Case Reports in Clinical Medicine
Vol.04 No.05(2015), Article ID:56675,4 pages
10.4236/crcm.2015.45037
Ataxia Telangiectasia Syndrome Revealed by Severe Pneumonia
Hind Serhane1, Nisserine Louhab2, Hafsa Sajiai1, Selma Aitbatahar1, Lamyae Amro1
1Department of Respiratory Medicine, Laboratory PCIM, Faculty of Medicine and Pharmacy, Cadi Ayyad University, Marrakesh, Morocco
2Department of Neurological Medicine, Faculty of Medicine and Pharmacy, Cadi Ayyad University, Marrakesh, Morocco
Email: hindserhane8@gmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 April 2015; accepted 22 May 2015; published 26 May 2015
ABSTRACT
Ataxia Telangiectasia (AT) is a rare autosomal recessive multisystem disease. The diagnosis is often made on a clinical triad that combines neurological signs dominated by a progressive cerebellar ataxia, oculocutaneous signs (telangiectasia, coffee stain milk), immunodeficiency (humoral and cellular) with sinopulmonary infections and elevated alphaphetoprotein. The diagnosis of AT is usually early, however, some forms may be revealed late. We reported a case of a 19-year-old patient, admitted for severe pneumonia with Klebsiella Pneumonia. In its history, it was found a notion of recurrent respiratory infections and bronchiectasis. In its clinical examination, it had been discovered cerebellar ataxia and occulocutaneous telangiectasia. The determination of plasmatic alphafoetoprotein was elevated, and the search of immunodeficiency showed a mixed deficit (humoral and cellular) suggesting the diagnosis of AT.
Keywords:
Ataxia Telangiectasia, Cerebellar Ataxia, Occulocutaneous Telangiectasia, Alphaphotoprotein, Immunodeficiency, Autosomal Recessive Disease

1. Introduction
Ataxia Telangiectasia (AT) is a rare autosomal recessive multisystem disease. Its prevalence is about 1/40,000 to 1/300,000 live births [1] [2] . Diagnosis of AT is often made on a clinical triad that combines neurological signs dominated by a progressive cerebellar ataxia, oculocutaneous signs (telangiectasia, coffee stain milk), immunodeficiency (humoral and cellular) with sinopulmonary infections and elevated alphaphetoprotein [3] . It usually begins around the age of 5 years old [4] , it is more apparent to the age of 10 years old [5] , but the mean age of diagnosis is on average 3 years old [3] . Furthermore, the later forms are possible [4] . We report the case of an AT syndrome revealed at the age of 19 years old during a severe pneumonia with Klebsiella pneumoniae.
2. Case Report
A 19-year-old North African patient, born of a non-consanguineous marriage, is the eldest in a family of three, his two brothers are healthy. He was admitted in an array of severe pneumonia consisting of nux and moderate hemoptysis and right chest pain in context of fever sensations and decline in general health. In its history, it was found a notion of recurrent and dragging respiratory infections on a plot of bronchiectasis with a psychomotor retardation discovered at the age of 7 years old. Clinical examination noted polypnea to 35 cycles/minute (normal respiratory rate is 16 - 20 cycles/minute), a blood pressure at 110/60 mm Hg (the normal rate is <120 mmHg for systolic and <80 mmHg for diastolic), a fever at 39.8˚C, an oxygen saturation of 96% (normal rate is between 95% and 100%), and incomplete condensation syndrome in right basi-thoracic, a finger clubbing and failure to thrive at −2 SD, with cerebellar ataxia, ocular bobbing in neurological examination. The mucocutaneous examination noted ocular telangiectasia and in the ear lobes. A Chest X-ray showed opacity with air fluid level in right basithoracic, associated with blunting ipsilateral pleural cul-de-sac (Figure 1). Chest computerized tomography (CT) was objectified tissue density opacity with excavation and signs for bronchiectasis (Figure 2). Laboratory tests showed leukocytosis at 14,200 cells/mm3. C-reactive protein was very high at 256.75 mg/l. Probabilistic antibiotic therapy was started with amoxicillin clavulanic acid intravenously at 1000 mg/8 hours, without improvement after 48 hours. The search for Mycobacterium tuberculosis (BK) on direct microscopic examination was negative. The cytobacteriological sputum had isolated a gram-positive cocci in favor of Klebsiella pneumoniae resistant to penicillin but susceptible to cephalosporin. The patient was put under 3rd generation of cephalosporin with good clinical, biological and radiological outcome. Before the notion of repeated respiratory infection, bronchectasis, cerebellar ataxia, cutaneous telangiectasia, diagnosis of Ataxia Telangiectasia syndrome was discussed. The α-fetoprotein was high at 570 ng/ml. The search of combined immunodeficiency showed rates of IgA, IgM and IgG respectively at 7 g/l; 1.76 g/l; 11.3 g/l. The CD4/CD8 ratio was down to 0.6. Brain Magnetic Resonance Imaging (MRI) showed cerebellar atrophy. Oral consent was obtained from the patient’s family to publish this case.
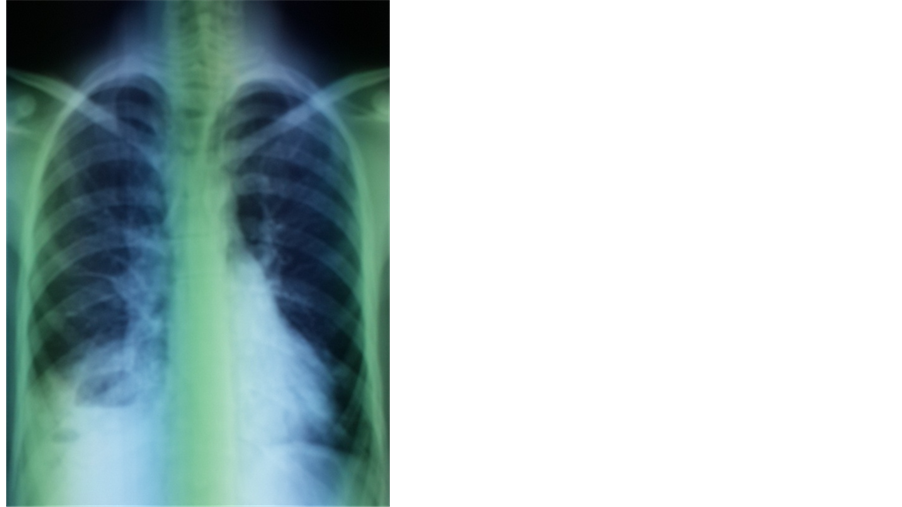
Figure 1. Opacity with air fluid level in right basithoracic, associated with blunting ipsilateral pleural cul-de-sac.
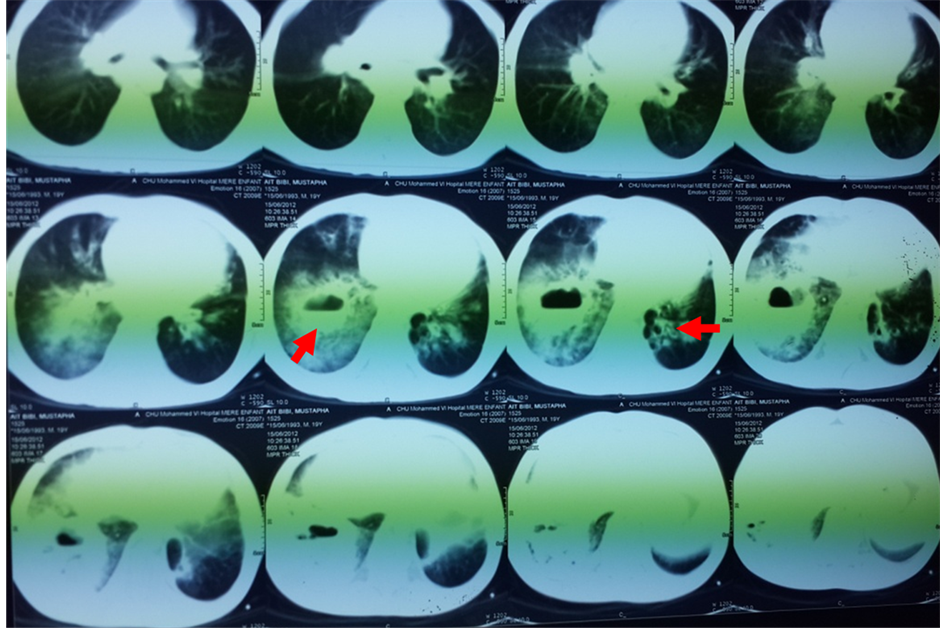
Figure 2. Tissue density opacity with excavation and signs for bronchiectasis.
3. Discussion
The ataxia telangiectasia (AT) is a rare autosomal recessive disease, relatively common in the Mediterranean area [6] . Described for the first time by Louis-Bar in 1941 [7] , it is a monogenic disease whose gene is located in 11q22-23 that was cloned in 1995 by Savitsky et al. [8] . The diagnosis of this disease is usually based on a clinical triad of neurologic signs, dominated by an evolutive static cerebellar syndrome, consisting of dyssynergia, intentional tremor, adiadochokinesia, and complete hypotonia leading to loss of ambulation generally diagnosed around the age of 2 years old and bedridden state in adolescence with stature-weight impact. The clinical array also includes oculocutaneous signs (telangiectasia, coffee stain milk), immunodeficiency responsible to repeated sinopulmonary infections and elevated alphaphetoprotein. It was the case of our patient, who sent to us for severe pulmonary infection and in whom we found on clinical examination cerebellar ataxia and oculo-cu- taneous telangiectasia reflecting an AT syndrome. Actually, the AT is considered as a disease entity in its own right contained in both serious constitutional immunodeficiencies that in the group of autosomal recessive cerebellar ataxias [6] . The neurological symptoms can be explained by the mutation of the gene responsible for ATM, in homozygous state, to systematic elimination of certain cell population. This is expressed by a progressive atrophy of cerebellar cortex, a destruction of the basal ganglia, a demyelination of the posterior columns and a loss of neurons in the anterior horn of the spinal cord [9] . Oculocutaneous signs [10] , as telangiectasia, are the second signs after neurological manifestations, these are most often inaugural. Telangiectasia are generally present but inconsistent. They are first in the eyes and then appear on the skin especially in the ear lobes. Other cutaneous signs as coffee stain milk, vitiligo, early skin atrophy, early onset of gray hair; warty lesions are also described [9] . The immunodeficiency is very common in AT, which is the case of our patient, who presented respiratory infections since childhood highly suggestive of immunodeficiency. This deficit is often mixed, progressively worsening, on cellular immunity (CD4 and CD8) but also on humoral immunity, reaching immunoglobulin A and subclasses of immunoglobulin G [3] . The IgM levels are normal or sometimes high. This immune deficiency is responsible for respiratory disorders that are common and may precede the onset of neurological signs. It is often repeated bronchial infections, diffuse lung diseases, bronchiectasis, and rarely interstitial chronic syndrome. It was noted that patients with AT also exhibited increased sensitivity to ionizing radiation, most notably γ and X. Thus, radiologic examination should be limited to the maximum in these patients [11] . A high of development cancer has been reported in patients homozygous (100 times greater in the normal population) [12] . This predisposition is partly due to the increased radiosensitivity but especially acquired chromosomal abnormalities [6] . For homozygous individuals, it is essentially lymphoma (50%), lymphoid leukemia (30%), and carcinomas (20% especially in adults) [13] [14] . Among females heterozygous for the gene mutation, breast cancers are more often seen than in the general population [3] .
The management of AT is multidisciplinary, requiring neurologically, motor physiotherapy and nutrition program (fight against inhalations and reflux) [3] [15] . Neurological drugs (dopaminergic) did not demonstrate their effectiveness [3] . Active respiratory management should also be considered (chest physiotherapy and antibiotics in case of infection). Regular pulmonary monitoring should be installed; hypoxemia requires the prescription of long-term oxygen therapy and radiological investigations should be limited to the maximum. Replacement therapy of polyvalent immunoglobulins can be started when a humoral deficiency is diagnosed [2] . Cancer treatment must be done in centers with experience in such patients, because of their sensitivity of ionizing radiation.
4. Conclusion
Ataxia Telangiectasia is a rare, multisystemic disease. Its management is multidisciplinary. Its prognosis is poor, depends primarily on respiratory infections and the susceptibility to cancer. Diagnosis is usually early, late forms of revelations have also been described, which is the case of our patient.
Conflicts of Interests
I declare that I have no conflict of interests here.
References
- Chun, H.H. and Gatti, R.A. (2004) Ataxia-Telangiectasia, an Evolving Phenotype. DNA Repair, 3, 1187-1196. http://dx.doi.org/10.1016/j.dnarep.2004.04.010
- Taylor, A.M. and Byrd, P.J. (2005) Molecular Pathology of Ataxia-Telangiectasia. Journal of Clinical Pathology, 58, 1009-1015. http://dx.doi.org/10.1136/jcp.2005.026062
- Bott, L., Thumerelle, C., Cuvellier, C., Deschildre, A., Vallée, L. and Sardet, A. (2006) Ataxie-télangiectasie: de la clinique à la physiopathologie. Archives de péDiatrie, 13, 293-298. http://dx.doi.org/10.1016/j.arcped.2005.11.022
- Tranchant, C. and Anheim, M. (2009) Ataxies cérébelleuses autosomiques récessives. Presse Med, 38, 1852-1859. http://dx.doi.org/10.1016/j.lpm.2009.01.025
- Perlman, S., Becker-Catania, S. and Gatti, R.-A. (2003) Ataxia-Telangiectasia: Diagnosis and Treatment. Seminars in Pediatric Neurology, 10, 173-182. http://dx.doi.org/10.1016/S1071-9091(03)00026-3
- Chaouki, S., Benjelloun Dakhama, B.S., Alaoui, K., Arqam, L., Atmani, S., Bouharrou, A., Hida, M. and Bounasse, M. (2008) Ataxie télangiectasie: cas cliniques et revue de la littérature. Journal de pédiatrie et de puériculture, 21, 98-101. http://dx.doi.org/10.1016/j.jpp.2007.12.001
- Louis-Bar, D. (1941) Sur un syndrome progressif comprenant des télangiectasies capillaires cutanées et conjonctivales symétriques, à disposition naevoïde et des troubles cérébelleux. Confin Neurol, 4, 32-42. http://dx.doi.org/10.1159/000106149
- Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., et al. (1995) A Single Ataxia-Telangiectasia Gene with a Product Similar to PI-3 Kinase. Science, 268, 1749-1753. http://dx.doi.org/10.1126/science.7792600
- Tavani, F., Zimmerman, R.A., Berry, G.T., Sullivan, K., Gatti, R. and Bin-gham, P. (2003) Ataxia-Telangiectasia: The Pattern of Cerebellar Atrophy on MRI. Neuroradiology, 45, 315-319.
- Farr, A.K., Shalev, B., Crawford, T.O., Lederman, H.M., Winkelstein, J.A. and Repka, M.X. (2002) Ocular Manifestations of Ataxia-Telangiectasia. American Journal of Ophtalmology, 134, 891-896. http://dx.doi.org/10.1016/S0002-9394(02)01796-8
- Kühne, M., Riballo, E., Rief, N., Rothkamm, K., Jeggo, P.A. and Löbrich, M. (2004) A Double-Strand Break Repair Defect in ATM-Deficiency Cells Contributes to Radiosensitivity. Cancer Research, 64, 500-508. http://dx.doi.org/10.1158/0008-5472.CAN-03-2384
- Thompson, D., Duedal, S., Kirner, J., McGuffog, L., Last, J., Reiman, A., et al. (2005) Cancer Risks and Mortality in Heterozygous ATM Carriers. Journal of the National Cancer Institute, 97, 813-822. http://dx.doi.org/10.1093/jnci/dji141
- De Laet, C., Casimir, G., DuchPteau, J., Vamos, E., Devaick, C., Sariban, E. and Feaster, A. (1996) Lymphome T leucémisé comme première manifestation d’une ataxie télangiectasie. Archives de péDiatrie r, 3, 681-684. http://dx.doi.org/10.1016/0929-693X(96)87089-8
- Cavaciuti, E., Lauge, A., Janin, N.,Ossian, K., Hall, J., Stoppa-Lyonnet, D. and Andrieu, N. (2005) Cancer Risk According to Type and Location of ATM Mutation in Ataxia-Telangiectasia Families. Genes, Chromosomes Cancer, 42, 1-9. http://dx.doi.org/10.1002/gcc.20101
- Lefton-Greif, M.A., Crawford, T.O., Winkelstein, J.A., et al. (2000) Oropharyngeal Dysphagia and Aspiration in Patients with Ataxia-Telangiectasia. The Journal of Pediatrics, 136, 225-231. http://dx.doi.org/10.1016/S0022-3476(00)70106-5