Buchete, V. N., Tycko, R. and Hummer, G. (2005) Mo- lecular dynamics simulations of Alzheimer’s b-Amyloid protofilaments. J. Mol. Biol. 353, 804–821. [53] JimeÂnez, L. J., Tennent, G., Pepys, M. and Saibil, R. H. (2001) Structural Diversity of ex vivo Amyloid Fibrils Studied by Cryo-electron Microscopy. J. Mol. Biol. 311, 241–247.  356 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE [54] Gilead, S. and Gazit, E. (2005) Self-organization of short peptide fragments: From amyloid fibrils to nanoscale su- pramolecular assemblies. Supramolecular Chemistry. 17, 87–92. [55] Pellarin, R., Guarnera, E. and Caflisch, A. (2007) Path- ways and intermediates of amyloid fibril formation. J. Mol. Biol. 374, 917–924. [56] Serpell, L. C. and Smith, J. M. (2000) Direct visualisa- tion of the b-sheet structure of synthetic Alzheimer's amyloid. J. Mol. Biol. 299, 225–231. [57] Klement, K., Wieligmann, K., Meinhardt, J., Hortschansky, P., Richter, W. and Fändrich, M. (2007) Effect of different salt ions on the propensity of aggrega- tion and on the structure of Alzheimer’s Aβ(1-40) amy- loid fibrils. J. Mol. Biol. 373, 1321–1333. [58] Ban, T., Hoshino, M., Takahashi, T., Hamada, D., Hase- gawa, K., Naiki, H. and Goto, Y. (2004) Direct observa- tion of Ab amyloid fibril growth and inhibition. J. Mol. Biol. 344, 757–767. [59] Idicula-Thomas, S. and Balaji, V. P. (2007) Protein ag- gregation: A perspective from amyloid and inclusion- body formation. CURRENT SCIENCE. 92, 758–767. [60] Li, L., Darden, A. T., Bartolotti, L., Kominos, D. and Pedersen, G. L. (1999) An atomic model for the pleated b-sheet structure of Ab- amyloid protofilaments. Biophys J. 76, 2871–2878. [61] Weinreb, P. H., Jarrett, J. T. and Lansbury, P. T. (1994) Peptide models of a hydrophobic cluster at the C- terminus of the amyloid protein. J. Am. Chem. Soc. 116, 10835–10836. [62] Williams, A. D., Portelius, E., Kheterpal, I., Guo, J., Cook, K. D., Xu, Y. and Wetzel, R. (2004) Mapping A- amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842. [63] Lazo, N. D. and Downing, D. T. (1999). Fibril formation by amyloid b-proteins may involve b-helical protofibrils. J. Pept. Res., 53, 633-640. [64] Thusnelda, S. and Louise C, S. (2005) Structure and morphology of the Alzheimer’s amyloid fibril, Micros- copy Research and Technique, 67, 210-217. [65] El-Agnaf, A. M. O., Mahil, S. D., Patel, P. B. and Austen, M. B. (2000) Oligomerization and Toxicity of b- Amyloid-42 Implicated in Alzheimer’s Disease. Bio- chemical and Biophysical Research Communications. 273, 1003–1007. [66] Grundman, M. (2000) Vitamin E and Alzheimer disease: the basis for additional clinical trials. Am Clin Nutr. 71, 630S–636S. [67] Tuszynski, H. M., Thal, L., Pay, M., Salmon, P. D., Sang U, H., Bakay, R., Patel, P., Blesch, A., Vahlsing, L. H., Ho, G., Tong, G., Potkin, G. S., Fallon, J., Hansen, L., Mufson, J. L., Kordower, H. J., Gall, C. and Conner, J. (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551– 555. [68] Martin, K. B., Meinert, L. C. and Breitner, S. C. J. (2002) Double placebo design in a prevention trial for Alz- heimer's disease. Cont. Clin. Trials. 23, 93–99. [69] Dı´az-Hernández, M., Torres-Peraza, J., Salvatori- Abarca, A., Morán, M.A., Gómez-Ramos, P., Alberch, J. and Lucas, J. J. (2005) Full motor recovery despite stri- atal neuron loss and formation of irreversible amyloid- like inclusions in a conditional mouse model of Hunting- ton’s Disease. The Journal of Neuroscience. 25(42), 9773–9781. [70] Gusella, J. F., Wexler, N. S., Conneally, P. M., Naylor, S. L., Anderson, M. A., Tanzi, R. E., Watkins, P. C., Ottina, K., Wallace, M. R., Sakaguchi, A. Y., Young, A. B., Shoulson, I., Bonilla, E. and Martin, J. B. (1983) A po- lymorphic DNA marker genetically linked to Huntington; s disease. Nature. 306, 234–238. [71] Anderson, K. E., Louis, E. D., Stern, Y. and Marder, K. S. (2001) Cognitive Correlates of Obsessive and Com- pulsive Symptoms in Huntington’s Disease. Am J Psy- chiatry. 158, 799–801. [72] Mitchell, I. J., Heims, H., Neville, E.A. and Rickards, H. (2005) Huntington’s disease patients show impaired per- ception of disgust in the gustatory and olfactory modali- ties. J. Neuropsychiatry Clin Neurosci. 17, 119–121. [73] Hirakura, Y., Azimov, R., Azimova, R. and Kagan, L. B. (2000) Polyglutamine-Induced Ion Channels: A Possible Mechanism for the Neurotoxicity of Huntington and Other CAG Repeat Diseases. Journal of Neuroscience Research. 60, 490–494. [74] Davies, W. S., Beardsall, K., Turmaine, M., DiFiglia, M., Aronin, N. and Bates, P. G. (1998) Are neuronal intranu- clear inclusions the common neuropathology of triplet- repeat disorders with polyglutamine-repeat expansions? THE LANCET. 351, 131–33. [75] Temussi, A. P., Masino, L. and Pastore, A. (2003) From Alzheimer to Huntington: why is a structural understand- ing so difficult? The EMBO Journal. 22, 355–361. [76] Karpug, V. M., Becher, W. M., Springer, E. J., Chabas, D., Youssef, S., Pedotti, R., Mitchell, D. and Steinman, L. (2002). Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cys- tamine. Nat. Med. 8, 143–149. [77] Karpuj, V. M., Becher, W. M., Springer, E. J., Chabas, D., Youssef, S., Pedotti, R., Mitchell, D. and Steinman, L. (2002) Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cys- tamine. Nature Medicine. 8, 143–149. [78] Hoffner, G. and Djian, P. (2000) Protein aggregation in Huntington’s disease. Biochimie. 84, 273–278. [79] Mcgowan, P. D., Vanroon-Mom, W., Holloway, H., Bates, P. G., Mangiarini, L., Cooper, S. J. G., R., F. L. and Snell, G. R. (2000) Amyloid-lioke inclusions in hunting- ton, s disease. Neuroscience. 100, 677–680. [80] Gutekunst, A. C., Li, H. S., Yi, H., Mulroy, S. J., Kuem- merle, S., Jones, R., Rye, D., Ferrante, J. R., Hersch, M.S. and Li, J.X. (1999) Nuclear and Neuropil Aggre- gates in Huntington’s Disease: Relationship to Neuropa- thology. The Journal of Neuroscience. 19(7), 2522–2534. [81] Ross, A. C. (2002) Polyglutamine pathogenesis: emer- gence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 35, 819–822. [82] Dahlgren, R. P., Karymov, A. M., Bankston, J., Holden, T., Thumfort, P., Ingram, V. M. and Lyubchenko, L. Y. (2005) Atomic force microscopy analysis of the Hunting-  A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 357 SciRes Copyright © 2009 JBiSE ton protein nanofibril formation. Nanomedicine: Nanotechnology, Biology and Medicine. 1, 52–57. [83] Scherzinger, E., Sittler, A., Schweiger, K., Heiser, V., Lurz, R., Hasenbank, R., Bates, P.G., Lehrach, H. and Wanker, E. E. (1999) Self-assembly of polyglutamine- containing huntingtin fragment into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. USA. 96, 4604–4609. [84] Borlongan, C. V., Koutouzis, T. K., Freeman, T. B., Ca- hill, D. W. and Sanberg, P. R. (1995) Behavioral pathol- ogy induced by repeated systemic injections of 3- nitropropionic acid mimics the motoric symptoms of Huntington's disease. Brain Res. 697, 254–257. [85] Spillantini, G. M., Crowther, A. R., Jakes, R., Hasegawa, M. and Goedertm, M. (1998) A-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA. 95, 6469–6473. [86] Recchia, A., Debetto, P., Negro, A., Guidolin, D., Skaper, D. S. and Giusti, P. (2004) A-Synuclein and Parkinson’s disease. The FASEB J. 18, 617–626. [87] Dauer, W. and Przedborski, S. (2003) Parkinson’s dis- ease: mechanisms and models. Neuron. 39, 889–909. [88] Hariz, G. and Hriz, I. M. (2000) Gender distribution in surgery for Parkinson's disease. Parkinsonism Relate Disord. 6, 155–157. [89] Iwaki, T., Wisniewski, T., Iwaki, A., Corbin, E., Tomo- kane, N., Tateishi, J. and Goldman, J. E. (1992) Accumu- lation of aB-crystallin in central nervous system glia and neurons in pathologic conditions. Am. J. Path. 140, 345– 356. [90] Serpell, C. L., Berriman, J., Jakes, R., Goedert, M. and Crowther, A. R. (2000) Fiber diffraction of synthetic a- synuclein filaments shows amyloid-like cross-b confor- mation. PNAS. 97, 4897-4902. [91] Mizutani, T., Inose, T., Nakajima, S., Kakimi, S., Uchi- gata, M. and Ikeda, K. (1998) Familial parkinsonism and dementia with ballooned neurons, argyrophilic neuronal inclusions, atypical neurofibrillary tangles, tau-negative strocytic fibrillary tangles, and Lewy bodies.. Acta Neu- ropathol. (Berl). 95, 15–27. [92] Sasaki, K., Doh-ura, K., Wakisaka, Y. and Iwaki, T. (2002) Clusterin/apolipoprotein J is associated with cor- tical Lewy bodies: immunohistochemical study in cases with alpha-synucleinopathies. Acta Neuropathol. (Berl). 104, 225–230. [93] McLean, P. J., Kawamata, H., Shariff, S., Hewett, J., Sharma, N. and Ueda, K. (2002) Torsin A and heat shock proteins act as molecular chaperones: suppression of al- pha-synuclein aggregation. J. Neuro. 83, 846–854. [94] Splllantini, G. M. a., Crowther, A. R., JAKES, R., Cairns, J. N., Lantos, L. P. and Goedert, M. (1998) Filamentous a-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neuroscience. 251, 205–208. [95] El-Agnaf, A. M. O., Jakes, R., Curran, D. M. and Wal- lace, A. (1998) E¡ects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological proper- ties of K-synuclein protein implicated in Parkinson's dis- ease. FEBS. 440, 67–70. [96] Spillantini, G. M. a., Crowther, A. R., JAKES, R., Cairns, J. N., Lantos, L. P. and Goedert, M. (1998) Filamentous a-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neuroscience. 251, 205–208. [97] El-Agnaf, A. M. O., JAKES, R., Curran, D. M., Middle- ton, D., Ingenito, R., Bianchi, E., Pessi, A., Neill, D. and Wallace, A. (1998) Aggregates from mutant and wild- type a-synuclein proteins and NAC peptide induce apop- totic cell death in human neuroblastoma cells by forma- tion of L-sheet and amyloid-like ¢laments. FEBS. 440, 71–75. [98] Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Ue´da, K., Ikeda, K. and Kawai, M. (1999) Cellular co-localization of phosphorylated tau- and NACPr a- synuclein-epitopes in Lewy bodies in sporadic Parkin- son’s disease and in dementia with Lewy bodies. Brain Research. 843, 53–61. [99] Arima, K., Ue´da, K., Sunohara, N., Hirai, S., Izumi- yama, Y., Tonozuka-Uehara, H. and Kawai, M. (1998) Immunoelectron-microscopic demonstration of NACPra- synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson’s disease and in dementia with Lewy bodies. Brain Research. 808, 93–100. [100] Conway, A. K., Lee, J. S., Rochet, C. J., Ding, T. T., Wil- liamson, E. R. and Lansbury, T. P. (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both a-synuclein mutations linked to early-onset Parkin- son’s disease: Implications for pathogenesis and therapy. PNAS. 97, 571–576. [101] Conway, A. K., Harper, J. D. and Lansbury, P. T. (2000) Fibrils Formed in Vitro from R-Synuclein and Two Mu- tant Forms Linked to Parkinson’s Disease are Typical Amyloid. Biochem. 39, 2552–2563. [102] Lu¨cking, B. C. and Brice, A. (2000) a-synuclein and Parkinson’s disease. CMLS, Cell. Mol. Life Sci. 57, 1894–1908. [103] Williams, A. D., Portelius, E., Kheterpal, I., Guo, J., Cook, K. D., Xu, Y. and Wetzel, R. (2004) Mapping A amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842. [104] Casalone, C., Zanusso, G., Acutis, P., Ferrari, S., Capucci, L., Tagliavini, F., Monaco, S. and Caramelli, M. (2004) Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. PNAS 101, 3065–3070. [105] Ishikawa, K., Doh-ura, K., Kudo, Y., Nishida, N., Mura- kami-Kubo, I., Ando, Y., Sawada, T. and Iwaki, T. (2004) Amyloid imaging probes are useful for detection of prion plaques and treatment of transmissible spongiform en- cephalopathies. Journal of General. 85, 1785–1790. [106] Aguzzi, A., Heikenwalder, M. and Miele, G. (2004) Pro- gress and problems in the biology, diagnostics, and therapeutics of prion diseases. J. Clin. Invest. 114, 153– 160. [107] Weissmann, C. and Aguzzi, A. (2005) Approaches to therapy of prion diseases. Ann. Rev. of Med. 56, 321– 344. [108] Nandi, P. K. and Nicole, J.-C. (2004) Nucleic acid and prion protein interaction produces spherical amyloids which can function in vivo as coats of spongiform en-  358 A. Ghahghaei et al. / J. Biomedical Science and Engineering 2 (2009) 345-358 SciRes Copyright © 2009 JBiSE cephalopathy agent. J. Mol. Biol. 344, 827–837. [109] Kourie, J I and A, S. A. (2000) Properties of cytotoxic peptide-induced ion channels. Am J Physiol Cell Physiol. 278, C1063–C1087. [110] Kourie, J. I. and L, H. C. (2002) Ion channel formation and membrane-linked pathologies of misfolded hydro- phobic proteins:the role of dangerous unchaperoned molecules. Clin Exp Pharmacol Physiol, 29, 741–753. [111] Volles, M. J. and T, L. P. (2001) Vesicle permeabilization by protofibrillar α-synuclein: comparison of wild-type with Parkinson’s disease linked mutants and insights in the mechanisms. Biochem. 40, 7812.7819. [112] Williams, A. D., Portelius, E., Kheterpal, I., Guo, J., Cook, K. D., Xu, Y. and Wetzel, R. (2004) Mapping A amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842. [113] Dobson. (2004) Principle of protein folding, Misfolding and aggregation. Semin. Cell. Dev. Biol. 15, 3–16. [114] Thomas, Q. and Pederson. (1995) Defective protein fold- ing as a basis of human diseases. TIBS. 20 (11), 456– 459. [115] Welch and Brown (1996) Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperone. 1(2), 109–115. Appendix APP Abbreviation: Amyloid precursor protein AD Alzheimer’s disease HD Huntington’s disease PD Parkinson’s disease LBs Lewy bodies TSEs Transmissible spongiform encephalopathies
|