Natural Science
Vol.6 No.5(2014), Article ID:43726,15 pages DOI:10.4236/ns.2014.65034
Towards a Comprehensive Search of Putative Chitinases Sequences in Environmental Metagenomic Databases
Towards a Comprehensive Search of Putative Chitinases Sequences in Environmental Metagenomic Databases
Aline S. Romão-Dumaresq1, Adriana M. Fróes1, Rafael R. C. Cuadrat1, Floriano P. Silva Jr.2,3, Alberto M. R. Dávila1,3*
1Laboratório de Biologia Computacional e Sistemas, Instituto Oswaldo Cruz (IOC), FIOCRUZ, Rio de Janeiro, Brazil
2Laboratório de Bioquímica de Proteínas e Peptídeos, Instituto Oswaldo Cruz (IOC), FIOCRUZ, Rio de Janeiro, Brazil
3Pólo de Biologia Computacional e Sistemas, Instituto Oswaldo Cruz (IOC), FIOCRUZ, Rio de Janeiro, Brazil
Email: *davila@fiocruz.br
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 26 October 2013; revised 26 November 2013; accepted 6 December 2013
ABSTRACT
Chitinases catalyze the hydrolysis of chitin, a linear homopolymer of β-(1,4)-linked N-acetylglucosamine. The broad range of applications of chitinolytic enzymes makes their identification and study very promising. Metagenomic approaches offer access to functional genes in uncultured representatives of the microbiota and hold great potential in the discovery of novel enzymes, but tools to extensively explore these data are still scarce. In this study, we develop a chitinase mining pipeline to facilitate the comprehensive search of these enzymes in environmental metagenomic databases and also to explore phylogenetic relationships among the retrieved sequences. In order to perform the analyses, UniprotKB fungal and bacterial chitinases sequences belonging to the glycoside hydrolases (GH) family-18, 19 and 20 were used to generate 15 reference datasets, which were then used to generate high quality seed alignments with the MAFFT program. Profile Hidden Markov Models (pHMMs) were built from each seed alignment using the hmmbuild program of HMMER v3.0 package. The best-hit sequences returned by hmmsearch against two environmental metagenomic databases (Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis—CAMERA and Integrated Microbial Genomes—IMG/M) were retrieved and further analyzed. The NJ trees generated for each chitinase dataset showed some variability in the catalytic domain region of the metagenomic sequences and revealed common sequence patterns among all the trees. The scanning of the retrieved metagenomic sequences for chitinase conserved domains/signatures using both the InterPro and the RPS-BLAST tools confirmed the efficacy and sensitivity of our pHMM-based approach in detecting putative chitinases sequences. These analyses provide insight into the potential reservoir of novel molecules in metagenomic databases while supporting the chitinase mining pipeline developed in this work. By using our chitinase mining pipeline, a larger number of previously unannotated metagenomic chitinase sequences can be classified, enabling further studies on these enzymes.
Keywords:Chitinase; Metagenome; pHMM; Sequence Search

1. Introduction
Enzymes are catalysts that support the development of environmental-friendly industrial processes. At present, most of the industrial enzymes of major importance are of microbial origin, so the search for novel of these catalysts is a key step towards the development of innovative bioprocesses. Chitinases are enzymes responsible for the hydrolysis of chitin, a linear homopolymer of β-(1,4)-linked N-acetylglucosamine, which is the second most abundant biopolymer in nature. A set of different enzymes are needed to drive the complete hydrolysis of chitin to free N-acetylglucosamine (GlcNAc), involving diverse mode of actions known to be synergistic and consecutive [1] [2] . The endochitinases (EC 3.2.1.14) randomly cleave the chitin chain at internal sites, whilst the exochitinases (EC 3.2.1.52) catalyze either the successive removal of sugar unit from the non-reducing end or the hydrolysis of terminal non-reducing sugar [3] [4] .
Based on amino acid sequence similarities these chitinolytic enzymes are classified into glycoside hydrolases (GH) family 18, 19 and 20 [5] [6] . GH family-18 and 20 are thought to have a common evolutionary ancestry, since they possess significant similarity in their tertiary structure, catalytic residues and mechanism. GH family- 18 exhibit considerable variability in evolutionary terms and comprises chitinases from bacteria, fungi, viruses, insect and plants [7] . GH family-19 contains plant, bacteria and some Streptomyces chitinases, and GH family- 20 includes the β-N-acetylhexosaminidases from bacteria, fungi, Streptomyces and humans [4] [7] . These enzymes have widespread applications, such as in bioremediation [8] , biological control [9] -[11] , production of chitooligosaccharides [12] -[14] , preparation of single-cell protein [15] and isolation of protoplasts from fungi [16] .
The low discovery rate of novel natural products from culturable microorganisms [17] coupled with the fact that only a small portion (estimated less than 1%) of the microbial community is capable of growing under artificial conditions [18] [19] has brought about the need to explore metagenomic approaches to speed up the finding of new biomolecules potentially useful in biotechnology [20] . To date, a great number of environmental metagenomic studies were performed, such as the extensive studies on the Sargasso Sea [21] and the Global Ocean Expedition [22] [23] , and as a result, a huge amount of sequence data has been generated but has not been entirely explored. Different projects have been implemented to provide an open infrastructure for metagenomic sequence data storage and analysis, as CAMERA (“Community Cyberinfrastructure for Advanced Microbial Ecology Research & Analysis”) [24] , MG-RAST (“Metagenomic Rapid Annotation using Subsystem Technology”) [25] , and IMG/M (“Integrated Microbial Genomes”) [26] . The current challenge is to fully exploit the metagenomic sequence information using appropriate data-management and data-analysis methods.
Typical metagenomic analyses rely on similarity search against some databases, followed by annotation of the output. The most frequently used similarity search tool is BLAST [27] , but as it requires significant computational capacity for large datasets, faster searching tools have been developed, such as Pattern-Hunter [28] [29] and BLAT [30] . However, comprehensive searches on specific genes or gene families require more sensitive tools to be used. Therefore, methods are needed to find subtler similarities between sequences and to assign putative structure and functional characterization to new proteins [31] . Pipelines based on Hidden Markov Model (HMM) [32] are very promising since this is a statistical representation of a protein family conservation pattern extracted from multiple alignment of sequences, which has been demonstrated to be very effective in detecting distantly related homologues [33] -[35] .
The aim of this work was to develop and validate a data mining strategy based on profile HMM (pHMM) in order to be able to broadly explore environmental metagenomic databases for putative chitinase sequences. The results confirmed the efficacy of our pipeline in detecting chitinase sequences and highlighted the power of pHMM-based strategies to identify remote homologues.
2. Methodology
2.1. Collection of Chitinase Reference Sequences
Fungal and bacterial curated amino acid sequences of chitinases belonging to the glycoside hydrolase (GH) families 18, 19 and 20 were retrieved from the UniprotKB version 2011-06 database (http://www.uniprot.org) on July 2011. A total of 170, 13 and 46 sequences were collected for GH family-18, GH family-19 and GH family- 20, respectively. GH family-18 sequences UniprotKB IDs were: P04067, P36912, P80036, F3Y8V4, D6ESW9, F3NDC4, O83008, Q6T6I1, P07254, A8G807, Q9ALZ0, Q8KKF5, Q9L5D5, Q43919, Q25BN2, Q8GHI4, P32823, A7M6A0, Q9WX41, Q9AMP1, C3LU56, D2YR61, D2YAB4, Q48373, Q5MYT4, Q9RCG5, Q56077, Q845S2, O30678, Q09IY6, Q6BCF8, A5YRG4, B7UB89, P20533, Q48494, Q9KHB3, C6IW88, B1VBB0, A6FD95, P96168, A6CVZ0, Q9CE95, Q7PC52, Q9Z493, Q59143, Q59924, Q9KY99, Q59141, D0VV10, D0VV09, Q81A65, D5TUL7, P11797, A6B8H6, A7LHM6, B2TQ75, B8DGV4, C1IAI6, D0ELI3, D0WTF8, D1RSJ9, D3YGV3, E3YUT8, F3RZA6, O50076, Q0MRC3, Q1EM71, Q547S1, Q5WKC0, Q8KWS2, Q99PX0, Q9KED7, Q9REI6, O69311, D5ZUF3, D6EQC0, O86826, Q9S5K1, Q05638, Q09WI7, A0Q8N1, F4BBA4, E2MRS9, B2SEL0, P36909, Q6A4C3, Q700B8, Q75ZW9, Q7PC51, Q8KVU8, Q9L8G0, Q9Z9M8, Q9ZIX2, Q8RQP6, B1W0A0, D6ANP5, A4GZI8, B5H9B1, D5ZXC4, P11220, P27050, Q9Z9M7, E3FMX3, Q099U8, Q1CZN0, Q092X1, C6J4E8, E8U3R7, D6EPZ7, D9XVU0, D9XI74, B5I3A2, A7UGE4, Q12735, Q9UV45, Q9UV49, A6YNL9, Q99006, P48827, Q9C1T8, Q9C1T7, Q9C1T6, O59928, Q9C1U0, Q65YQ7, Q9C1T4, Q9C1T9, O14456, Q9Y841, A9LI60, Q8J042, Q5MNU1, A6Y9S8, P32470, Q870C0, Q873X9, Q06HA3, Q3YLC5, Q9HGU5, Q92222, Q9HEW6, Q9P4Q1, Q5YLC0, A6YJX1, Q4FCX2, Q92270, Q7Z8C9, Q8J1Y3, Q96VR2, E5KCK8, A5JV26, A3RLY3, A5X8W3, Q96UW2, A2VEC4, Q8NJQ4, D6N0Y7, D6N0Y8, F6MIV5, E5LEW9, A2SW11, E9F7R6, P29026, P29027, P29025, P54197, P40954, P40953, P29029, P46876. GH family-19 sequences UniprotKB IDs were: Q9WXI9, Q59I46, Q9LBM0, Q8GI53, Q9S6T0, Q8CK55, B3XZQ2, O50152, Q9Z4P2, Q5J1K1, Q9RHU4, Q9RHU5, Q25BT4. GH family-20 sequences UniprotKB IDs were: Q9F9B4, Q75V90, A7M7B5, Q9LC82, Q7WUL4, Q9L448, Q9ZN69, Q9WXH9, D2KW09, P49008, A1XNE6, P49007, Q8VUM1, Q9R6Y9, Q9FAC5, Q9ZH38, Q7PC48, Q7PC49, Q54468, P49610, Q9ACN7, O85361, Q83WL6, Q9RHV6, Q84FS9, P96155, D9ISD9, D9ISE0, P13670, Q60081, Q04786, C8VMN3, Q8J2T0, A2SW08, P43077, Q309C3, P13723, Q643Y1, Q9URR8, E3NYM0, P87258, Q0ZLH7, P78738, P78739, Q8NIN7, Q8NIN6. The great sequence diversity found in the GH family-18 required the partitioning of it into nine subsets of bacterial sequences and three subsets of fungal sequences. This division was carried out taking into account both the existing chitinase subfamilies and a Neighbor-Joining guide tree topology. The retrieved sequences were then used to generate 15 multi-fasta chitinase reference sets (with 12 GH family-18, one GH family-19 and two GH family-20 sets).
2.2. Environmental Metagenomic Databases
Two environmental metagenomic databases were selected to test our chitinase mining strategy. The first one was CAMERA v2.0 [36] , available at http://camera.calit2.net/, which contains 84 unannotated metagenomic datasets with 135,704,056,943 nucleotide sequences. Six-frame translation of the nucleotide sequences was performed using the EMBOSS Transeq tool available at http://www.ebi.ac.uk/Tools/st/ and a total of 75 Gb of sequences were generated. The second database was IMG/M [26] , available at http://img.jgi.doe.gov/cgi-bin/m/main.cgi/, which includes 364 automatically annotated metagenomic datasets containing 119,059,610 amino acid sequences, making a total of 20 Gb. Database sequences were downloaded to a local server by June 2011.
2.3. Construction of Profiles HMM and Search for Putative Chitinase Homologues
First, multiple sequence alignments were generated for each chitinase reference set (seed alignments) using the default settings (“-auto”) of MAFFT v6.717b program [37] [38] . Alignment visualizations were carried out in Jalview version 2 [39] . The quality of each seed alignment was controlled by manual checking and, in a few cases, manual editing was necessary. Profile HMMs (pHMMs) were then built from each seed alignment using the hmmbuild program of HMMER v3.0 package (http://hmmer.janelia.org/). The 15 pHMMs generated were used to perform sequence database searches with the hmmsearch program also of the HMMER v3.0 package and an e-value threshold of 1.0E−05 against the two environmental databases CAMERA and IMG/M.
2.4. Mining Strategy Validation
The resulting sequence database searches (described in detail in Section 2.3) were used to extract the best-hit sequences of each metagenomic dataset, that is, the hits which presented the lowest e-value parameter among all the sequences of a metagenomic project. Best-hit sequences were retrieved in a fasta format using fastacmd program of BLAST package [27] [40] and then scanned for the occurrence of chitinase conserved domains/ signatures using both InterPro v4.7 (http://www.ebi.ac.uk/interpro/) and RPS-BLAST v.2.2.21 resources, with a evalue threshold of 1.0E−05. InterPro v4.7 combines predictive models and protein signatures from 10 member databases (Gene3D, PANTHER, Pfam, PIRSF, PRINTS, ProDom, PROSITE, SMART, SUPERFAMILY and TIGRFAMs) [41] and RPS-BLAST v2.2.21 integrates seven conserved domain databases (CDD v2.25, Pfam v.24.0, Smart v.5.1, COG v1.0, KOG, TigrFam v9.0 and Prk v.5.0). These conserved domain and protein signature databases were downloaded from EBI and NCBI on October 2010. InterPro and RPS-BLAST search results were parsed into spreadsheets using an in-house ruby script, and the frequency of the different chitinase conserved domain/signatures was calculated.
2.5. Phylogenetic Analysis of Putative Chitinase Sequences
Best-hit sequences (described in detail in section 2.4) were selected to perform phylogenetic reconstructions using the Neighbor-Joining (NJ) algorithm from MEGA 5.05 [42] , p-distance model and 1000 bootstrap tests. Catalytic domain amino acid sequences from the chitinase reference sets and the selected best hit sequences were concatenated to generate a multiple sequence alignment using MAFFT v6.717b [37] , which was used as query to build the NJ trees with MEGA 5.05.
3. Results
The construction of chitinase-reference sequence sets was a key step in the success of the mining strategy applied in this work. The collection and grouping of chitinase sequences on subsets allowed the generation of 15 chitinase groups covering all the three chitinase GH families, in which 9 were fungal GH family-18, three were bacterial GH family-18, one was bacterial GH family-19, one was fungal GH family-20 and one was bacterial GH family-20 (Figure 1). The use of these chitinase-reference subsets enabled the production of high quality multiple sequence alignments and, consequently, the properly construction of chitinase pHMMs.
The hmmsearch analysis performed against CAMERA and IMG/M metagenomic environmental databases retrieved a total of 708, 104 and 256 best-hit sequences putative of GH family-18, 19 and 20, respectively. The scanning of these sequences using a RPS-BLAST search revealed the presence of chitinase conserved domains in 74.6%, 97.1% and 97.7% of the GH family-18, GH family-19 and GH family-20 sequences, respectively (Figures 2(a)-(c)). Only a small portion of the sequences presented hits with conserved domains other than the chitinase ones (4.8% of GH family-18 and 0.8% of GH family-20). No hits sequences were 20.6% of GH family-18, whilst just 2.9% of GH family-19 and 1.6% of GH family-20 (Figures 2(a)-(c)). The InterPro search inferred the occurrence of chitinase signatures in 81.7%, 89.4% and 98.8% of the metagenomic sequences belonging to GH family-18, 19 and 20, respectively (Figures 2(d)-(f)). Compared to the RPS-BLAST search, the InterPro analysis revealed a higher percentage of sequences hosting protein signatures other than the chitinase ones (10.3% of GH family-18, 8.7% of GH family-19 and 0.4% of GH family-20) and a smaller percentage of sequences presenting no hits against the databases examined (8.0% of GH family-18, 1.9% of GH family 19 and 0.8% of GH family 20) (Figures 2(d)-(f)).
A large difference in diversity among all the three chitinase GH families was revealed in the RPS-BLAST and the InterPro analysis. That is, GH family-19 and GH family-20 presented no more than 12 types of conserved domains, and most of the sequences shared the same conserved domain hits (Tables 1 and 2). In contrast, GH family-18 displayed up to 34 different sorts of conserved domains and there was not a predominant set of conserved domains to the majority of the sequences (at most, half of the sequences shared the same conserved domain hits) (Tables 1 and 2). In addition, the scanning of IMG/M sequences has showed that some sequences annotated as hypothetical protein exhibited chitinase conserved domain hits, showing the sensitivity of our mining pipeline.
The phylogenetic analysis generated NJ trees corresponding to each chitinase dataset. All datasets showed some variability in the amino acid sequence of the catalytic domain region, except for the two active site residues (aspartate and glutamate in GH family-18 and 20, and two glutamates in the case of GH family-19), which
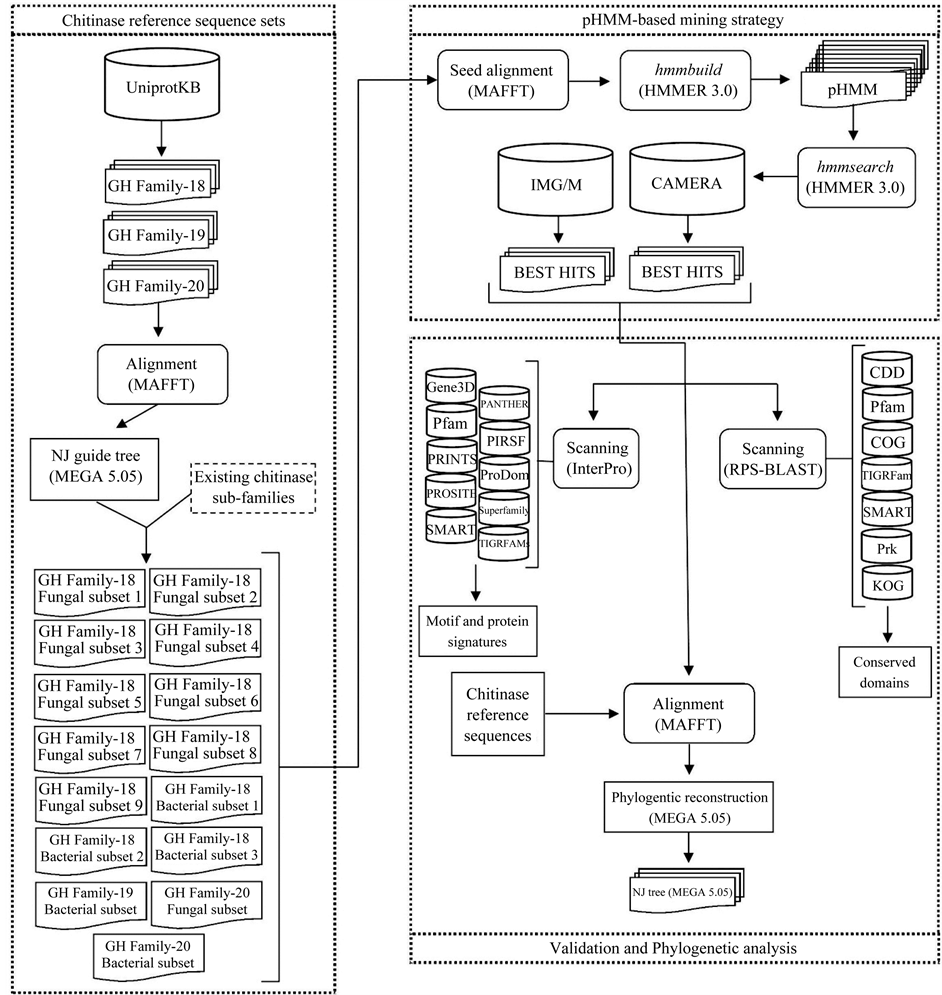
Figure 1. Workflow of the methodology applied in this study. The first step was to generate fungal and bacterial chitinase reference sets for the glycoside hydrolase (GH) families 18, 19 and 20. Fifteen subsets were created, in which 9 were fungal GH family-18, 3 were bacterial GH family-18, one was bacterial GH family-19, one was fungal GH family-20 and one was bacterial GH family-20. The second stage consisted of the generation of profile Hidden Markov Models (pHMM) for each chitinase reference sequence subset, followed by a sequence database search against CAMERA and IMG/M. The best-hit sequences of each metagenomic project were retrieved and used in the last step of our analysis. The validation of the mining strategy was carried out by performing both an InterPro and a RPS-BLAST search against protein signatures, conserved domains and motifs databases. The phylogenetic analysis of the metagenomic sequences together with the chitinase reference sequences generated NJ trees for each chitinase subset.
were conserved in almost all sequences examined (data not shown). In addition, the NJ tree analysis also revealed two common sequence patterns, that is, all the trees presented metagenomic sequences phylogenetically related to characterized chitinases; and all these trees also displayed metagenomic sequences which did not cluster with any characterized chitinase (Figures 3-6). Interestingly, some metagenomic sequences annotated as

Figure 2. Pie charts representing the percentage of metagenomic sequences (the hmmsearch best hits sequences) which exhibited chitinase-related domain and/or signatures after RPS-BLAST ((a), (b), (c)) and InterPro ((d), (e), (f)) searches against different conserved domain databases. The plots represent each GH family separately: GH family-18 results are presented in (a) and (d); GH family-19 in (b) and (e), and GH family-20 in (c) and (f). *Percentage of metagenomic sequences showing conserved domains other than the ones found in the representative chitinase sequences; **Percentage of metagenomic sequences which did not find any hit in these searches against conserved domain databases.
“hypothetical protein” in the IMG/M database were retrieved after running our mining pipeline and were grouped with chitinase GH family-18 reference sequences in the NJ phylogenetic analysis (Figure 4), indicating they are putative chitinase sequences.
4. Discussion
The broad range of applications of chitinolytic enzymes makes their identification and study very promising. Metagenomic approaches offer access to functional genes in uncultured representatives of the microbiota and hold great potential in the discovery of novel enzymes, but tools to extensively explore these data are still scarce. This study aimed the development of a chitinase mining pipeline to facilitate the comprehensive search of these enzymes in metagenomic databases. The use of a pHMM-based strategy allowed sensitive and efficient detection of putative chitinase sequences.
The generation of representative seed alignments and the selection of the homology detection method are key steps in sequence mining pipelines. The quality of an alignment is critical to its utility in different approaches, such as functional analysis, evolutionary studies and structure prediction [43] . For instance, the quality of a query and template sequence alignment is a major determinant of model quality in comparative modeling studies [44] . In fact, the higher an alignment quality, the higher the sensitivity in detecting homologous sequences [43] . However, the assignment of a high quality alignment depends on the relatedness of the sequences being aligned. Alignments of sequences sharing high levels of similarity, or about 50% identity, are generally unambiguous and easier to be automatically generated, but alignments of more distant sequences, as for some family of proteins (sharing 30% identity or less), usually will need to be manually checked for higher qualities. For most alignment methods, the quality increases significantly at about 20% identity [45] . The algorithm implemented in the MAFFT program is considered to be faster though still accurate compared to other methods, such as ClustalW and T-Coffee [38] , thus making this program to be considered one of the best global alignment tools currently available [46] [47] and justifying the decision for using it in our mining pipeline. In this study we put some effort on properly generating chitinase reference sets representative of the different subgroups of sequences belonging to the GH families-18, 19 and 20. Basically, well-characterized chitinase sequences were chosen and organized in subsets of at least five sequences. Seed alignments were generated and manually checked, and then used to build reliable pHMMs.
pHMMs are statistical models that use multiple alignments of homologous sequences to quantify amino acids frequencies and the position-specific probabilities for inserts and deletions along the alignment [32] [48] . They are broadly used for modeling conserved motifs of protein families since they contain more information about

Table 1 . Conserved domains hits recovered after a RPS-BLAST search using the metagenomic sequences (hmmsearch best hit sequences) against seven conserved domain databases (CDD, COG, KOG, Pfam, Prk, SMART and TIGRfam).
aOnly the conserved domains hits found in more than 10% of the sequences analyzed were displayed in table; bPercentage of sequences which showed hit with that conserved domain.
Table 2. Conserved domains hits recovered after an InterPro search using the metagenomic sequences (hmmsearch best hit sequences) against ten conserved domain databases (Gene3D, PANTHER, Pfam, PIRSF, PRINTS, ProDom, PROSITE, SMART, SUPERFAMILY and TIGRfams).
aOnly the conserved domains hits found in more than 10% of the sequences analyzed were displayed in table; bPercentage of sequences which showed hit with that conserved domain.
the sequence family than a single sequence [32] [48] [49] . These pHMMs have been described as very efficient to detect conserved patterns in multiple sequences [35] [50] [51] and to perform better than simple profilesequence methods such as PSI-BLAST [48] [49] . This higher sensitivity found with pHMMs is very promising when performing comprehensive searches to find remote homologues, as is such the case in our study. Two software packages are frequently used to build pHMMs and to perform profile-sequence searches, SAM [33] and HMMER [52] , but the last one has been reported as more suitable for large sequence dataset searches [53] and then was used in the analyses of the present work.
The scanning for the presence of chitinase conserved domains and motifs/signatures in the best hit sequences (the ones retrieved after the hmmsearch analysis) was carried out in order to evaluate the performance of our chitinase mining pipeline on detecting true putative chitinase sequences. Many annotation pipelines use searches against conserved domain databases since these regions are evolutionarily conserved units in proteins [54] . The recognition of a conserved domain footprint in a protein sequence usually indicates its cellular or molecular function [55] and provides more reliable protein classification than sequence similarity analysis. The RPS-Blast and InterPro searches performed in this work found high percentages of chitinase-related domains and motifs in

Figure 3. Phylogenetic tree of fungal chitinases subset-3 (GH family-18) using the Neighbor-Joining (NJ) algorithm from MEGA 5.05, p-distance model and 1000 bootstrap tests. The “□” symbol indicates CAMERA metagenomic sequences; “Δ” indicates IMG/M metagenomic sequences; “♦” indicates UniprotKB chitinase reference sequences and “●” indicates the outgroup sequences.
the best hit metagenomic sequences, validating our chitinase mining pipeline. The presence of best hit metagenomic sequences showing no hits to any conserved domain may represent putative novel chitinases that possibly would not be identified using sequence-sequence similarity searches. Furthermore, some IMG/M metagenomic sequences annotated as hypothetical proteins resulted in hits with chitinase conserved domains in our analysis, indicating that our pipeline may have high sensitivity and it is able to detect remote homologues.
The results obtained in the RPS-Blast and InterPro analyses emphasized the large differences in diversity among the three chitinases GH families-18, 19 and 20. As described in previous reports, GH family-18 holds higher variability in evolutionary terms and contains the greatest number protein members [4] [7] . The diversity observed in the GH family-18, 19 and 20 was also assessed in the phylogenetic reconstructions for the metagenomic and the chitinase reference sequences. Indeed, interpreting phylogenetic relationships among sequences is particularly important since it allows to infer gene function [56] , genetic variability and protein evolution. Phylogeny-based classification systems have been used before to identify enzymes in metagenomic sequence datasets [57] [58] . Based on the phylogenetic relationships observed in the NJ trees generated in this study, two common sequence patterns were identified, one including metagenomic sequences phylogenetically related to characterized chitinases—which may help to understand their origin and classification; and the other comprising metagenomic sequences which did not cluster with any characterized chitinase—suggesting a great reservoir of putative new chitinases to be exploited in these metagenomic databases. Our results reinforced the sensitivity and efficiency of our mining pipeline in detecting putative chitinase sequences from metagenomic databases.
5. Conclusion
Traditional sequence search pipelines frequently are not able to extensively exploit metagenomic databases. The current flood of sequence data from metagenomic studies and the wide range of applications of chitinases brought about the need to develop a new data search pipeline. The chitinase mining pipeline developed in this work was based on the generation of high quality seed alignments from reliable chitinase reference sets, which

Figure 4. Phylogenetic tree of endo-β-N-acetylglucosaminidase (GH family-18) using the Neighbor-Joining (NJ) algorithm from MEGA 5.05, p-distance model and 1000 bootstrap tests. The “□” symbol indicates CAMERA metagenomic sequences; “Δ” indicates IMG/M metagenomic sequences; “♦” indicates UniprotKB chitinase reference sequences and “●” indicates the outgroup sequences.
were then used on the construction of chitinase pHMMs. The searches using these pHMMs were able to retrieve high percentages of putative chitinase sequences, which were confirmed in silico by a scanning for chitinase conserved domains and motif/signatures and in NJ phylogenetic reconstructions. The results confirmed the efficacy of our pipeline in detecting chitinase sequences and highlighted the sensitivity of pHMM-based strategies to identify remote homologues. These analyses provide insight into the potential reservoir of novel molecules in

Figure 5. Phylogenetic tree of bacterial chitinases (GH family-19) using the Neighbor-Joining (NJ) algorithm from MEGA 5.05, p-distance model and 1000 bootstrap tests. The “□” symbol indicates CAMERA metagenomic sequences; “Δ” indicates IMG/M metagenomic sequences; “♦” indicates UniprotKB chitinase reference sequences and “●” indicates the outgroup sequences.

Figure 6. Phylogenetic tree of fungal chitinases (GH family-20) using the Neighbor-Joining (NJ) algorithm from MEGA 5.05, p-distance model and 1000 bootstrap tests. The “□” symbol indicates CAMERA metagenomic sequences; “Δ” indicates IMG/M metagenomic sequences; “♦” indicates UniprotKB chitinase reference sequences and “●” indicates the outgroup sequences.
metagenomic databases while supporting the in silico chitinase mining pipeline developed in this work and identifying phylogenetic relationships among the chitinase sequences. By using our chitinase mining pipeline, a larger number of previously unannotated metagenomic chitinase sequences can be classified, enabling further exploration of these enzymes.
Acknowledgements
This research was supported by CAPES/PNPD.
References
References
Deshpande, M.V. (1986) Enzymatic Degradation of Chitin and Its Biological Applications. Journal of Scientific & Industrial Research, 45, 273-281.
Shaikh, S.A. and Deshpande, M.V. (1993) Chitinolytic Enzymes: Their Contribution to Basic and Applied Research. World Journal of Microbiology and Biotechnology, 9, 468-475. http://dx.doi.org/10.1007/BF00328035
Sahai, A.S. and Manocha, M.S. (1993) Chitinases of Fungi and Plants: Their Involvement in Morphogenesis and Host Parasite Interaction. FEMS Microbiology Reviews, 11, 317-338. http://dx.doi.org/10.1111/j.1574-6976.1993.tb00004.x
Patil, R.S., Ghormade, V. and Deshpande, M.V. (2000) Chitinolytic Enzymes: An Exploration. Enzyme and Microbial Technology, 26, 473-483. http://dx.doi.org/10.1016/S0141-0229(00)00134-4
Henrissat, B. (1991) A Classification of Glycosyl Hydrolases Based on Amino Acid Sequence Similarities. Biochemical Journal, 280, 309-316.
Henrissat, B. and Bairoch, A. (1993) New Families in the Classification of Glycosyl Hydrolases Based on Amino Acid Sequence Similarities. Biochemical Journal, 293, 781-788.
Dahiya, N., Tewari, R. and Hoondal, G.S. (2006) Biotechnological Aspects of Chitinolytic Enzymes: A Review. Applied Microbiology and Biotechnology, 71, 773-782. http://dx.doi.org/10.1007/s00253-005-0183-7
Deane, E.E., Whipps, J.M., Lynch, J.M. and Peberdy, J.F. (1999) Transformation of Trichoderma reesei with a Constitutively Expressed Heterologous Fungal Chitinase Gene. Enzyme and Microbial Technology, 24, 419-424. http://dx.doi.org/10.1016/S0141-0229(98)00155-0
Wiwat, C., Lertcanawanichakul, M., Siwayapram, P., Pantuwatana, S. and Bhumiratana, A. (1996) Expression of Chitinase-Encoding Genes from Aeromonas hydrophila and Pseudomonas maltophila in Bacillus thuringiensis spp. isrealiensis. Gene, 179, 119-126. http://dx.doi.org/10.1016/S0378-1119(96)00575-6
Romão-Dumaresq, A.S., Araújo, W.L., Talbot, N.J. and Thornton, C.R. (2012) RNA Interference of Endochitinases in the Sugarcane Endophyte Trichoderma virens 223 Reduces Its Fitness as a Biocontrol Agent of Pineapple Disease. PLoS One, 7, Article ID: e47888. http://dx.doi.org/10.1371/journal.pone.0047888
Tantimavanich, S., Pantuwatana, S., Bhumiratana, S. and Panbangred, W. (1997) Cloning of a Chitinase Gene in to Bacillus thuringiensis spp. aizawai for Enhanced Insecticidal Activity. Journal of General and Applied Microbiology, 43, 341-347. http://dx.doi.org/10.2323/jgam.43.341
Murao, S., Kawada, T., Itoh, H., Oyama, H. and Shin, T. (1992) Purification and Characterization of a Novel Type of Chitinase from Vibrio alginolyticus. Bioscience, Biotechnology, and Biochemistry, 56, 368-369. http://dx.doi.org/10.1271/bbb.56.368
Terayama, H., Takahashi, S. and Kuzuhara, H. (1993) Large-Scale Preparation of N,N’-Diacetylchitobiose by Enzymic Degradation of Chitin and Its Chemical Modifications. Journal of Carbohydrate Chemistry, 12, 81-93. http://dx.doi.org/10.1080/07328309308018542
Friedman, S.J. and Skehan, P. (1980) Membrane-Active Drugs Potentiate the Killing of Tumor Cells by D-Glucosamine. Proceedings of the National Academy of Sciences, 77, 1172-1176. http://dx.doi.org/10.1073/pnas.77.2.1172
Revah-Moiseev, S. and Carroad, P.A. (1981) Conversion of the Enzymatic Hydrolysate of Shellfish Waste Chitin to Single-Cell Protein. Biotechnology and Bioengineering, 23, 1067-1078. http://dx.doi.org/10.1002/bit.260230514
Kelkar, H.S., Shankar, V. and Deshpande, M.V. (1990) Rapid Isolation and Regeneration of Sclerotium rolfsii Protoplasts and Their Potential Application for Starch Hydrolysis. Enzyme and Microbial Technology, 12, 510-514. http://dx.doi.org/10.1016/0141-0229(90)90067-Z
Davies, J. (2011) How to Discover New Antibiotics: Harvesting the Parvome. Current Opinion in Chemical Biology, 15, 5-10. http://dx.doi.org/10.1016/j.cbpa.2010.11.001
Torsvik, V. and Ovreas, L. (2002) Microbial Diversity and Function in Soil: From Genes to Ecosystems. Current Opinion in Microbiology, 5, 240-245. http://dx.doi.org/10.1016/S1369-5274(02)00324-7
Torsvik, V., Goksoyr, J. and Daae, F.L. (1990) High Diversity in DNA of Soil Bacteria. Applied and Environmental Microbiology, 56, 782-787.
Uchiyama, T. and Miyazaki, K. (2009) Functional Metagenomics for Enzyme Discovery: Challenges to Efficient Screening. Current Opinion in Biotechnology, 20, 616-622. http://dx.doi.org/10.1016/j.copbio.2009.09.010
Venter, J.C., Remington, K., Heidelberg, J.F., Halpern, A.L., Rusch, D., Eisen, J.A., et al. (2004) Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science, 304, 66-74. http://dx.doi.org/10.1126/science.1093857
Rusch, D.B., Halpern, A.L., Sutton, G., Heidelberg, K.B., Williamson, S., Yooseph, S., Wu, D., et al. (2007) The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology, 5, e77. http://dx.doi.org/10.1371/journal.pbio.0050077
Yooseph, S., Sutton, G., Rusch, D.B., Halpern, A.L., Williamson, S.J., Remington, K., et al. (2007) The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families. PLoS Biology, 5, e16. http://dx.doi.org/10.1371/journal.pbio.0050016
Seshadri, R., Kravitz, S.A., Smarr, L., Gilna, P. and Frazier, M. (2007) CAMERA: A Community Resource for Metagenomics. PLoS Biology, 5, e75. http://dx.doi.org/10.1371/journal.pbio.0050075
Meyer, F., Paarman, D., D’Souza, M., Olson, R., Glass, E. M., Kubal, M. et al. (2008) The Metagenomics RAST Server—A Public Resource for the Automatic Phylogenetic and Functional Analysis of Metagenomes. BMC Bioinformatics, 9, 386. http://dx.doi.org/10.1186/1471-2105-9-386
Markowitz V.M., Ivanova, N.N., Szeto, E., Palaniappan, K., Chu, K., Dalevi, D., et al. (2008) IMG/M: A Data Management and Analysis System for Metagenomes. Nucleic Acids Research, 36, D534-538. http://dx.doi.org/10.1093/nar/gkm869
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic Local Alignment Search Tool. Journal of Molecular Biology, 215, 403-410.
Li, M., Ma, B., Kisman, D. and Tromp, J. (2004) PatternHunter II: Highly Sensitive and Fast Homology Search. Journal of Bioinformatics and Computational Biology, 2, 417-439. http://dx.doi.org/10.1142/S0219720004000661
Ma, B., Tromp, J. and Li, M. (2002) PatternHunter: Faster and More Sensitive Homology Search. Bioinformatics, 18, 440-445.
Kent, W.J. (2002) BLAT—The Blast-Like Alignment Tool. Genome Research, 12, 656-664. http://dx.doi.org/10.1101/gr.229202. Article published online before March 2002
Karplus, K., Barrett, C. and Hughey, R. (1998) Hidden Markov Models for Detecting Remote Protein Homologies. Bioinformatics, 14, 846-856. http://dx.doi.org/10.1093/bioinformatics/14.10.846
Krogh, A., Brown, M., Mian, I.S., Sjolander, K. and Haussler, D. (1994) Hidden Markov Models in Computational Biology: Applications to Protein Modeling. Journal of Molecular Biology, 235, 1501-1531. http://dx.doi.org/10.1006/jmbi.1994.1104
Hughey, R. and Krogh, A. (1996) Hidden Markov Models for Sequence Analysis: Extension and Analysis of the Basic Method. Computer Applications in the Biosciences, 12, 95-107. http://dx.doi.org/10.1093/bioinformatics/12.2.95
Baldi, P., Chauvin, Y., Hunkapillar, T. and McClure, M. (1994) Hidden Markov Models of Biological Primary Sequence Information. Proceedings of the National Academy of Sciences of the United States of America, 91, 1059-1063. http://dx.doi.org/10.1073/pnas.91.3.1059
Karplus, K., Sjölander, K., Barrett, C., Cline, M., Haussler, D., Hughey, R. Holm, L. and Sander, C. (1997) Predicting Protein Structure Using Hidden Markov Models. Proteins: Structure, Function, and Bioinformatics, 29, 134-139. http://dx.doi.org/10.1002/(SICI)1097-0134(1997)1+<134::AID-PROT18>3.0.CO;2-P
Sun, S., Chen, J., Li, W., Altintas, I., Lin, A., Peltier, S. Stocks, K., Allen, E.E., Ellisman, M., Grethe, J. and Wooley, J. (2011) Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: The CAMERA Resource. Nucleic Acids Research, 39, D546-D551. http://dx.doi.org/10.1093/nar/gkq1102
Katoh, K. and Toh, H. (2008) Recent Developments in the MAFFT Multiple Sequence Alignment Program. Briefings in Bioinformatics, 9, 276-285. http://dx.doi.org/10.1093/bib/bbn013
Katoh, K., Misawa, K., Kuma, K.I. and Miyata, T. (2002) MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Research, 30, 3059-3066. http://dx.doi.org/10.1093/nar/gkf436
Waterhouse, A.M., Procter, J.B., Martin, D.M.A., Clamp, M. and Barton, G.J. (2009) Jalview Version 2: A Multiple Sequence Alignment and Analysis Workbench. Bioinformatics, 25, 1189-1191. http://dx.doi.org/10.1093/bioinformatics/btp033
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research, 1, 3389-3402. http://dx.doi.org/10.1093/nar/25.17.3389
Hunter, S., Apweiler, R., Attwood, T., Bairoch, A., Bateman, A., Binns, D. et al. (2009) InterPro: The Integrative Protein Signature Database. Nucleic and Acids Research, 37, D211-D215. http://dx.doi.org/10.1093/nar/gkn785
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. (2011) MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Molecular Biology and Evolution, 28, 2731-2739. http://dx.doi.org/10.1093/molbev/msr121
Söding, J. (2005) Protein Homology Detection by HMM-HMM Comparison. Bioinformatics, 21, 951-960. http://dx.doi.org/10.1093/bioinformatics/bti125
Venclovas, Č. (2003) Comparative Modeling in CASP5: Progress Is Evident, but Alignment Errors Remain a Significant Hindrance. Proteins: Structure, Function and Bioinformatics, 53, 380-388. http://dx.doi.org/10.1002/prot.10591
Elofsson, A. (2002) A Study on Protein Sequence Alignment Quality. Proteins: Structure, Function and Bioinformatics, 46, 330-339. http://dx.doi.org/10.1002/prot.10043
Edgar, R.C. and Batzoglou, S. (2006) Multiple Sequence Alignment. Current Opinion in Structural Biology, 16, 368- 373. http://dx.doi.org/10.1016/j.sbi.2006.04.004
Liu, K., Raghavan, S., Nelesen, S., Linder, C.R. and Warnow, T. (2009) Rapid and Accurate Large-Scale Coestimation of Sequence Alignments and Phylogenetic Trees. Science, 324, 1561-1564. http://dx.doi.org/10.1126/science.1171243
Eddy, S.R. (1998) Profile Hidden Markov Models. Bioinformatics, 14, 755-763. http://dx.doi.org/10.1093/bioinformatics/14.9.755
Karplus, K., Karchin, R., Barrett, C., Tu, S., Cline, M., Diekhans, M. Grate, L., Casper, J. and Hughey, R. (2001) What Is the Value Added by Human Intervention in Protein Structure Prediction? Proteins: Structure, Function and Bioinformatics, 45, 86-91. http://dx.doi.org/10.1002/prot.10021
Eddy, S.R., Mitchison, G. and Durbin, R. (1995) Maximum Discrimination Hidden Markov Models of Sequence Consensus. Journal of Computational Biology, 2, 9-23. http://dx.doi.org/10.1089/cmb.1995.2.9
Karchin, R. and Hughey, R. (1998) Weighting Hidden Markov Models for Maximum Discrimination. Bioinformatics, 14, 772-782. http://dx.doi.org/10.1093/bioinformatics/14.9.772
Eddy, S. (2001) HMMER: Profile Hidden Markov Models for Biological Sequence Analysis. http://hmmer.org/
Ocaña, K.A.D.C.S. (2006) Detecção e caracterização de Elementos Móveis Genéticos usando HMMs (Hidden Markov Models). Msc. Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de Janeiro.
Fong, J.H. and Marchler-Bauer, A. (2008) Protein Subfamily Assignment Using the Conserved Domain Database. BMC Research Notes, 1, 114. http://dx.doi.org/10.1186/1756-0500-1-114
Marchler-Bauer, A., Lu, S., Anderson, J.B., Chitsaz, F., Derbyshire, M.K., DeWeese-Scott, C. et al. (2011) CDD: A Conserved Domain Database for the Functional Annotation of Proteins. Nucleic Acids Research, 39, D225-D229. http://dx.doi.org/10.1093/nar/gkq1189
Eisen, J.A. (1998) A Phylogenomic Study of the Muts Family of Proteins. Nucleic Acids Research, 26, 4291-4300. http://dx.doi.org/10.1093/nar/26.18.4291
Foerstner, K.U., Doerks, T., Creevey, C.J., Doerks, A. and Bork, P. (2008) A Computational Screen for Type I Polyketide Synthases in Metagenomics Shotgun Data. PLoS ONE, 3, Article ID: e3515. http://dx.doi.org/10.1371/journal.pone.0003515
Ziemert, N., Podell, S., Penn, K., Badger, J.H., Allen, E. and Jensen, P.R. (2012) The Natural Product Domain Seeker NaPDoS: A Phylogeny Based Bioinformatic Tool to Classify Secondary Metabolite Gene Diversity. PLoS ONE, 7, Article ID: e34064. http://dx.doi.org/10.1371/journal.pone.0034064
NOTES

*Corresponding author.