 American Journal of Anal yt ical Chemistry, 2011, 2, 422-428 doi:10.4236/ajac.2011.24051 Published Online August 2011 (http://www.SciRP.org/journal/ajac) Copyright © 2011 SciRes. AJAC Rapid HPLC Method for Determination of Parachloroaniline in Chlorhexidine Antiseptic Agent in Mouthrinses, Ophthalmic and Skin Solution Alain Nicolay, Estelle Wolff, Marie-France Vergnes, Jacques Kaloustian, Henri Portugal Faculty of Pharmacist, Institut National de la Recherche Agronomique, Marseille, France E-mail: alain.nicolay@univmed.fr Received January 18, 2011; revised February 19, 2011; accepted May 30, 2011 Abstract We described a simple and rapid method to quantify simultaneously chlorhexidine (CHD) and its major me- tabolite, para Chloroaniline (pCA) by HPLC with UV detection without the additional need of mobile-phase amine modifiers or ion-pairing reagents, with good resolution between pCA and CHD, symmetry peak of the compound and short run time. HPLC-UV analyses were performed using a Dionex® Summit liquid chromato- graph (Dionex Corp, Sunnyvale, CA, USA). Chromatographic separations were carried out on a Luna® 150 mm×3 mm i.d. column packed with 3 µm CN (cyano) particles (Phenomenex®), guarded by an on-line filter. Mobile phase consist of methanol:water with sodium chloride with 0.02% of formic acid (55:45). Wavelengths for pCA and for CHD are 238 and 255 nm respectively. Influence of methanol and of sodium chloride content in the eluant has been studied. Linearity of CHD is very good, from 0.5 up to 21.2 µg/l while linearity of pCA is in the range of 0.05 to 10 µg/l with correlation coefficients above 0.999. Resolution between the components is above 4, asymmetry is about 1.3 and 1.7 for pCA and CHD respectively and the run time is less than 5 min- utes. This method has been applied to CHD solution of different medical devices. No interference has been re- ported, and the analysis of direct injection of solution, without any treatment is achieved in less than five min- utes.In conclusion, we present a validated method for dosage of CHD and its major impurity pCA, known to be carcinogen, available into medical products or medicinal device for in-vitro diagnostic. Keywords: Chlorhexidine, Chloroaniline, HPLC 1. Introduction Chlorhexidine [CHD; 1,1’-hexamethylenebis[5-(4-chlo- rophenyl) biguanide]] has a wide spectrum of bacteri- cidal and antiviral activity and is a common ingredient in various formulations ranging from skin disinfectants in healthcare products to antiplaque agents in dentistry [1,2]. The presence of two symmetrically positioned basic chlorophenyl guanide groups attached to a lipophilic hexamethylene chain (Figure 1) aid in rapid absorption through the outer bacterial cell wall, causing irreversible bacterial membrane injury, cytoplasmic leakage, and enzyme inhibition [3]. This molecule exists as various forms of salts: diacetate, dihydrochloride, or digluconate, mainly differing by their solubilizing abilities in aqueous or oily media. CHD di- gluconate (or gluconate), as most soluble in water or al- cohol, is the most used form in topical dermatology or cosmetic preparations. Aqueous solutions of CHD are most stable within the pH range of 5-8. Above pH 8.0 CHD base is precipitated and in more acid conditions there is gradual deterioration of activity because the compound is less stable. Hydrolysis yields p-chloroan- Cl NN NH N NH N NH N N NH Cl Cl NH2 Figure 1. Chemical structure of CHD and pCA.  A. NICOLAY ET AL.423 iline (pCA); the amount is insignificant at room tempera- ture, but is increased by heating above 100°C, especially at alkaline pH [4]. CHD diacetate is soluble in alcohol, glycerol, propylene glycol, polyethylene glycols. Ac- cording to the manufacturer, 1 part CHD is soluble in 15 parts of 96% ethanol. This cationic molecule (positively charged species) is thus generally compatible with other cationic materials, although compatibility will depend on the nature and relative concentration of the second cationic species. It is, however, possible for a reaction to occur between CHD and the counter-ion (anion) of a cationic molecule which is negatively charged, resulting in the formation of a less soluble CHD salt, which then may precipitate. CHD is incompatible with inorganic anions in all but extremely dilute solutions. CHD is also incompatible with organic anions, such as soaps, sodium lauryl sulphate, sodium carboxymethyl cellulose, alginates, and many pharma- ceutical dyes. In certain instances, there will be no visi- ble signs of incompatibility, but the antimicrobial activ- ity may be significantly reduced because of the CHD being incorporated into micelles (ionic clusters) [5]. pCA is very toxic if inhaled, swallowed or absorbed through the skin. It may act as a human carcinogen. It is readily absorbed through the skin and it may act as a sensitizer. The lethal dose 50 percent kill is 310 mg·kg–1 and 100 mg·kg–1 for rat and mouse respectively. Liquid chromatography is the most widely used me- thod for analysis of CHD [6]; UV detection around 250 nm is used for quantitative assays [7-9], while for the detec- tion of impurities, mass spectrometry or photodiode array detectors are employed [10,11]. Other methods reported in the literature include fluorometry [12] and direct UV spectroscopy [13]; both of them have several disadvan- tages: lack of sensitivity, serious interference. There are many reports about the determination of CHD in biological fluids using high performance liquid chromatography with a UV detector (HPLC-UV) [8,9, 14-18] and gas chromatography mass spectrometry (GC- -MS) [19-23]. Column used are generally C18 [11,14,15,24], Amide [11] or phenyl [25]. Furthermore, reversed-phase ion- -pair system is frequently used. However, as pCA is the principal product degradation, and because of his toxicity and to be in line with actual recommendation for genotoxic impurities [26], it is im- portant to quantify pCA in CHD solution. Therefore, we describe hereafter a new very sensible HPLC-UV method without the need of additional mobile-phase amine modi- fiers or ion-pairing reagents, with good resolution be- tween pCA and CHD, symmetry peak of the compo- unds and short run time. To prove the reliability of this method, CHD and pCA were analyzed in medical products. 2. Materials and Methods 2.1. Chemicals and Reagents CHD gluconate solution (20%, with a density of 1.06) was purchased from Sigma (St Quentin-Fallavier, France) and stored at 4˚C in the dark. Methanol and formic acid was of analytical grade from Carlo Erba (Val de Reuil, France). Other chemicals used were of analytical grade. Seven pharmaceutical specialities: Prexidine 0.12%® mouthrinses (Expanscience, France), Septisol® eye drops (Merck, France), Correctol 0.1%® eye drops (Alcon, France), Sophtal® eye drops (Alcon, France), Septeal® for topical application (Pierre Fabre Dermatologie, France), Chlorhexidine Aqueuse Gilbert à 0.05%® for topical application (Gilbert, France) and Dacrine® eye drops (McNeil, France) were purchased in pharmacy. CHD is used as active substance except in Septisol®, Correctol 0.1%® and Sophtal® where it is used as ex- cipient. 2.2. Apparatus and Chromatographic Conditions HPLC-UV analyses were performed using a Dionex® Summit liquid chromatograph (Dionex Corp, Sunnyvale, CA, USA), equipped with a vacuum solvent degassing unit, a binary high pressure gradient pump P680, an ASI100 automated sample injector, an UVD340U vari- able wavelength UV–VIS detector. Chromatographic separations were carried out on a Luna® 150 mm × 3 mm i.d. column packed with 3 µm CN (cyano) particles (Phenomenex®), guarded by an on-line filter. Data were collected and analyzed using the Chromeleon® software v6.50SP4 from Dionex®. Since it is known that amines tend to form stable salts with acetic acid, ammonium acetate and phosphate salts, the mobile phases for chlorhexidine quantitative assay consisting of methanol:water (55:45, v/v) with sodium chloride with 0.02% of formic acid (pH = 3) and pumped at a flow rate of 0.5 ml/min. Wavelengths for pCA and for CHD are 238 and 255 nm respectively. 2.3. Sample Preparation Pharmaceutical specialities were directly injected (20 µl) into the chromatograph system (without any extraction) after or not dilution. 2.4. Mobile Phase Published HPLC assays also suffer from quantitation Copyright © 2011 SciRes. AJAC  A. NICOLAY ET AL. Copyright © 2011 SciRes. AJAC 424 problems caused by irreversible adsorption of CHD onto the silica-based reversed phase (RP)-HPLC columns [27, 28]. These problems can be overcome by adding NaCl to the mobile phase [28] and explain the presence of this compound in our mobile phase. The effect of NaCl can be explained by the two simultaneously occurring pro- cesses: the salting out effect, and the electrostatic inter- actions between polar molecules and salt ions in sample solution [29]. Chlorhexidine is a dicationic compound with Ka1 = 6.3 × 10−3 and Ka2 = 5.0 × 10−11 and is almost com- pletely ionized (1+) at pH = 3, pH of our mobile phase. Because chlorhexidine and some of its degradation products are strongly basic, they are intensely retained on most silica-based reversed phase columns [30]; the dif- ficulty was overcome by using a cyano column and an acid mobile phase (pH = 3), without the need to use ion-pairing reagents in the mobile phase. It is known that chlorhexidine is quite stable at this low pH (some HPLC methods use pH = 2.5) [9]. 2.5. Statistics Results are presented as the mean SD and results were analyzed using Excel® 10, WinStat® v2003.1 and Chr- omeleon®. A p value < 0.05 was considered as signifi- cant. 2.6. Validation Criteria 2.6.1 Linearity Working standards were prepared by dilution in water and taking account of density for CHD gluconate. Height reference samples were used for calibration curves of CHD (0.53 to 21.20 mg/l) and pCA (0.01 to 10.0 ng/ml). Each determination was done in triplicate. The calibra- tion factors were calculated according to least-squares linear regression . 2.6.2 Precision and Accuracy Precision was determined for both inter- and intra-day variability. These measurements were made by HPLC analysis of CHD (5.3 and 12.72 mg/l) and pCA (0.50, and 2.0 mg/l), on three consecutive days (inter-day varia- tion or reproducibility) or during the same day for intra- day variation determination (repeatability). 2.6.3. Limit of Detection (LOD) and Limit of Quantification (LOQ) Standard curves were prepared for the two analytes and the following equation were used to calculate the LOD and the LOQ for each compound [31]: LOD = 3.3σ / S; LOQ = 10σ / S where σ is the standard deviation of the response (esti- mated from the standard deviation of y-intercepts of re- gression lines) and S is the slope of the standard curve. At the LOQ, bias and precision should not exceed 12% [32]. 3. Results To optimise the mobile phase, the influence of concen- tration of methanol and sodium chloride was first stud- ied. 4. Discussion Based on the results presented in Figures 2 and 3 and in Table 1, the concentration of methanol in the mobile phase was chosen as 55% in order to have a short run time (less than 5 minutes) and a good resolution (about 4.1) (Figure 4) between the two components. There is few influence of sodium chloride concentra- tion on retention time and peak asymmetry of pCA and CHD. It is noted that asymmetry for CHD (1.7) and for pCA (1.3) is very good with this cyano column from Phenomenex® (Table 2). However, height of CHD de- crease, when concentration of sodium chloride increase, while pCA height peak increase too. A concentration of 12‰ of sodium chloride was chosen, the goal is to avoid an important decrease of CHD and to increase the sensi- bility of pCA detection, especially because of its toxicity. Linearity of CHD is very good, from 0.5 up to 21.2 µg/l (Table 3). This range is in the same order of that de- scribes by Hebert et al. [7], but with a run time of 22 minutes. Some authors [9,11,14,30] present better sensi- bility for CHD (LOD is 0.05 mg/l) quantitation but they don’t quantify pCA. But for Usui [33] which works with LC/MS, it was difficult to estimate the reliable values at the concentration range below 0.1 mg/l. Only Below and Kramer [16] present a dosage method for both pCA and CHD, with a run time of 20 minutes. Linearity of pCA is very good (range from 0.05 to 10 mg/l), for a product known to be difficult to be ana- lysed [9,30] and with LOD and LOQ very low. This method has been applied to a CHD solution in- cluded into pharmaceutical specialities (Table 4) (Figure 5). In conclusion, we present a validated method for dos- age of CHD and its major impurity pCA. No interference has been reported, and the analysis of direct injection of solution, without any treatment except an eventually dilu- tion is achieved in less than five minutes. In all cases, pCA was less than LOD, and CHD is in the range gener- ally accepted in pharmaceutical products (95% - 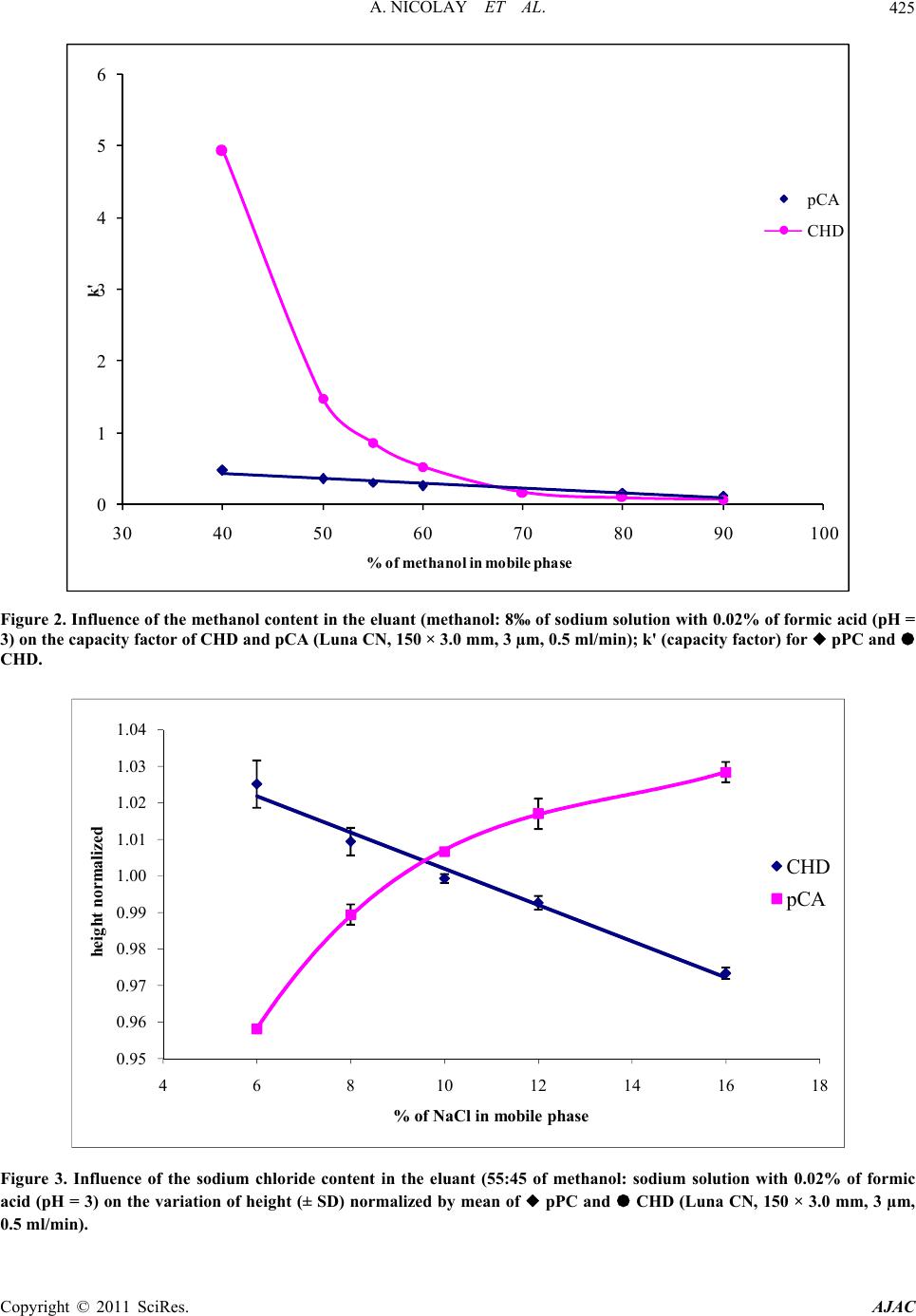 A. NICOLAY ET AL.425 0 1 2 3 4 5 6 30 40 50 60 70 80 90100 k' % of m eth an ol in mob il e p h as e pCA CHD Figure 2. Influence of the methanol content in the eluant (methanol: 8‰ of sodium solution with 0.02% of formic acid (pH = 3) on the capacity factor of CHD and pCA (Luna CN, 150 × 3.0 mm, 3 µm, 0.5 ml/min); k' (capacity factor) for pPC and CHD. 0.95 0.96 0.97 0.98 0.99 1.00 1.01 1.02 1.03 1.04 468101214161 heig ht normalize d % of NaCl in mobile phase 8 CHD pCA Figure 3. Influence of the sodium chloride content in the eluant (55:45 of methanol: sodium solution with 0.02% of formic acid (pH = 3) on the variation of height (± SD) normalized by mean of pPC and CHD (Luna CN, 150 × 3.0 mm, 3 µm, 0.5 ml/min). Copyright © 2011 SciRes. AJAC 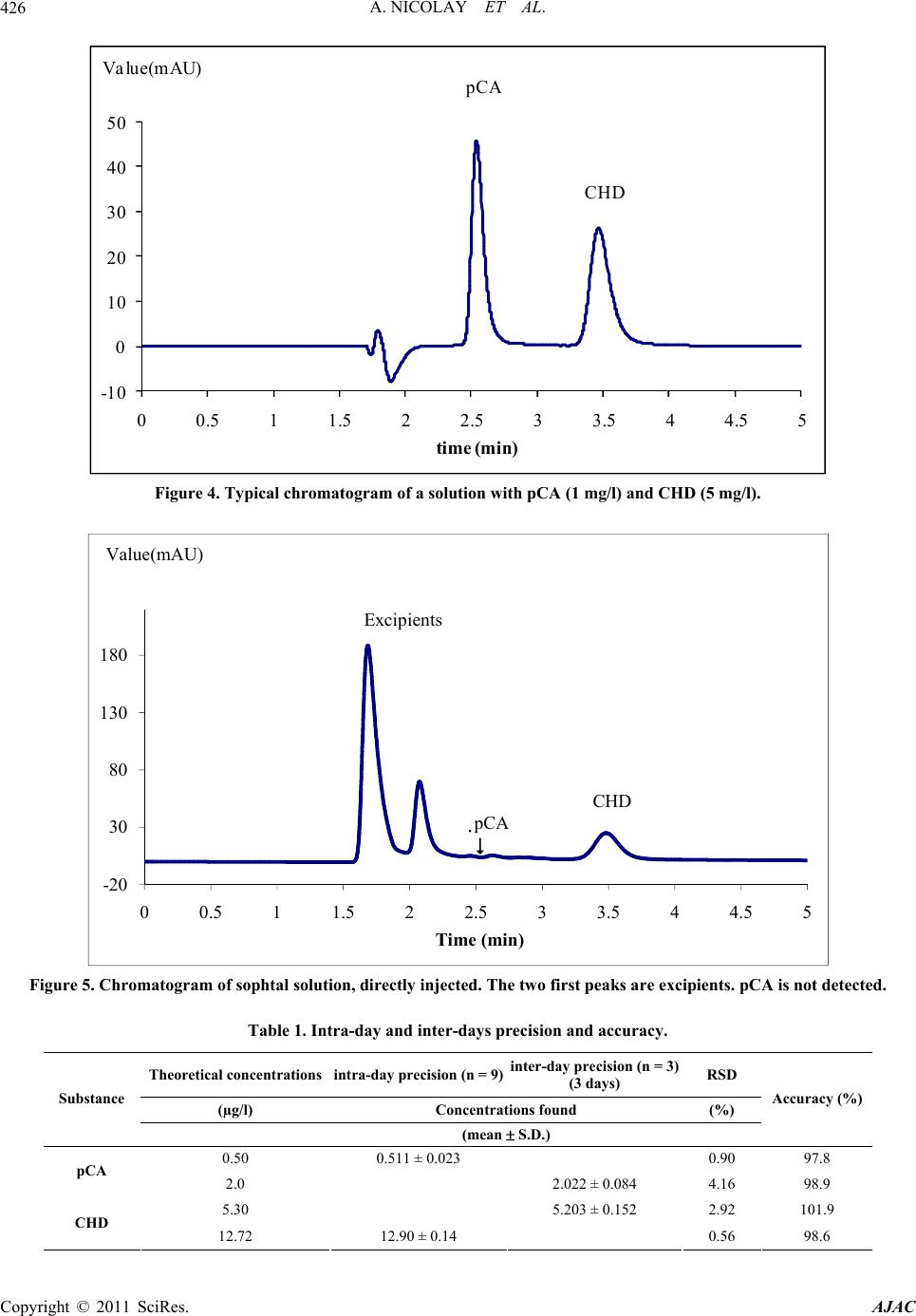 A. NICOLAY ET AL. 426 -10 0 10 20 30 40 50 00.511.522.533.544.55 time (min) Va lue(mAU) pCA CHD Figure 4. Typical chromatogram of a solution with pCA (1 mg/l) and CHD (5 mg/l). -20 30 80 130 180 00.511.522.533.544.55 Time (min) Valu e (m AU) CHD Excipients pCA Figure 5. Chromatogram of sophtal solution, directly injected. The two first peaks are excipients. pCA is not detected. Table 1. Intra-day and inter-days precision and accuracy. Theoretical concentrations intra-day precision (n = 9)inter-day precision (n = 3) (3 days) RSD (µg/l) Concentrations found (%) Substance (mean S.D.) Accuracy (%) 0.50 0.511 ± 0.023 0.90 97.8 pCA 2.0 2.022 ± 0.084 4.16 98.9 5.30 5.203 ± 0.152 2.92 101.9 CHD 12.72 12.90 ± 0.14 0.56 98.6 Copyright © 2011 SciRes. AJAC 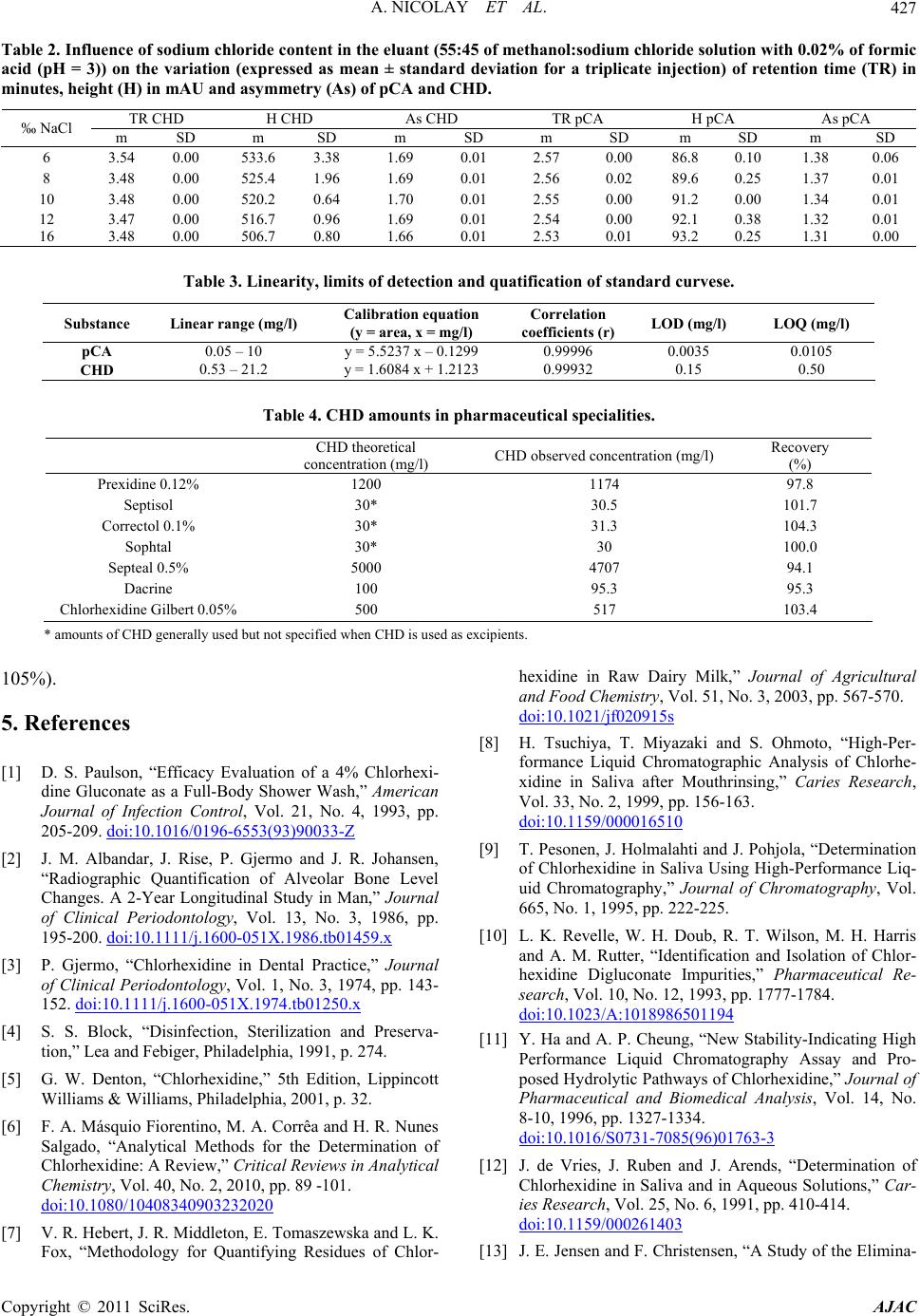 A. NICOLAY ET AL. Copyright © 2011 SciRes. AJAC 427 Table 2. Influence of sodium chloride c ontent in the eluant (55:45 of methanol:sodium chloride solution with 0.02% of formic acid (pH = 3)) on the variation (expressed as mean ± standard deviation for a triplicate injection) of retention time (TR) in minutes, height (H) in mAU and asymmetry (As) of pCA and CHD. TR CHD H CHD As CHD TR pCA H pCA As pCA ‰ NaCl m SD m SD m SD m SD m SD m SD 6 3.54 0.00 533.6 3.38 1.69 0.01 2.57 0.00 86.8 0.10 1.38 0.06 8 3.48 0.00 525.4 1.96 1.69 0.01 2.56 0.02 89.6 0.25 1.37 0.01 10 3.48 0.00 520.2 0.64 1.70 0.01 2.55 0.00 91.2 0.00 1.34 0.01 12 3.47 0.00 516.7 0.96 1.69 0.01 2.54 0.00 92.1 0.38 1.32 0.01 16 3.48 0.00 506.7 0.80 1.66 0.01 2.53 0.01 93.2 0.25 1.31 0.00 Table 3. Linearity, limits of detection and quatification of standard curvese. Substance Linear range (mg/l) Calibration equation (y = area, x = mg/l) Correlation coefficients (r)LOD (mg/l) LOQ (mg/l) pCA 0.05 – 10 y = 5.5237 x – 0.1299 0.99996 0.0035 0.0105 CHD 0.53 – 21.2 y = 1.6084 x + 1.2123 0.99932 0.15 0.50 Table 4. CHD amounts in pharmaceutical specialities. CHD theoretical concentration (mg/l) CHD observed concentration (mg/l) Recovery (%) Prexidine 0.12% 1200 1174 97.8 Septisol 30* 30.5 101.7 Correctol 0.1% 30* 31.3 104.3 Sophtal 30* 30 100.0 Septeal 0.5% 5000 4707 94.1 Dacrine 100 95.3 95.3 Chlorhexidine Gilbert 0.05%500 517 103.4 * amounts of CHD generally used but not specified when CHD is used as excipients. 105%). 5. References [1] D. S. Paulson, “Efficacy Evaluation of a 4% Chlorhexi- dine Gluconate as a Full-Body Shower Wash,” American Journal of Infection Control, Vol. 21, No. 4, 1993, pp. 205-209. doi:10.1016/0196-6553(93)90033-Z [2] J. M. Albandar, J. Rise, P. Gjermo and J. R. Johansen, “Radiographic Quantification of Alveolar Bone Level Changes. A 2-Year Longitudinal Study in Man,” Journal of Clinical Periodontology, Vol. 13, No. 3, 1986, pp. 195-200. doi:10.1111/j.1600-051X.1986.tb01459.x [3] P. Gjermo, “Chlorhexidine in Dental Practice,” Journal of Clinical Periodontology, Vol. 1, No. 3, 1974, pp. 143- 152. doi:10.1111/j.1600-051X.1974.tb01250.x [4] S. S. Block, “Disinfection, Sterilization and Preserva- tion,” Lea and Febiger, Philadelphia, 1991, p. 274. [5] G. W. Denton, “Chlorhexidine,” 5th Edition, Lippincott Williams & Williams, Philadelphia, 2001, p. 32. [6] F. A. Másquio Fiorentino, M. A. Corrêa and H. R. Nunes Salgado, “Analytical Methods for the Determination of Chlorhexidine: A Review,” Critical Reviews in Analytical Chemistry, Vol. 40, No. 2, 2010, pp. 89 -101. doi:10.1080/10408340903232020 [7] V. R. Hebert, J. R. Middleton, E. Tomaszewska and L. K. Fox, “Methodology for Quantifying Residues of Chlor- hexidine in Raw Dairy Milk,” Journal of Agricultural and Food Chemistry, Vol. 51, No. 3, 2003, pp. 567-570. doi:10.1021/jf020915s [8] H. Tsuchiya, T. Miyazaki and S. Ohmoto, “High-Per- formance Liquid Chromatographic Analysis of Chlorhe- xidine in Saliva after Mouthrinsing,” Caries Research, Vol. 33, No. 2, 1999, pp. 156-163. doi:10.1159/000016510 [9] T. Pesonen, J. Holmalahti and J. Pohjola, “Determination of Chlorhexidine in Saliva Using High-Performance Liq- uid Chromatography,” Journal of Chromatography, Vol. 665, No. 1, 1995, pp. 222-225. [10] L. K. Revelle, W. H. Doub, R. T. Wilson, M. H. Harris and A. M. Rutter, “Identification and Isolation of Chlor- hexidine Digluconate Impurities,” Pharmaceutical Re- search, Vol. 10, No. 12, 1993, pp. 1777-1784. doi:10.1023/A:1018986501194 [11] Y. Ha and A. P. Cheung, “New Stability-Indicating High Performance Liquid Chromatography Assay and Pro- posed Hydrolytic Pathways of Chlorhexidine,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 14, No. 8-10, 1996, pp. 1327-1334. doi:10.1016/S0731-7085(96)01763-3 [12] J. de Vries, J. Ruben and J. Arends, “Determination of Chlorhexidine in Saliva and in Aqueous Solutions,” Car- ies Research, Vol. 25, No. 6, 1991, pp. 410-414. doi:10.1159/000261403 [13] J. E. Jensen and F. Christensen, “A Study of the Elimina-  A. NICOLAY ET AL. 428 tion of Chlorhexidine from the Oral Cavity Using a New Spectrophotometric Method,” Journal of Periodontal Research, Vol. 6, No. 4, 1971, pp. 306-311. doi:10.1111/j.1600-0765.1971.tb00622.x [14] K. Kudo, N. Ikeda, A. Kiyoshima, Y. Hino, N. Nishida and N. Inoue, “Toxicological Analysis of Chlorhexidine in Human Serum Using HPLC on a Polymer-Coated ODS Column,” Journal of Analytical Toxicology, Vol. 26, No. 2, 2002, pp. 119-122. [15] L. R. Brougham, H. Y. Cheng and K. A. Pittman, “Sensi- tive High-Performance Liquid Chromatographic Method for the Determination of Chlorhexidine in Human Serum and Urine,” Journal of Chromatography, Vol. 383, No. 2, 1986, pp. 365-373. [16] H. Below, N. Lehan and A. Kramer, “HPLC Determina- tion of the Antiseptic Agent Chlorhexidine and Its Deg- radation Products 4-Chloroaniline and 1-Chloro-4-Ni- trobenzene in Serum and Urine,” Microchimica Acta, Vol. 146, No. 2, 2004, pp. 129-135. [17] Y. W. Lam, D. C. Chan, S. Y. Rodriguez, J. H. Lintakoon and T. H. Lam, “Sensitive High-Performance Liquid Chr- omatographic Assay for the Determination of Chlorhexi- dine in Saliva,” Journal of Chromatography, Vol. 612, No. 1, 1993, pp. 166-171. [18] P. Wainwright and M. Cooke, “Direct Determination of Chlorhexidine in Urine by High-Performance Liquid Chr- omatography,” The Analyst, Vol. 111, No. 11, 1986, pp. 1343-1344. doi:10.1039/an9861101343 [19] H. Matsushima, K. Sugimoto, K. Shibata and N. Sakurai, “Determination by Mass Fragmentography of the Chlor- hexidine in Biological Samples,” Japanese Journal of Hygiene, Vol. 37, No. 5, 1982, pp. 762-767. [20] V. G. Alder, D. Burman, R. A. Simpson, J. Fysh and W. A. Gillespie, “Comparison of Hexachlorophane and Chl- orhexidine Powders in Prevention of Neonatal Infection,” Archives of Disease in Childhood, Vol. 55, No. 4, 1980, pp. 277-280. doi:10.1136/adc.55.4.277 [21] E. Read, “A Note of Hibitane Assay with Final Iodina- tion,” Methodological Surveys in Biochemistry, Chich- ester Ellis, Horwwod, 1978. [22] W. K. Gavlick and P. K. Davis, “Gas Chromatographic Determination of p-Chloroaniline in a Chlorhexidine Di- gluconate-Containing Alcohol Foam Surgical Scrub Product,” Journal of AOAC International, Vol. 77, No. 3, 1994, pp. 583-586. [23] A. Ono, “Gas-Liquid Chromatographic Separation of Toluidine, Chloroaniline and Dichloroaniline Isomers on Various Stationary Phases Including Heteroaromatic Compounds,” Analyst, Vol. 107, No. 1275, 1982, pp. 600-605. [24] Y. Zhu, Y. Yang and Q. Qi, “Determination of Chlor- hexidine Acetate in Disinfectant by High Performance Liquid Chromatography,” Wei Sheng Yan Jiu, Vol. 32, No. 1, 2003, pp. 51-87. [25] L. Havlíková, L. Matysová, L. Nováková, R. Hájková and P. Solich, “HPLC Determination of Chlorhexidine Gluconate and p-Chloroaniline in Topical Ointment,” Vol. 43, No. 3, 2007, pp. 1169-1173. [26] European Medicines Agency, “Guideline on the Limits of Genotoxic Impurities,” EMEA/CHMP/QWP/251344/2006, 2006, pp. 1-8. [27] A. P. Cheung, R. Mavar, C. Carlson and W. K. Chiang, “Problems Affecting the Liquid Chromatographic Quan- titation of Chlorhexidine Digluconate in Ophthalmic So- lutions,” Journal of Pharmaceutical and Biomedical Ana- lysis, Vol. 9, No. 1, 1991, pp. 41-45. doi:10.1016/0731-7085(91)80235-2 [28] L. E. Stevens, J. R. Durrwachter and D. O. Helton, “Ana- lysis of Chlorhexidine Sorption in Soft Contact Lenses by Catalytic Oxidation of [14C]Chlorhexidine and by Liquid Chromatography,” Journal of Pharmaceutical Sciences, Vol. 75, No. 1, 1986, pp. 83-86. doi:10.1002/jps.2600750120 [29] L. Zhao, L. Zhu and H. K. Lee, “Analysis of Aromatic Amines in Water Samples by Liquid-Liquid-Liquid Mi- croextraction with Hollow Fibers and High-Performance Liquid Chromatography,” Journal of Chromatography, Vol. 963, No. 1-2, 2002, pp. 239-248. doi:10.1016/S0021-9673(02)00544-7 [30] F. M. Musteata and J. Pawliszyn, “Assay of Stability, Free and Total Concentration of Chlorhexidine in Saliva by Solid Phase Microextraction,” Journal of Pharmaceu- tical and Biomedical Analysis, Vol. 37, No. 5, 2005, pp. 1015-1024. doi:10.1016/j.jpba.2004.09.055 [31] ICH Harmonised Tripartite Guideline, “Validation of Analytical Procedures: Methodology: ICH Topic Q2B,” CPMP/ICH/281/95, 1996, pp. 1-15. [32] J. Vial and A. Jardy, “Experimental Comparison of the Different Approaches to Estimate LOD and LOQ of an HPLC Method,” Analytical Chemistry, Vol. 71 No. 14, 1999, pp. 2672-2677. [33] K. Usui, T. Hishinuma, H. Yamaguchi, N. Tachiiri and J. Goto, “Determination of Chlorhexidine (CHD) and Non- ylphenolethoxylates (NPEOn) Using LC-ESI-MS Method and Application to Hemolyzed Blood,” Journal of Chro- matography, Vol. 831, No. 1-2, 2006, pp. 105-109. Copyright © 2011 SciRes. AJAC
|