Health
Vol.4 No.11A(2012), Article ID:24987,11 pages DOI:10.4236/health.2012.431175
Alpha-synuclein truncation and disease
![]()
Department of Physiology, University of Texas Southwestern Medical Center, Dallas, USA; *Corresponding Author: philip.thomas@utsouthwestern.edu
Received 1 October 2012; revised 4 November 2012; accepted 10 November 2012
Keywords: Alpha-Synuclein; Neurodegeneration; Parkinson’s Disease; Lewy Body; Proteasome; Truncation; Degradation; Aggregation; Protease
ABSTRACT
Alpha-synuclein is the major component of Lewy bodies, insoluble protein aggregates, found in patients with Parkinson’s disease, diffuse Lewy body disease, and the Lewy body variant of Alzheimer’s disease. Alpha-synuclein has been found within Lewy bodies to contain many different modifications, including nitration, phosphorylation, ubiquitination, and truncation. C-terminally truncated forms of alpha-synuclein aggregate faster than the full-length protein in vitro, and are thus believed to play a role in Lewy body formation and disease progression. Pathological studies of post mortem brain tissue and the generation of transgenic mouse models further support a role of C-terminally truncated forms of alpha-synuclein in disease. Several enzymes, some of which function extracellularly, have been implicated in the production of these C-terminally truncated forms of alpha-synuclein. However, the enzymes responsible for alphasynuclein truncation in vivo have not yet been firmly established.
1. INTRODUCTION
Lewy bodies are insoluble, predominantly cytoplasmic, protein aggregates located in the brain that are characteristic of a group of neurological diseases. Lewy body diseases include Parkinson’s disease (PD), diffuse Lewy body disease (DLBD), and the Lewy body variant of Alzheimer’s disease (LBV). No single event has been shown to cause Lewy body diseases, yet all of these diseases result in similar pathological and physiological characteristics. Lewy body diseases are all pathologically defined by the accumulation of cytoplasmic protein deposits and neuronal cell death (reviewed in [1]). Physiological effects include an increase in cellular oxidative damage [2] and inflammation ([3], reviewed in [4]). These diseases are all progressive, and Lewy body formation correlates with a decline in motor and cognitive functions, and eventual fatality.
2. MAIN BODY
2.1. Lewy Body Diseases
The major component of Lewy bodies is alpha-synuclein [5] (Figure 1). Before the identification of alpha-synuclein, Lewy bodies were characterized by the presence of ubiquitin and hyper-ubiquitinated proteins [6]. In addition to alpha-synuclein and ubiquitin, Lewy bodies have been found to contain a plethora of other protein components, but not all of these components are present in every Lewy body in every patient. Other proteins that play a role in the UPS have been identified
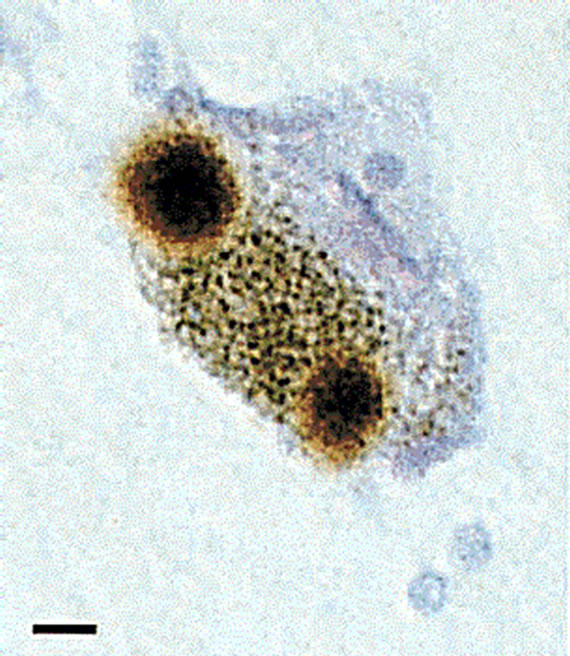
Figure 1. Alpha-synuclein-positive Lewy bodies. Post mortem tissue from the substantia nigra of Parkinson’s disease patients was stained for alpha-synuclein (brown). A single nerve cell containing two Lewy bodies is shown, with a scale bar of 8 µm. Reprinted by permission from Macmillan Publishers Ltd: Nature [16].
in Lewy bodies, and these include dorfin [7], Nub1 [8], and p62 [9,10]. Other proteins identified in Lewy bodies include microtubule-associated proteins [11-14] and protein kinases [15]. The mechanism by which these proteins co-aggregate with alpha-synuclein and the significance of their aggregation is unknown.
While a vast amount of information has been generated about Lewy body diseases in the past two decades, many questions remain about the cause and progression of these diseases. One such question is whether the alpha-synuclein-containing protein aggregates are harmful or if they are merely a mechanism by which the cell sequesters individual protein components that might otherwise be toxic. Recent results suggest that the alphasynuclein-containing inclusions may not be toxic themselves, but that the intermediate, oligomeric species along the aggregation pathway might be responsible for proteotoxicity [17,18]. The factors that initiate this aggregation pathway are unknown. Several factors, however, have been shown to contribute to the development and progression of Lewy body diseases, including both genetic and environmental factors. In the case of Parkinson’s disease (PD), three independent missense mutations in alpha-synuclein (A30P, E46K, and A53T) have been shown to cause early-onset forms of the disease [19-21]. Duplications [22] and triplications [23] of the alpha-synuclein gene, and, thus, overexpression of the protein, also cause early-onset forms of PD. Mutations in other genes, such as LRRK2 [24,25], PARK2 [26], PINK1 [27], DJ-1 [28], and ATP13A2 [29] are linked to familial forms of PD; however, these monogenic forms of PD only account for about 5% of all cases. Interestingly, the other 95% of cases have the same pathological hallmarks as the monogenic forms of disease. No mutations in the SCNA gene encoding for alpha-synuclein have been found to cause other Lewy body diseases, yet these diseases exhibit alpha-synuclein pathology similar to that found in PD patients. These observations suggest that a common mechanism of pathogenesis exists in all Lewy body diseases, but that the initiation of pathogenesis may vary.
The most prevalent risk factor for Lewy body diseases is age. Many physiological processes are altered as an organism ages. Some of the age-related processes that have been correlated with Lewy body diseases include an increase in oxidative damage to cellular components (reviewed in [30]), dysfunction of the mitochondria (reviewed in [31]), and the long-term exposure to environmental toxins [32]. Additionally, many studies have aimed to understand the effects of aging on the cellular protein degradation machinery. Proteolysis offers an alternative mechanism for reduction in levels of the presumptive cytotoxic protomer. Changes in both major pathways of protein degradation in the cell, the UPS and autophagy, have been observed with age. While effects on the trypsinand chymotrypsin-like activities of the proteasome with age have been inconclusive [33-36], the PGPH-like activity of the proteasome has consistently been shown to decrease with age [37,38]. Additionally, gene expression studies have indicated a change in proteasomal subunit expression patterns with age in both murine muscle [39] and human fibroblasts [40]. A decline in autophagy function was observed in rats and human fibroblasts, through a decrease in both substrate binding and transport to lysosomes [41].
With these wide-ranging physiological alterations that occur with age, it is reasonable to hypothesize that proteins could be modified over time, leading to enhanced aggregation propensity, and the possible initiation of disease. Lewy body diseases might be the result of the failing protein degradation pathways being unable to compensate for the buildup of damaged proteins. Certain combinations of variables or specific genetic backgrounds may yield an individual more susceptible to these alterations and the lack of compensatory mechanisms, explaining why some individuals succumb to Lewy body diseases while others do not. A further study of each of these processes will allow for a more complete understanding of disease pathogenesis and the generation of targeted therapeutics to slow progression or prevent these diseases altogether.
2.2. Alpha-Synuclein Structure
Alpha-synuclein is a 140-amino acid protein that is paralogous to two other nervous system proteins, referred to as betaand gamma-synuclein. Alpha-, beta-, and gamma-synuclein are similar in sequence, with most of the similarity lying within the N-terminus of the proteins. The N-terminal portion of alpha-synuclein includes seven imperfect repeats of 11 residues containing the KTKEGV consensus sequence, while the C-terminus contains many acidic residues and is, thus, negativelycharged (Figure 2). Residues 61 - 95 encompass many hydrophobic residues, and a peptide corresponding to this region of the protein (referred to as the NAC region)
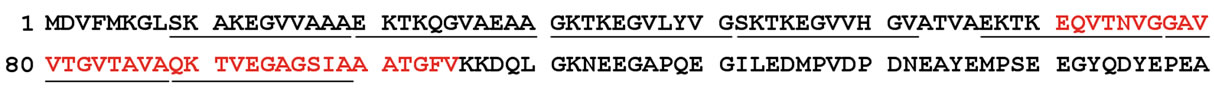
Figure 2. Human alpha-synuclein sequence. The sequence of the 140-amino acid protein (accession number P37840.1) is shown. Underlined regions are the seven imperfect 11-residue repeats and the sequence in red is the amyloidogenic NAC (Non-Aβ Component) region of the protein.
has been found to colocalize with Aβ plaques in Alzheimer’s disease [42].
Alpha-synuclein is among the increasing number of proteins recognized as an intrinsically-disordered protein (IDP), a class of proteins characterized by their lack of a defined structure in the native state (reviewed in [43]). Upon binding to negatively-charged vesicles, alphasynuclein adopts a conformation that has a high alpha-helical propensity [44,45], and a fraction of neuronal alpha-synuclein has been found to associate with membranes in vivo [46]. Structural studies aimed at understanding the membrane-bound form of alpha-synuclein have relied on nuclear magnetic resonance (NMR) spectroscopy. A structure of the full-length, membranebound form of alpha-synuclein (Figure 3) reveals a conformation in which the N-terminal two-thirds of the protein forms a broken, amphipathic alpha-helix [47,48]. This structured portion of the protein is responsible for membrane binding, and residues at the very N-terminus are essential for this process [49]. In the NMR structure of alpha-synuclein, the negatively-charged C-terminal tail remains flexible and disordered [47,48].
Structural studies aimed at understanding the unbound state of alpha-synuclein have relied on molecular dynamics (MD) simulations and more complicated NMR techniques, such as residual dipolar coupling (RDC) and paramagnetic relaxation enhancement (PRE). These techniques have produced results suggesting that alphasynuclein adopts several thousand structurally distinct conformations, many of which are more compact than expected for a random coil [50]. Many of these conformations include long-range (15 Å to 20 Å) interactions between the C-terminus and both the N-terminus and central portion of the protein [50-52].
Several observations suggest that disruption of these long-range interactions facilitates aggregation of the protein. In one study, spermine (a polyamine that has been shown to interact with the acidic C-terminus of alpha-synuclein) was shown to disrupt these long-range interactions while simultaneously promoting in vitro aggregation [52]. A similar result was observed when temperature was increased [52]. Additionally, studies have shown that the PD-causing A30P and A53T alpha-synu-
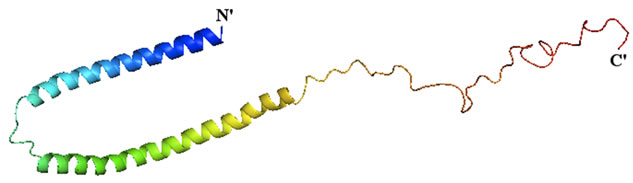
Figure 3. Structure of alpha-synuclein bound to lipid vesicles. The structure of the full-length human protein was determined by NMR (PDB ID: 1XQ8) and image was rendered in PyMol. The structure is colored from blue (N-terminus) to red (C-terminus).
clein mutations both have decreased propensity for these long-range interactions [53]. A disruption of these intramolecular long-range interactions and increased aggregation propensity of the protein may serve as a mechanism by which these two point mutations cause Lewy body formation and disease pathogenesis.
2.3. Alpha-Synuclein Physiology and Function
Alpha-synuclein is a protein expressed in all vertebrates. Homology of alpha-synuclein across species is greater at the N-terminus of the protein, with more variability in sequence located toward the C-terminus. Alpha-synuclein is expressed predominantly in the central nervous system and localizes to presynaptic terminals [54]. Expression of alpha-synuclein is quite high, consisting of up to 1% of the total protein in certain regions of the brain [54]. Within neuronal cells, alpha-synuclein has been detected in both the cytoplasm and nucleus [55]. Studies utilizing fractionated rat brains revealed that about 15% of alpha-synuclein is membrane-bound [56], and the protein was recently found associated with mitochondrial membranes in normal dopaminergic neurons [57,58]. The relative subcellular distribution of alphasynuclein varies among different neuronal cell populations [58,59].
While the function of alpha-synuclein has not been clearly established, observations related to the protein’s cellular localization have provided clues to its function. Studies aimed directly at establishing a role for alpha-synuclein have relied on mammalian cell culture and animal models. Alpha-synuclein knockout mice have been generated in several laboratories and these mice are viable, suggesting that other proteins might play a redundant role in the cell. Deletion of alpha-synuclein in mice causes only mild phenotypes including defects in presynaptic vesicles [60], synaptic transmission [61], and the trafficking [62] and metabolism [63,64] of fatty acids. Additionally, mice lacking alpha-synuclein are protected from the changes in cellular morphology and cell death caused by exposure to MPTP (an inhibitor of mitochondrial complex 1) that are observed in wild-type mice [65]. The levels of striatal dopamine were also less affected by MPTP-treatment in alpha-synuclein knockout mice than wild-type mice [66].
Alpha-synuclein has been shown to play a role in neurotransmitter release, as studied by neuronal cell lines expressing alpha-synuclein [67], knockout mice [68], and mice overexpressing alpha-synuclein [67]. Alphasynuclein has also been shown to exhibit a non-classical chaperone activity that plays a role in SNARE complex assembly [69]. Both the Nand C-termini of alphasynuclein play a role in this process, as the N-terminus of the protein binds to phospholipids while the C-terminus is necessary for synaptobrevin-2 binding [69]. Alpha-, beta-, and gamma-synuclein triple knockout mice showed an age-dependent decrease in SNARE complex assembly [69], indicating that other members of the synuclein family may serve roles that are similar to, or possibly overlap with, those of alpha-synuclein.
While neuronally-expressed alpha-synuclein has been the focus of most studies, many other tissues have been found to express the protein. These tissues include muscle [70], cerebral blood vessels [71], red blood cells [72], plasma [73], and blood cells of the immune system [74]. The function of alpha-synuclein in these tissues has not been elucidated. An investigation of alpha-synuclein expression in human fetuses revealed that alpha-synuclein is expressed throughout fetal tissue; however, expression in most of these tissues is reduced in adulthood [75], suggesting that alpha-synuclein might also play a role in development.
3. DISCUSSION
3.1. Alpha-Synuclein Truncation and Disease
Wild-type and the three PD-causing missense mutations of alpha-synuclein were the primary focus of early studies related to alpha-synuclein aggregation and disease; however, recent studies have acknowledged posttranslational modifications of alpha-synuclein and the role that these forms of the protein might play in disease. Within Lewy bodies, alpha-synuclein has been found to exist with several modifications (Table 1). These modifications include phosphorylation [76-78], nitration [79], and ubiquitination [80,81]. In addition to full-length alpha-synuclein, truncated forms of alpha-synuclein have
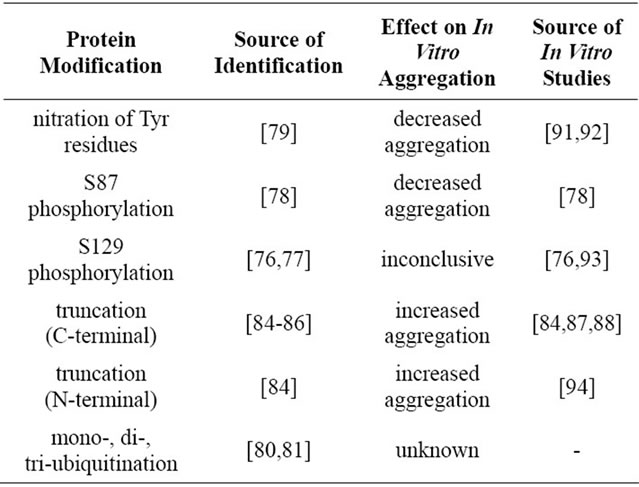
Table 1. Modifications of alpha-synuclein identified in Lewy bodies and their effects on in vitro aggregation. The identification of alpha-synuclein modifications that are present in Lewy bodies might provide insight into disease progression.
been identified in pathological aggregates [82,83]. These truncations have been found to occur from either the C-terminus or both the N-terminus and C-terminus in patient samples [84,85] and transgenic mouse models of PD [84,86]. The C-terminus of alpha-synuclein is negatively-charged, and truncating the protein to remove this terminus produces species that are more prone to aggregation in vitro [84,87,88]. Notably, truncated alphasynuclein can facilitate aggregation of the full-length protein in vitro [84] and in vivo [89]. A truncated form of alpha-synuclein, the NAC region, accumulates in Alzheimer’s disease patients [90], suggesting a role for alpha-synuclein truncation in the pathogenesis of multiple diseases.
Transgenic mice have recently been generated that overexpress truncated forms of alpha-synuclein. These mice exhibit physiological and pathological similarities to patients with Lewy body diseases [95-97]. The three different models generated expressed the human alphasynuclein (residues 1 - 120) on a mouse alpha-synuclein null background [95], human A53T alpha-synuclein (residues 1 - 130) on an endogenous mouse alpha-synuclein background [96], and human alpha-synuclein (residues 1 - 119) on an endogenous mouse alpha-synuclein background [97]. The mice expressing human A53T alpha-synuclein (residues 1 - 130) exhibited loss of dopaminergic neurons in the substantia nigra, lower levels of striatal dopamine, and an alteration in spontaneous locomotor activities [96]. Mice expressing human alphasynuclein (residues 1 - 119) showed a similar loss of striatal dopamine [97]. The expression of truncated alphasynuclein also led to a greater susceptibility to stress [95].
The identification of truncated forms of alpha-synuclein that are both prone to aggregation and capable of cross-seeding aggregation of the full-length protein, suggests that alpha-synuclein truncation is a mechanism that contributes to the progression of Lewy body diseases. Some reports have indicated that truncation of alphasynuclein is a natural process, and that truncated forms of alpha-synuclein are detectable in the brain of healthy individuals [98]. However, expression of the diseasecausing A30P and A53T alpha-synuclein leads to enhanced production of these C-terminally truncated species and faster aggregation [86]. These results, in addition to the transgenic mice studies described previously [95-97], suggest that truncated alpha-synuclein species might lead to the development of clinical and pathological features if expression exceeds a certain level. In individuals with disease, the amount of truncated alphasynuclein species generated might have reached a threshold that can no longer be tolerated by the cell. The mechanism by which these truncated forms of alpha-synuclein are produced and accumulated in the cell is unknown.
3.2. Degradation of Alpha-Synuclein by the 20S Proteasome
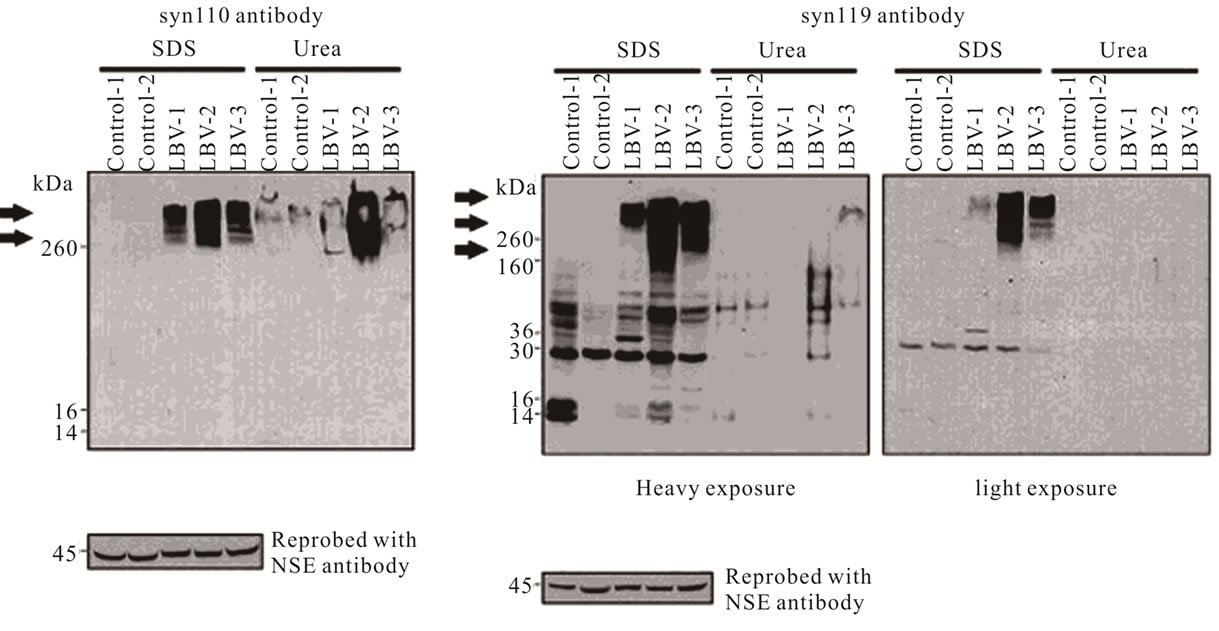
Several in vitro studies have shown that alpha-synuclein can be degraded by the 20S proteasome in a ubiquitin-independent manner [84,99,100]. While degradation of alpha-synuclein by the 20S proteasome has not been established in an animal model, several in vivo observations support a role for the 20S proteasome in alpha-synuclein truncation and disease. C-terminally truncated forms of alpha-synuclein isolated from A53T alpha-synuclein transgenic mice were identified by mass spectrometry, and some species were identical to those produced by the 20S proteasome in vitro [84,86]. Follow-up studies, in which antibodies were generated to specifically recognize the C-terminus of truncated forms of alpha-synuclein, revealed that two C-terminally-truncated alpha-synuclein species, residues 1 - 110 (syn110) and residues 1 - 119 (syn119) are present at much higher levels in patients with Lewy body diseases than in agesimilar controls (Figure 4) [85]. Additionally, it was shown that these truncated species are not always colocalized within the same cell [85], hinting at a mechanism by which their production may be regulated.
3.3. Roles of Other Enzymes in Alpha-Synuclein Degradation
While many independent laboratories have shown that alpha-synuclein can be degraded by the 20S proteasome in vitro [99-101], other studies have implicated different enzymes in the cleavage and degradation of alpha-synuclein [102-115]. These enzymes are different in their activities, their cellular localization, and their regulation. It is possible that more than one of these enzymes works in concert to produce truncated forms of alpha-synuclein that promote Lewy body formation and disease progression. Understanding these processes and their cooperativity in normal physiological processes and in disease progression is essential to the understanding and treatment of Lewy body diseases.
Calpain 1 is a calcium-dependent cysteine protease [116,117]. In in vitro assays, the monomeric form of alpha-synuclein is predominantly cleaved by calpain 1 after residue 57, while fibrillar forms of alpha-synuclein are degraded at the C-terminus, specifically after residues 114 and 122 [102]. Another study by the same group revealed that the cleavage products produced by calpain 1-mediated degradation of soluble alpha-synuclein inhibited aggregation of the full-length protein, while cleavage products produced by calpain 1-mediated degradation of fibrillar forms of alpha-synuclein were aggregation-prone and capable of cross-seeding aggregation of full-length, monomeric alpha-synuclein [103]. It has also been reported that the activities of calpain I and the 20S proteasome may act in a concerted manner in producing aggregation-prone C-terminally truncated forms
 (a) (b)
(a) (b)
Figure 4. C-terminally truncated forms of alpha-synuclein are present in post mortem brain tissue. Antibodies specific for alpha-synuclein truncated after residue 110, syn110, (panel A) and after residue 119, syn119, (panel B) recognize high molecular weight aggregated species (arrows) in patients with the Lewy body variant of Alzheimer’s disease (LBV) at higher levels than in age-similar controls. Reprinted from American Journal of Pathology, Volume 177, Karen A. Lewis, et al., Abnormal Neurites Containing C-Terminally Truncated α-Synuclein Are Presented in Alzheimer’s Disease without Conventional Lewy Body Pathology, pp. 3037-3050, 2010 (reference [85]), with permission from Elsevier.
of alpha-synuclein [104]. In this study, a product of calpain 1-mediated degradation that was resistant to further degradation by the enzyme, was able to enhance the degradation of full-length alpha-synuclein by the 20S proteasome and this enhancement was specific for alpha-synuclein, as no enhancement was observed for the degradation of azocasein or peptide substrates [104].
Another protein that has been suggested to play a role in the accumulation of truncated forms of alpha-synuclein is cathepsin D. Cathepsin D is a lysosomal protease that has been shown to cleave alpha-synuclein both in vitro and in the lysosomal fraction of cells expressing alpha-synuclein [105]. In one study, reduced proteasomal activity was observed in cathepsin D-deficient mice [106], implicating a connection between these two mechanisms of proteolysis. Additionally, RNAi knockdown of cathepsin D in a mammalian cell culture model [105] and cathepsin D knockout mice [106] exhibit alpha-synuclein accumulations. Another study, utilizing Drosophila [107], suggests that this enzyme might play a role in the normal clearance of alpha-synuclein.
In light of recent reports suggesting that alpha-synuclein is found extracellularly [108,118-120], proteases that might act on these extracellular forms of alpha-synuclein might also play a role in this process. One such enzyme is neurosin, a serine protease that is highly expressed in the nervous system [121]. Neurosin has been shown to co-aggregate with alpha-synuclein in Lewy bodies [109] and, in in vitro assays, specific cleavage products of alpha-synuclein were produced in which the protein was cleaved in the NAC region and at several sites within the C-terminus [110]. A recent study has revealed that neurosin-mediated cleavage of alpha-synuclein can only occur extracellularly, once neurosin is activated upon secretion [111].
Matrix metalloproteases (MMPs) are a class of enzymes that are secreted, and they are known to play a role in the degradation of extracellular and membranebound proteins (reviewed in [122]). Several studies have implicated MMPs in the cleavage and aggregation of alpha-synuclein. In one study, a dopaminergic neuronal cell line was transfected with alpha-synuclein, and the overexpression of alpha-synuclein led to its secretion [112]. Additionally, when these transfected cells were subjected to oxidative stress, the expression of matrix metalloprotease-3 (MMP-3) was increased and alphasynuclein fragments were observed in the media. Generation of alpha-synuclein fragments was blocked by pre-incubation with a matrix metalloprotease inhibitor. In addition, results from this study showed that alphasynuclein can be cleaved at several positions, and that the products generated facilitate aggregation and cell toxicity. Other studies have also shown that matrix metalloproteases can cleave alpha-synuclein, and cleavage by both MMP-1 and MMP-3 was shown to increase aggregation propensity [113]. Another study showed Cterminal cleavage by MMP-3, and found that MMP-3 cleavage of the disease-causing A53T mutation of alpha-synuclein resulted in an increased number of degradation products [114]. By analyzing post mortem brain tissue from PD patients, the authors reported that over 50% of Lewy bodies contain MMP-3 [121]. Recently, plasmin, a serine protease in the blood, was also implicated in alpha-synuclein degradation and disease pathogenesis [115].
4. CONCLUSION
These in vitro and in vivo results reveal that the formation of truncated alpha-synuclein species is a complex process that likely plays a role in disease. The major goal of studying these diseases is to develop therapeutics to halt or slow down the progression of the disease. Whereas truncation of alpha-synuclein is correlated with accelerated disease progression, interference in this process may have therapeutic benefit. Elucidating the mechanism by which these enzymes produce partially-truncated and aggregation-prone alpha-synuclein cleavage products is an initial step in identifying relevant therapeutic targets. Considering the large number of enzymes that have been shown to produce truncated alpha-synuclein species in vivo, it is likely that alpha-synuclein degradation is the result of a combination of enzymes that either work independently or together to produce specific aggregation-prone species.
![]()
![]()
REFERENCES
- Trojanowski, J.Q., et al. (1998) Fatal attractions: Abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death and Differentiation, 5, 832-837. doi:10.1038/sj.cdd.4400432
- Norris, E.H. and Giasson, B.I. (2005) Role of oxidative damage in protein aggregation associated with Parkinson’s disease and related disorders. Antioxidants & Redox Signaling, 7, 672-684. doi:10.1089/ars.2005.7.672
- Mackenzie, I.R. (2000) Activated microglia in dementia with Lewy bodies. Neurology, 55, 132-134. doi:10.1212/WNL.55.1.132
- Qian, L., Flood, P.M. and Hong, J.-S. (2010) Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. Journal of Neural Transmission, 117, 971-979. doi:10.1007/s00702-010-0428-1
- Spillantini, M.G., et al. (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proceedings of the National Academy of Sciences of the United States of America, 95, 6469-6473. doi:10.1073/pnas.95.11.6469
- Kuzuhara, S., et al. (1988) Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathologica, 75, 345-353. doi:10.1007/BF00687787
- Ito, T., et al. (2003) Dorfin localizes to Lewy bodies and ubiquitylates synphilin-1. Journal of Biological Chemistry, 278, 29106-29114. doi:10.1074/jbc.M302763200
- Tanji, K., et al. (2011) Synphilin-1-binding protein NUB1 is colocalized with nonfibrillar, proteinase K-resistant α-synuclein in presynapses in Lewy body disease. Journal of Neuropathology and Experimental Neurology, 70, 879-889. doi:10.1097/NEN.0b013e3182303745
- Kuusisto, E., Salminen, A. and Alafuzoff, I. (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport, 12, 2085-2090. doi:10.1097/00001756-200107200-00009
- Zatloukal, K. and Stumptner, C. (2002) p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. The American Journal of Pathology, 160, 255-263. doi:10.1016/S0002-9440(10)64369-6
- Gai, W.P., Blumbergs, P.C. and Blessing, W.W. (1996) Microtubule-associated protein 5 is a component of Lewy bodies and Lewyneurites in the brainstem and forebrain regions affected in Parkinson’s disease. Acta Neuropathologica, 91, 78-81. doi:10.1007/s004010050395
- Jensen, P.H., et al. (2000) Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. The Journal of Biological Chemistry, 275, 21500-21507. doi:10.1074/jbc.M000099200
- D'Andrea, M.R., Ilyin, S. and Plata-Salaman, C.R. (2001) Abnormal patterns of microtubule-associated protein-2 (MAP-2) immunolabeling in neuronal nuclei and Lewy bodies in Parkinson’s disease substantianigra brain tissues. Neuroscience Letters, 306, 137-140. doi:10.1016/S0304-3940(01)01811-0
- Duda, J.E., et al. (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathologica, 104, 7-11. doi:10.1007/s00401-002-0563-3
- Arawaka, S., et al. (2006) The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. The Journal of Neuroscience, 26, 9227-9238. doi:10.1523/JNEUROSCI.0341-06.2006
- Spillantini, M.G., et al. (1997) Alpha-synuclein in Lewy bodies. Nature, 388, 839-840. doi:10.1038/42166
- Volles, M.J. and Lansbury, P.T. (2003) Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry, 42, 7871-7878. doi:10.1021/bi030086j
- Winner, B., et al. (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proceedings of the National Academy of Sciences of the United States of America, 108, 4194-4199. doi:10.1073/pnas.1100976108
- Krüger, R., et al. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nature Genetics, 18, 106-108. doi:10.1038/ng0298-106
- Zarranz, J.J., et al. (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology, 55, 164-173. doi:10.1002/ana.10795
- Polymeropoulos, M.H., et al. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045-2047. doi:10.1126/science.276.5321.2045
- Chartier-Harlin, M.-C., et al. (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet, 364, 1167-1169. doi:10.1016/S0140-6736(04)17103-1
- Singleton, A.B., et al. (2003) Alpha-synuclein locus triplication causes Parkinson’s disease. Science, 302, 841. doi:10.1126/science.1090278
- Paisán-Ruíz, C., et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595-600. doi:10.1016/j.neuron.2004.10.023
- Zimprich, A., et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601-607. doi:10.1016/j.neuron.2004.11.005
- Kitada, T., et al. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605-608. doi:10.1038/33416
- Valente, E.M., et al. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304, 1158-1160. doi:10.1126/science.1096284
- Bonifati, V., et al. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science, 299, 256-259. doi:10.1126/science.1077209
- Ramirez, A., et al. (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature Genetics, 38, 1184-1191. doi:10.1038/ng1884
- Sohal, R.S. and Orr, W.C. (2012) The redox stress hypothesis of aging. Free Radical Biology and Medicine, 52, 539-555. doi:10.1016/j.freeradbiomed.2011.10.445
- Bratic, I. and Trifunovic, A. (2010) Mitochondrial energy metabolism and ageing. Biochimicaet Biophysica Acta, 1797, 961-967. doi:10.1016/j.bbabio.2010.01.004
- Masalha, R., et al. (1997) Selective dopamine neurotoxicity by an industrial chemical: An environmental cause of Parkinson’s disease? Brain Research, 774, 260-264. doi:10.1016/S0006-8993(97)81717-9
- Shibatani, T., Nazir, M. and Ward, W.F. (1996) Alteration of rat liver 20S proteasome activities by age and food restriction. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 51, B316-B322. doi:10.1093/gerona/51A.5.B316
- Hayashi, T. and Goto, S. (1998) Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mechanisms of Ageing and Development, 102, 55-66. doi:10.1016/S0047-6374(98)00011-6
- Andersson, M., Sjøstrand, J. and Karlsson, J.O. (1998) Proteolytic cleavage of N-Succ-Leu-Leu-Val-Tyr-AMC by the proteasome in lens epithelium from clear and cataractous human lenses. Experimental Eye Research, 67, 231-236. doi:10.1006/exer.1998.0519
- Ponnappan, U., Zhong, M. and Trebilcock, G.U. (1999) Decreased proteasome-mediated degradation in T cells from the elderly: A role in immune senescence. Cellular Immunology, 192, 167-174. doi:10.1006/cimm.1998.1418
- Conconi, M., et al. (1996) Age-related decline of rat liver multicatalytic proteinase activity and protection from oxidative inactivation by heat-shock protein 90. Archives of Biochemistry and Biophysics, 331, 232-240. doi:10.1006/abbi.1996.0303
- Hayashi, T. and Goto, S. (1998) Age-related changes in the 20S and 26S proteasome activities in the liver of male F344 rats. Mechanisms of Ageing and Development, 102, 55-66. doi:10.1016/S0047-6374(98)00011-6
- Lee, C.K., et al. (1999) Gene expression profile of aging and its retardation by caloric restriction. Science, 285, 1390-1393. doi:10.1126/science.285.5432.1390
- Ly, D.H., et al. (2000) Mitotic misregulation and human aging. Science, 287, 2486-2492. doi:10.1126/science.287.5462.2486
- Cuervo, A.M. and Dice, J.F. (2000) Age-related decline in chaperone-mediated autophagy. The Journal of Biological Chemistry, 275, 31505-31513. doi:10.1074/jbc.M002102200
- Uéda, K., et al. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 90, 11282- 11286. doi:10.1073/pnas.90.23.11282
- Dunker, A.K., et al. (2001) Intrinsically disordered protein. Journal of Molecular Graphics and Modelling, 19, 26-59. doi:10.1016/S1093-3263(00)00138-8
- Davidson, W.S., et al. (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. Journal of Biological Chemistry, 273, 9443-9449. doi:10.1074/jbc.273.16.9443
- Eliezer, D., et al. (2001) Conformational properties of alpha-synuclein in its free and lipid-associated states. Journal of Molecular Biology, 307, 1061-1073. doi:10.1006/jmbi.2001.4538
- Leng, Y., Chase, T.N. and Bennett, M.C. (2001) Muscarinic receptor stimulation induces translocation of an alpha-synuclein oligomer from plasma membrane to a light vesicle fraction in cytoplasm. The Journal of Biological Chemistry, 276, 28212-28218. doi:10.1074/jbc.M011121200
- Chandra, S., et al. (2003) A broken alpha-helix in folded alpha-synuclein. Journal of Biological Chemistry, 278, 15313-15318. doi:10.1074/jbc.M213128200
- Ulmer, T.S., et al. (2005) Structure and dynamics of micelle-bound human alpha-synuclein. Journal of Biological Chemistry, 280, 9595-9603. doi:10.1074/jbc.M411805200
- Vamvaca, K., Volles, M.J. and Lansbury, P.T. (2009) The first N-terminal amino acids of alpha-synuclein are essential for alpha-helical structure formation in vitro and membrane binding in yeast. Journal of Molecular Biology, 389, 413-424. doi:10.1016/j.jmb.2009.03.021
- Dedmon, M.M., et al. (2005) Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. Journal of the American Chemical Society, 127, 476-477. doi:10.1021/ja044834j
- Bernadó, P., et al. (2005) Defining long-range order and local disorder in native alpha-synuclein using residual dipolar couplings. Journal of the American Chemical Society, 127, 17968-17969. doi:10.1021/ja055538p
- Bertoncini, C.W., Jung, Y.-S., et al. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America, 102, 1430-1435. doi:10.1073/pnas.0407146102
- Bertoncini, C.W., Fernandez, C.O., et al. (2005) Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. The Journal of Biological Chemistry, 280, 30649-30652. doi:10.1074/jbc.C500288200
- Iwai, A., et al. (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron, 14, 467-475. doi:10.1016/0896-6273(95)90302-X
- Yu, S., et al. (2007) Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience, 145, 539-555. doi:10.1016/j.neuroscience.2006.12.028
- Lee, H.-J., Choi, C. and Lee, S.-J. (2002) Membranebound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. The Journal of Biological Chemistry, 277, 671-678. doi:10.1074/jbc.M107045200
- Li, W.-W., et al. (2007) Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport, 18, 1543-1546. doi:10.1097/WNR.0b013e3282f03db4
- Zhang, L., et al. (2008) Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: an immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Research, 1244, 40-52. doi:10.1016/j.brainres.2008.08.067
- Liu, G., et al. (2009) alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neuroscience Letters, 454, 187-192. doi:10.1016/j.neulet.2009.02.056
- [61] Cabin, D., Shimazu, K. and Murphy, D. (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. The Journal of Neuroscience, 22, 8797- 8807.
- [62] Martín, E.D., et al. (2004) Stressor-related impairment of synaptic transmission in hippocampal slices from alpha-synuclein knockout mice. The European Journal of Neuroscience, 20, 3085-3091. doi:10.1111/j.1460-9568.2004.03801.x
- [63] Castagnet, P.I., et al. (2005) Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. Journal of Neurochemistry, 94, 839- 849. doi:10.1111/j.1471-4159.2005.03247.x
- [64] Golovko, M.Y., et al. (2007) Alpha-synuclein gene ablation increases docosahexaenoic acid incorporation and turnover in brain phospholipids. Journal of Neurochemistry, 101, 201-211. doi:10.1111/j.1471-4159.2006.04357.x
- [65] Barceló-Coblijn, G., et al. (2007) Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. Journal of Neurochemistry, 101, 132-141. doi:10.1111/j.1471-4159.2006.04348.x
- [66] Dauer, W., et al. (2002) Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences of the United States of America, 99, 14524-14529. doi:10.1073/pnas.172514599
- [67] Drolet, R.E., et al. (2004) Mice lacking alpha-synuclein have an attenuated loss of striatal dopamine following prolonged chronic MPTP administration. Neurotoxicology, 25, 761-769. doi:10.1016/j.neuro.2004.05.002
- [68] Nemani, V.M., et al. (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron, 65, 66-79. doi:10.1016/j.neuron.2009.12.023
- [69] Abeliovich, A., et al. (2000) Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron, 25, 239-252. doi:10.1016/S0896-6273(00)80886-7
- [70] Burré, J., et al. (2010) Alpha-synuclein promotes SNAREcomplex assembly in vivo and in vitro. Science, 329, 1663-1667. doi:10.1126/science.1195227
- [71] Askanas, V., et al. (2000) Novel Immunolocalization of [alpha]-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. Journal of Neuropathology and Experimental Neurology, 59, 592.
- [72] Tamo, W., et al. (2002) Expression of alpha-synuclein, the precursor of non-amyloid beta component of Alzheimer’s disease amyloid, in human cerebral blood vessels. Neuroscience Letters, 326, 5-8. doi:10.1016/S0304-3940(02)00297-5
- [73] Barbour, R., et al. (2008) Red blood cells are the major source of alpha-synuclein in blood. Neuro-Degenerative Diseases, 5, 55-59. doi:10.1159/000112832
- [74] Tinsley, R.B., et al. (2010) Sensitive and specific detection of α-synuclein in human plasma. Journal of Neuroscience Research, 88, 2693-2700.
- [75] Kim, S., et al. (2004) Alpha-synuclein induces apoptosis by altered expression in human peripheral lymphocyte in Parkinson’s disease. FASEB Journal, 18, 1615-1617.
- [76] Ltic, S., et al. (2004) Alpha-synuclein is expressed in different tissues during human fetal development. Journal of Molecular Neuroscience, 22, 199-204. doi:10.1385/JMN:22:3:199
- [77] Fujiwara, H., et al. (2002) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology, 4, 160-164.
- [78] Anderson, J.P., et al. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. The Journal of Biological Chemistry, 281, 29739-29752. doi:10.1074/jbc.M600933200
- [79] Paleologou, K.E., et al. (2010) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synucleinoligomerization, and influences synuclein-membrane interactions. The Journal of Neuroscience, 30, 3184- 3198. doi:10.1523/JNEUROSCI.5922-09.2010
- [80] Giasson, B.I., et al. (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science, 290, 985-989. doi:10.1126/science.290.5493.985
- [81] Sampathu, D., Giasson, B. and Pawlyk, A. (2003) Ubiquitination of α-synuclein is not required for formation of pathological inclusions in α-synucleinopathies. The American Journal of Pathology, 163, 91-100. doi:10.1016/S0002-9440(10)63633-4
- [82] Tofaris, G.K., et al. (2003) Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. The Journal of Biological Chemistry, 278, 44405-44411. doi:10.1074/jbc.M308041200
- [83] Baba, M., et al. (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. The American Journal of Pathology, 152, 879-884.
- [84] Campbell, B. and McLean, C. (2001) The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson’s disease. Journal of Neurochemistry, 76, 87-96. doi:10.1046/j.1471-4159.2001.00021.x
- [85] Liu, C-W., et al. (2005) A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: Implications for pathogenesis of Parkinson disease. The Journal of Biological Chemistry, 280, 22670- 22678. doi:10.1074/jbc.M501508200
- [86] Lewis, K.A., et al. (2010) Abnormal neurites containing C-terminally truncated alpha-synuclein are present in Alzheimer's disease without conventional Lewy body pathology. The American Journal of Pathology, 177, 3037- 3050. doi:10.2353/ajpath.2010.100552
- [87] Li, W., et al. (2005) Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s diseaselinked mutations. Proceedings of the National Academy of Sciences of the United States of America, 102, 2162- 2167. doi:10.1073/pnas.0406976102
- [88] Murray, I., et al. (2003) Role of α-synucleincarboxyterminus on fibril formation in vitro. Biochemistry, 42, 8530-8540. doi:10.1021/bi027363r
- [89] Hoyer, W., et al. (2004) Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry, 43, 16233-16242. doi:10.1021/bi048453u
- [90] Ulusoy, A., et al. (2010) Co-expression of C-terminal truncated alpha-synuclein enhances full-length alphasynuclein-induced pathology. The European Journal of Neuroscience, 32, 409-422. doi:10.1111/j.1460-9568.2010.07284.x
- [91] Uéda, K., et al. (1993) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 90, 11282- 11286. doi:10.1073/pnas.90.23.11282
- [92] Yamin, G., Uversky, V.N. and Fink, A.L. (2003) Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Letters, 542, 147- 152. doi:10.1016/S0014-5793(03)00367-3
- [93] Uversky, V.N., et al. (2005) Effects of nitration on the structure and aggregation of alpha-synuclein. Brain research. Molecular Brain Research, 134, 84-102. doi:10.1016/j.molbrainres.2004.11.014
- [94] Paleologou, K.E., et al. (2008) Phosphorylation at Ser- 129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. The Journal of Biological Chemistry, 283, 16895-16905. doi:10.1074/jbc.M800747200
- [95] Kessler, J.C., Rochet, J.-C. and Lansbury, P.T. (2003) The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry, 42, 672-678. doi:10.1021/bi020429y
- [96] Michell, A.W., et al. (2007) The effect of truncated human alpha-synuclein (1-120) on dopaminergic cells in a transgenic mouse model of Parkinson’s disease. Cell Transplantation, 16, 461-474.
- [97] Wakamatsu, M., et al. (2008) Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiology of Aging, 29, 574-585. doi:10.1016/j.neurobiolaging.2006.11.017
- [98] Daher, J., Ying, M. and Banerjee, R. (2009) Conditional transgenic mice expressing C-terminally truncated human α-synuclein (αSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Molecular Neurodegeneration, 4, 34. doi:10.1186/1750-1326-4-34
- [99] Muntané, G., Ferrer, I. and Martinez-Vicente, M. (2012) α-synuclein phosphorylation and truncation are normal events in the adult human brain. Neuroscience, 200, 106- 119. doi:10.1016/j.neuroscience.2011.10.042
- [100] Tofaris, G.K., Layfield, R. and Spillantini, M.G. (2001) alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Letters, 509, 22-26. doi:10.1016/S0014-5793(01)03115-5
- [101] Liu, C-W., et al. (2003) Endoproteolytic activity of the proteasome. Science, 299, 408-411. doi:10.1126/science.1079293
- [102] Machiya, Y., et al. (2010) Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. The Journal of Biological Chemistry, 285, 40732-40744. doi:10.1074/jbc.M110.141952
- [103] Mishizen-Eberz, A.J., et al. (2003) Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. Journal of Neurochemistry, 84, 836- 847. doi:10.1046/j.1471-4159.2003.01878.x
- [104] Mishizen-Eberz, A.J., et al. (2005) Cleavage of alphasynuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry, 44, 7818-7829. doi:10.1021/bi047846q
- [105] Kim, H., et al. (2006) Calpain-resistant fragment (s) of [alpha]-synuclein regulates the synuclein-cleaving activiity of 20S proteasome. Archives of Biochemistry and Biophysics, 455, 40-47. doi:10.1016/j.abb.2006.08.019
- [106] Sevlever, D. and Jiang, P. (2008) Cathepsin D Is the main lysosomal enzyme involved in the degradation of α- synuclein and generation of its carboxy-terminally truncated species. Biochemistry, 47, 9678-9687. doi:10.1021/bi800699v
- [107] Qiao, L., et al. (2008) Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Molecular Brain, 1, 17. doi:10.1186/1756-6606-1-17
- [108] Cullen, V., et al. (2009) Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Molecular Brain, 2, 5. doi:10.1186/1756-6606-2-5
- [109] Sung, J., et al. (2005) Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. The Journal of Biological Chemistry, 280, 25216-25224. doi:10.1074/jbc.M503341200
- [110] Iwata, A., et al. (2003) Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Human Molecular Genetics, 12, 2625- 2635. doi:10.1093/hmg/ddg283
- [111] Kasai, T., et al. (2008) Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neuroscience Letters, 436, 52-56. doi:10.1016/j.neulet.2008.02.057
- [112] Tatebe, H., et al. (2010) Extracellular neurosin degrades α-synuclein in cultured cells. Neuroscience Research, 67, 341-346. doi:10.1016/j.neures.2010.04.008
- [113] Sung, J., et al. (2005) Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. The Journal of Biological Chemistry, 280, 25216-25224. doi:10.1074/jbc.M503341200
- [114] Levin, J., et al. (2009) Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Experimental Neurology, 215, 201-208. doi:10.1016/j.expneurol.2008.10.010
- [115] Choi, D.-H., et al. (2011) Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. The Journal of Biological Chemistry, 286, 14168-14177. doi:10.1074/jbc.M111.222430
- [116] Kim, K.S., et al. (2012) Proteolytic cleavage of extracellular α-synuclein by plasmin: Implications for Parkinson’s disease. The Journal of Biological Chemistry, 287, 24862- 24872. doi:10.1074/jbc.M112.348128
- [117] Ohno, S., et al. (1984) Evolutionary origin of a calcium-dependent protease by fusion of genes for a thiol protease and a calcium-binding protein? Nature, 312, 566-570. doi:10.1038/312566a0
- [118] Suzuki, K., et al. (1988) Regulation of activity of calcium activated neutral protease. Advances in Enzyme Regulation, 27, 153-169. doi:10.1016/0065-2571(88)90015-5
- [119] Borghi, R., et al. (2000) Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neuroscience Letters, 287, 65-67. doi:10.1016/S0304-3940(00)01153-8
- [120] El-Agnaf, O.M.A., et al. (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB Journal, 17, 1945-1947.
- [121] Lee, H.-J., Patel, S. and Lee, S.-J. (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. The Journal of Neuroscience, 25, 6016-6024. doi:10.1523/JNEUROSCI.0692-05.2005
- [122] Yamashiro, K., et al. (1997) Molecular cloning of a novel trypsin-like serine protease (neurosin) preferentially expressed in brain. Biochimicaet Biophysica Acta, 1350, 11-14. doi:10.1016/S0167-4781(96)00187-X
- [123] Blobel, C.P. (2000) Remarkable roles of proteolysis on and beyond the cell surface. Current Opinion in Cell Biology, 12, 606-612. doi:10.1016/S0955-0674(00)00139-3