Pharmacology & Pharmacy
Vol.4 No.3(2013), Article ID:32557,9 pages DOI:10.4236/pp.2013.43044
Motor Effects of 1,3-Disubstituted 8-Styrylxanthines as A1 and A2 Adenosine-Receptor Antagonists in Rats
![]()
1Facultad de Ciencias Químicas, Benemérita Universidad Autónoma de Puebla, Puebla, México; 2Instituto Nacional de Rehabilitación, Unidad de Ingeniería de Tejidos, Terapia Celular y Medicina Regenerativa, México DF, México.
Email: *ilhlimon@yahoo.com.mx
Copyright © 2013 Ilhuicamina Daniel Limón-Pérez de León et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 2nd, 2013; revised May 7th, 2013; accepted May 16th, 2013
Keywords: Xantines; Adenosine Receptors Antagonists; Turning Behavior; Anti-Parkinsonian Drugs
ABSTRACT
A series of 1,3-substituted 8-styrylxanthines (11a-d) was synthesized, under chemoand regioselective conditions, in a good overall yield. The compounds showed affinity towards both A1 and A2A-adenosine receptors by radioligand binding by means of in vitro assays. The (E)-3-ethyl-1-propyl-8-styrylxanthine (11a) showed the greatest affinity towards the A2A receptor, whereas (E)-3-pentyl-1-propyl-8-styrylxanthine (11d) showed the greatest affinity for the A1 receptor. When the 8-styrylxanthines 11a (A15Et) and 11c (A15Bu) were administrated in rats, which were previously injured with 6-hydroxydopamine at the substantia nigra pars compacta (SNc), the turning behavior decreased 50%. Based on these results we propose to A15Et as a potential compound to treat some symptoms of Parkinson’s disease.
1. Introduction
The nucleoside adenosine exerts a modulatory influence on the central nervous system by the activation of the G-protein-coupled receptors, through a mechanism involving vasodilatation, inhibition of lipolysis, and insulin release [1,2]. Most of the actions of adenosine are mediated by four extra-cellular receptors [3] termed A1, A2A, A2B and A3. The activation of A1 and A3 receptors inhibits adenylate cyclase through Gi coupling, and activation of A2A and A2B receptors stimulates adenylate cyclase through Gs coupling [4]. The A1 receptors are widely distributed throughout the brain, where they regulate neurotransmitter release and neuronal firing by activation of potassium channels. The distribution of A2A is heterogeneous with high levels of expression in the nucleus accumbens, olfactory tubercle, globus pallidus and striatum [5,6] where A2A receptors are co-localized with dopamine D2 receptors in the striato-pallidal terminal [7]. The A2A-D2 interaction seems to be more potent in striatal dopamine denervation with supersensitive D2 receptors [8]. This fact has been confirmed in the course of the binding assay of the selective agonists and antagonists [6,8,9]. When considered together with the important role of dopamine in the control of motor activity and in the etiology and management of Parkinson’s disease (PD), this observation suggested that adenosine A2A receptors could be a novel target for drugs that manage movement disorders [9-11].
Results from recent PD studies on animal models suggest that antagonism of the A2A receptor may also protect against underlying neuronal degenerative processes [9, 12].
The blockade of A2A receptor reverses damage by excitotoxicity [13] and counteracts the motor deficits produced by dopaminergic toxin [14].
Recent preclinical and clinical data suggest that A2A antagonist may provide a complementary therapy for PD [11,15,16]. Furthermore the A2A receptor participates as a neuroprotector agent [12,14,15]. The (E)-8-(3-chlorostyryl)caffeine (CSC) has been employed as a selective inhibitor of the A2A subtype of adenosine receptor [17]. However several analogues of CSC act as inhibitors of monoamino oxidase B (MAOB) [18-23]. Because of their role in the metabolism of monoamines and the inhibition of MAOB, CSC is being used in preclinical studies for treatment of symptoms of PD [20-23].
Therefore the protective effect may be related to the ability of the A2A antagonists to antagonize the increase of toxicity of glutamate induction by quinolinic acid. Recently, some researchers have proposed that the inhibitors of MAOB, caffeine and their derivatives reduce the risk of developing PD [23] and produce an anxiolytic effect [22]. Current data suggest that adenosine acts on dopamine regulation and dopaminergic function, and that the antagonists of the adenosinergic receptor decrease the dopaminergic activity [23].
For the above mentioned reasons, A2A receptors have become important targets for development of drugs for the treatment of PD [10,23]. Some structures have shown affinity and selectivity towards A2A receptors [9,19,24, 25], as xanthines, adenines, 1,2,4-triazolo[1,5-a]quinoxalines and pyrazolo[3,4-d]-pyrimidines (Figure 1). Pharmacological studies suggested that derivates of 8-styrylxanthine have biological activity as an anti-Parkinsonian drug [26-28].
In this paper we report the synthesis, binding studies and pharmacology properties of 8-styrylxanthines. We found that (E)-3-ethyl-1-propyl-8-styrylxanthine (A15Et, compound 11a) showed the greatest affinity for the A2A receptor, also the biological experiments showed a decrease in the turning behavior, so we suggest that this compound represent a potential drug for treatment of Parkinson’s disease.
2. Materials and Methods
2.1. Synthesis of Chemical Compounds
The melting points were measured with a Melt-temp apparatus and were not corrected. The NMR spectra were recorded on a Varian spectrometer; 1H spectra at 400 MHz and 13C at 100 MHz. DMSO-d6 and CDCl3 were used as solvents. All chemical shifts are quoted in ppm and the coupling constants are expressed in Hertz. The chemical shift of the remaining no deuterated protons of DMSO-d6 served as an internal standard (d 1H: 2.50, 13C: 39.7). The IR spectra were obtained from a Shimadzu FT-IR 8400 spectrophotometer, using KBr pellets; wavenumbers of maximum absorption peaks are reported as cm−1. The UV spectra were recorded in a Perkin Elmer Lambda Bio 40 apparatus, using ethanol as solvent; lmax is described in nm. TLC was performed on 60 F254 silica gel plates.
2.1.1. Synthesis of 6-Amino-3-propyluracil (6)
A suspension of 6-aminouracil (5) (5.08 g, 40 mmol) and (NH4)2SO4 (250 mg) in hexamethyldisilazane (14 mL, 60 mmol), was refluxed for 45 min. In this time the mixture became transparent indicating that all 5 was trisilylated. The crude was allowed to cool to 45˚C; then, the npropyl iodide (1.9 mL, 20 mmol) was added. The reaction was refluxed for 2 h. The crude was treated with an aqueous solution of Na2S2O3 (6 g/20 mL). Additionally, a saturated aqueous solution of NaHCO3 (20 mL) was added drop wise until effervescence ceased. The suspension obtained was filtered, and the white precipitate was washed with cold water, diethyl ether, and hexane. Yield: 92%; mp 267˚C - 269˚C (lit. 275˚C). 1H-NMR (DMSO-d6) d: 6.32 (2H, b, NH2), 4.53 (1H, s, H-5), 3.59 (2H, t, EtCH2N, J = 7.3 Hz), 1.46 (2H, m, MeCH2CH2N), 0.80, (3H, t, CH3(CH2)2N, J = 7.3 Hz); 13C-NMR (DMSO-d6) d: 163.7 (C-4), 154.5 (C-2), 151.9 (C-6), 74.7 (C-5), 40.2 (EtCH2N), 21.5 (MeCH2CH2N), 11.7 (CH3CH2CH2N); IR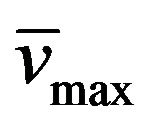 : 2930, 1741, 1631; UV lmax(e): 261(3500).
: 2930, 1741, 1631; UV lmax(e): 261(3500).
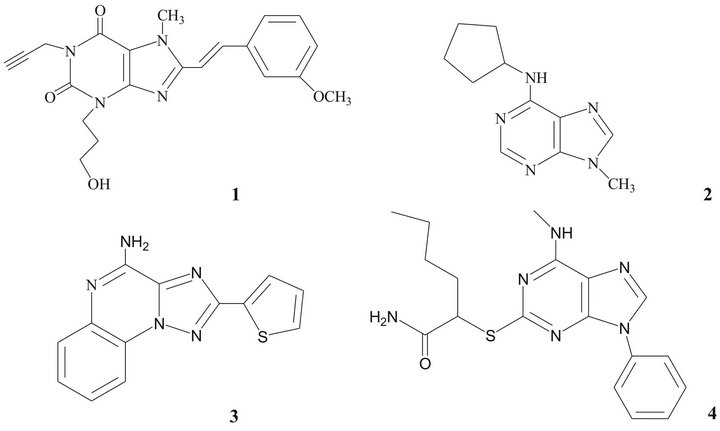
Figure 1. Compounds showing selectivity towards A2A receptors.
2.1.2. Synthesis of 6-Amino-5-nitroso-3-propyluracil (7)
The uracil 6 (0.84 g, 5 mmol) was dissolved in 80% aq AcOH (10 mL)/MeOH (15 mL) warm solution. The solution was cooled down to room temperature and a solution of NaNO2 (0.69 g, 10 mmol) in water (5 mL) was added drop-wise. The yellow precipitate was collected by filtration and washed with water (2 × 20 mL) and diethyl ether (2 × 20 mL). Yield: 85%; mp > 300˚C; 1H-NMR (DMSO-d6) d: 11.37 (1H, b, H-N), 3.79 (2H, t, EtCH2N, J = 7.3), 1.60 (2H, m, MeCH2CH2N), 0.89 (3H, t, CH3CH2CH2N, J = 7.3); NMR 13C (DMSO-d6) d: 161.8 (C-4), 150.3 (C-2), 145.5 (C-6), 140.3 (C-5), 40.2 (EtCH2N), 21.4 (MeCH2CH2N), 11.8 (CH3CH2CH2N); IR `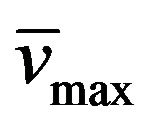 : 3294, 3185, 1511; UV lmax(e): 320(5920).
: 3294, 3185, 1511; UV lmax(e): 320(5920).
2.1.3. Synthesis of 5,6-Diamino-3-propyluracil (8)
The nitroso compound 7 (10 mmol, 1.98 g) was dissolved in 6% aq NH4OH (40 mL) at 80˚C, then Na2S2O3 (30 mmol, 3.5 g) was added in small portions. The solution decolorizes rapidly. The warm crude was concentrated under reduced pressure; a precipitate is produced. After filtration, the white precipitate was washed with a small amount of cold water (20 mL). Compound 8 is very sensitive to air and to CDCl3 solutions, so it was immediately used in the following step. Yield: 69%.
2.1.4. Synthesis of (E)-6-amino-3-propyl-5- styrylcarboxamidouracil (9)
A suspension of the diamino compound 8 (7 mmol, 1.28 g), EDCl (7 mmol, 1.34 g), and (E)-cinnamic acid (7 mmol, 1.03 g) in CH3OH (20 mL) was stirred at room temperature overnight. The precipitate obtained was collected by filtration, washed with MeOH, water, and diethyl ether. Yield: 76%; mp > 300˚C; 1H-NMR (DMSOd6) d: 8.7 (1H, b, HN in styrylamido group), 7.41 (6H, m, aromatics and H-vinylic), 6.85 (1H, d, vinylic, Jtrans = 16), 6.11 (2H, b, -NH2), 3.6 (2H, t, CH3CH2CH2N, J = 7.3), 1.5 (2H, m, CH3CH2CH2N), 0.81 (3H, t, CH3CH2CH2N, J = 7.3); 13C-NMR (DMSO-d6) d: 165.6 (C=O in styrylamido group), 161.2 (C-4), 150.5 (C-6), 150.4 (C-2), 139.4 (Ph-HC=), 135.5 (C-ipso), 130.0 (C-meta), 129.6 (C-orto), 128.0 (C-para), 122.9 (=CH-), 87.7 (C-5), 40.2 (EtCH2N), 21.5 (MeCH2CH2N), 11.8 (CH3CH2CH2N); IR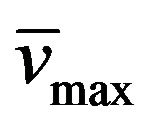 : 3318, 3187, 1648; UV lmax(e): 280(6670).
: 3318, 3187, 1648; UV lmax(e): 280(6670).
2.1.5. General Procedure for the Preparation of 1-Substituted (E)-6-amino-3-propyl-5- styrylcarboxamidouracils 10a-d
Compound 9 (1 mmol, 0.314 g) was dissolved in DMF (3.5 mL), then K2CO3 (1.1 mmol, 0.16 g) and the alkyl halide (1.1 mmol) were added. The mixture was stirred at room temperature for 48 h and the progress of the reaction was monitored by TLC (eluent: acetone/hexane, 2:3). After addition of water (30 mL) a precipitate was produced, which was collected by filtration and washed with water (2 × 20 mL) and dried in a vacuum desiccator.
1) (E)-6-amino-1-ethyl-3-propyl-8-styrylcarboxa-midouracil (10a)
Yield: 78%; mp 284˚C - 286˚C; 1H-NMR (CDCl3) d: 8.04 (1H, b, NH), 7.63 (1H, d, -CH=), 7.36 (5H, m, aromatics), 6.70 (1H, d, -CH=), 5.83 (2H, b, -NH2), 4.28, (2H, q, CH3CH2N), 4.12, (2H, t, CH3CH2CH2N), 1.79 (2H, m, CH3CH2CH2N), 1.42 (3H, t, CH3CH2N); 0.99 (3H, t, CH3CH2CH2N); 13C NMR (CDCl3) d: 166.0 (C=O styrylamido), 160.3 (C-4), 152.5 (C-6), 150.7 (C-2), 141.6 and 119.8 (-CH=CH-), 134.3, 129.6, 128.5, 127.7 (aromatics), 95.4 (C-5), 43.4 (CH3CH2CH2N), 39.1 (CH3CH2N), 21.1 (CH3CH2CH2N), 13.5 (CH3CH2N), 11.7 (CH3CH2CH2N); IR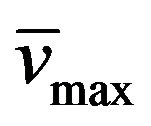 : 3165, 1734, 1651, 968; UV (EtOH) lmax(e): 278(11620).
: 3165, 1734, 1651, 968; UV (EtOH) lmax(e): 278(11620).
2) (E)-6-amino-1,3-dipropyl-8-styrylcarboxami-douracil (10b)
Yield: 75%; mp 272˚C - 273˚C; 1H-NMR (CDCl3) d: 8.40 (1H, b, NH styrylamido), 7.54 and 6.74 (d, 1H, -CH=CH-, Jtrans = 16.4 Hz), 7.31 (m, 5H, aromatics), 6.11 (b, 2H, -NH2), 4.14 (m, 4H, both CH3CH2CH2N), 1.86 (m, 2H, CH3CH2CH2N), 1.78 (t, 2H, CH3CH2CH2N, J = 7.6 Hz), 1.02 (t, 3H, CH3CH2CH2N, J = 7.6 Hz), 0.96 (t, 3H, CH3CH2CH2N, J = 7.6 Hz); 13C NMR (CDCl3) d: 166.0 (C=O styrylamido), 159.8 (C-4), 149.9 (C-6), 147.1 (C-2), 141.7 and 119.9 (-CH=CH-), 134.5, 129.7, 128.8, and 127.8 (aromatics), 89.7 (C-5), 43.8 (CH3CH2CH2N), 43.5 (CH3CH2CH2N), 21.6 (CH3CH2CH2N), 20.2 (CH3CH2CH2N), 13.9 (CH3CH2CH2N), 11.5 (CH3CH2CH2N); IR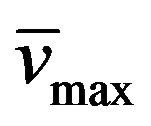 : 3024, 2962, 1735, 1705, 1651, 972; UV (EtOH) lmax(e): 279(9130).
: 3024, 2962, 1735, 1705, 1651, 972; UV (EtOH) lmax(e): 279(9130).
3) (E)-6-amino-1-butyl-3-propyl-8-styryluracil (10c)
Yield: 66%; mp 273˚C - 274˚C; 1H-NMR (CDCl3) d: 8.14 (b, 1H, N-H styrylamido), 7.55 (d, 1H, Ph-CH=, J = 15.3 Hz), 6.76 (d, 1H, =CH-, J = 15.3 Hz), 7.30 (m, 5H, arom.), 5.82 (b, 2H, -NH2), 3.78, (m, 4H, CH3CH2CH2N and CH3CH2CH2CH2N), 1.58 (m, 4H, CH3CH2CH2N and CH3CH2CH2CH2N), 1.36 (m, 2H, CH3CH2CH2CH2N), 0.91 (t, 3H, CH3CH2CH2N, J = 7.3 Hz), 0.88 (t, 3H, CH3CH2CH2CH2N, J = 7.3 Hz); 13C NMR (CDCl3) d: 165.9 (C=O styrylamido), 160.1 C-4), 160.1 (C-4), 150.1 (C-6), 148.5 (C-2), 142.2 and 119.9 (CH=CH), 134.5, 130.1, 128.9, and 128.0 (aromatics), 92.1 (C-5), 43.4 (CH3CH2CH2CH2N), 43.2 (CH3CH2CH2N), 30.1 (CH3CH2CH2CH2N), 21.6 (CH3CH2CH2CH2N), 21.2 (CH3CH2CH2N), 13.8 (CH3CH2CH2N), 11.4 (CH3CH2CH2CH2N); IR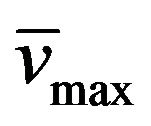 : 3319, 1731, 1652, 969; UV (EtOH) lmax(e): 279(8525).
: 3319, 1731, 1652, 969; UV (EtOH) lmax(e): 279(8525).
4) (E)-6-amino-1-pentyl-3-propyl-8-styryluracil (10d)
Yield: 70%; mp 258˚C - 260˚C; 1H-NMR (CDCl3) d: 8.04 (b, 1H, N-H styrylamido), 7.63 y 6.70 (d, 1H, CH=CH, Jtrans = 15.6 Hz), 7.35 (m, 5H, aromatics), 5.08 (b, 2H, -NH2), 4.28, (t, 2H, BuCH2N, J = 7.2 Hz), 4.12 (t, 2H, CH3CH2CH2N, J = 7.6 Hz), 1.79 (m, 4H, CH3CH2CH2N and PrCH2CH2N, J = 7.6 Hz), 1.42 (t, 3H, CH3CH2CH2N, J = 7.2 Hz); 0.97 (sx, 2H, MeCH2CH2CH2N), 0.87 (t, 3H, CH3CH2CH2CH2N, J = 7.6 Hz); 13C NMR (CDCl3) d: 166.0 (C=O styrylamido), 160.35 (C-4), 152.5 (C-6), 150.8 (C-2), 141.7 and 119.8 (CH=CH), 134.3, 129.6, 128.5, and 127.7 (aromatics), 95.4 (C-5), 43.4 (CH3CH2CH2N), 39.2 (BuCH2N), 21.1 (CH3CH2CH2N), 18.5 (PrCH2CH2N), 13.5 (EtCH3CH2CH2N), 11.7 (CH3CH2CH2N) 11.4 (CH3CH2CH2CH2CH2N); IR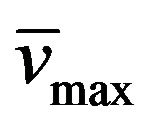 : 3165, 1734 1651, 968; UV (EtOH) lmax(e): 278(11620).
: 3165, 1734 1651, 968; UV (EtOH) lmax(e): 278(11620).
2.1.6. General Procedure for the Preparation of 3-Substituted (E)-1-propyl-8-styrylxanthines (11a-d)
Compounds 10a-d (1 mmol) were dissolved in a mixture of CH3OH (15 mL) and 20% aq KOH (5 mL) and solutions were refluxed for 3 h. The reaction was followed by TLC (eluent: acetone: hexane 2:3) and UV spectroscopy. After addition of conc. HCl (10 mL) a precipitate was obtained. The suspension was concentrated under vacuum. The precipitate was collected by filtration and washed with a small amount of cold water and MeOH.
1) (E)-3-ethyl-1-propyl-8-styrylxanthine (11a)
Yield: 65%; mp 277˚C - 278˚C (MeOH); 1H-NMR (CDCl3) d: 13.60 (bs, 1H, N-H), 7.67 and 7.07 (d, 1H, CH=CH, Jtrans = 16.4 Hz), 7.42 (m, 5H, aromatics), 4.07 (q, 2H, MeCH2N, J = 7.4 Hz), 3.85 (t, 3H, EtCH2N, J = 7 Hz), 1.58 (m, 2H, MeCH2CH2N), 1.25 (t, 3H, CH3CH2N, J = 7 Hz), 0.87 (t, 3H, CH3CH2CH2N, J = 7 Hz); 13C NMR (CDCl3) d: 154.5 (C-6), 151.0 (C-4), 150.2 (C-8), 148.6 (C-2), 135.9 and 116.2 (CH=CH), 135.7, 129.7, 129.6, and 127.7 (aromatics), 107.7 (C-5), 42.7 (EtCH2N), 38.8 (MeCH2N), 21.4 (MeCH2CH2N), 13.7 (CH3CH2N), 11.8 (CH3CH2CH2N); IR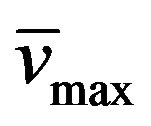 : 3318, 3187, 1648; UV lmax(e): 343(8660). Anal. Calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27; Found: C, 66.51; H, 6.09; N, 17.01.
: 3318, 3187, 1648; UV lmax(e): 343(8660). Anal. Calcd for C18H20N4O2: C, 66.65; H, 6.21; N, 17.27; Found: C, 66.51; H, 6.09; N, 17.01.
2) (E)-1,3-dipropyl-8-styrylxanthine (11b)
Yield: 72%; mp 267˚C - 269˚C (MeOH); 1H-NMR (CDCl3) d: 13.10 (bs, 1H, N-H), 7.80 and 7.13 (d, 1H, CH=CH, Jtrans = 16.4 Hz), 7.40 (m, 5H, aromatics), 4.14 (m, 4H, both CH3CH2CH2N), 1.85 (m, 2H, CH3CH2CH2N1), 1.76 (m, 2H, CH3CH2CH2N3), 0.99 (m, 6H, both CH3CH2CH2N); 13C NMR (CDCl3) d: 155.9 (C-6), 151.1 (C-4), 150.2 (C-8), 145.7 (C-2), 137.1 and 115.3 (CH=CH), 135.7, 129.4, 128.9, and 127.3 (aromatics), 107.2 (C-5), 45.5 (CH3CH2CH2N1), 38.8 (CH3CH2CH2N3), 21.4 (both CH3CH2CH2N), 11.5 (CH3CH2CH2N1), 11.3 (CH3CH2CH2N3); IR (KBr)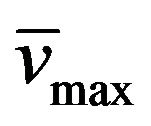 : 3318, 3187, 2961, 1702, 1651; UV (EtOH) lmax(e): 342(3585); Anal. Calcd for C19H22N4O2: C, 67.44; H, 6.55; N, 16.56; Found: C, 66.28; H, 6.48; N,16.37.
: 3318, 3187, 2961, 1702, 1651; UV (EtOH) lmax(e): 342(3585); Anal. Calcd for C19H22N4O2: C, 67.44; H, 6.55; N, 16.56; Found: C, 66.28; H, 6.48; N,16.37.
3) (E)-3-butyl-1-propyl-8-styrylxanthine (11c)
Yield: 69%; mp 278˚C - 279˚C (MeOH); 1H-NMR (CDCl3) d: 13.20 (bs, 1H, N-H), 7.77 and 7.09 (d, 1H, CH=CH, Jtrans = 16.4 Hz), 7.36 (m, 5H, aromatics), 4.19 (t, 2H, PrCH2N, J = 7.4 Hz), 4.11 (t, 2H, EtCH2N, J = 7.4 Hz), 1.80 (m, 4H, EtCH2CH2N and MeCH2CH2N), 1.43 (m, 2H, MeCH2CH2CH2N), 0.99 (m, 6H, both CH3CH2CH2CH2N); 13C NMR (CDCl3) d: 156.0 (C-6), 151.4 (C-4), 150.9 (C-8), 149.9 (C-2), 137.0 and 115.4 (CH=CH), 135.7, 129.4, 128.9, and 127.3 (aromatics), 107.4 (C-5), 43.8 (PrCH2N), 43.6 (EtCH2N), 30.2 (EtCH2CH2N), 21.5 (MeCH2CH2CH2N), 20.1 (MeCH2CH2N), 13.9 (CH3CH2CH2CH2N), 11.5 (CH3CH2CH2N); IR (KBr)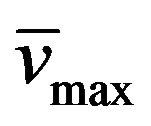 : 3318, 2958, 1702, 1649; UV (EtOH) lmax(e): 342(8785); Anal. Calcd for C20H24N4O2: C, 68.16; H, 6.86; N, 15.90; Found: C, 68.03; H, 6.89; N, 15.77.
: 3318, 2958, 1702, 1649; UV (EtOH) lmax(e): 342(8785); Anal. Calcd for C20H24N4O2: C, 68.16; H, 6.86; N, 15.90; Found: C, 68.03; H, 6.89; N, 15.77.
4) (E)-3-pentyl-1-propyl-8-styrylxanthine (11d)
Yield: 66%; 224˚C - 226˚C; 1H-NMR (CDCl3) d: 13.57 (bs, 1H, N-H), 7.63 and 7.06 (d, 1H, CH=CH, Jtrans = 16.4 Hz), 7.40 (m, 5H, aromatics), 4.01 (t, 2H, BuCH2N, J = 7 Hz), 3.85 (t, 2H, EtCH2N, J = 7 Hz), 1.70 (m, 2H, PrCH2CH2N), 1.56 (m, 2H, MeCH2CH2N), 1.31 (m, 4H, MeCH2CH2CH2CH2N), 0.87 (m, 6H, CH3(CH2)4N and CH3(CH2)2N); 13C-NMR (CDCl3) d: 154.4 (C-6), 151.2 (C-4), 150.1 (C-8), 148.9 (C-2), 138.9 and 116.4 (CH=CH), 135.7, 129.7, 129.5, and 127.7 (aromatics), 107.6 (C-5), 43.4 (BuCH2N), 42.7 (EtCH2N), 28.8 (PrCH2CH2N), 27.7 (EtCH2(CH2)2N), 22.3 (MeCH2(CH2)3N), 21.4 (MeCH2CH2N), 14.4 (CH3CH2CH2N), 11.7 (CH3(CH2)4N); IR (KBr)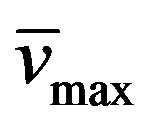 : 3318, 2958, 1702, 1649; UV (EtOH) lmax(e): 347(5520); Anal. Calcd for C21H26N4O2: C, 68.82; H, 7.15; N, 15.28; Found: C, 68.70; H, 7.01; N, 15.15.
: 3318, 2958, 1702, 1649; UV (EtOH) lmax(e): 347(5520); Anal. Calcd for C21H26N4O2: C, 68.82; H, 7.15; N, 15.28; Found: C, 68.70; H, 7.01; N, 15.15.
2.2. Binding Studies
Radioligand binding studies to determine the relative affinity of compounds 11a-d to A1 and A2A adenosine receptors were made using crude membrane preparations of rat cortical membrane and rat striatal membrane as previously described [29]. [3H]-2-chloro-N6-cyclopentyladenosine ([3H]CCPA) was used as A1 receptor ligand and [3H]-(E)-3-(3-hydroxypropyl)-8-(3-methoxystyryl)- 7-ethyl-1-propargylxanthine (MSX-2) as A2A receptor ligand. Radiolabelled compounds were purchased from NEN Life Sciences, Dreieich, Germany.
2.3. Rotational Behavior in Rats with a Unilateral Lesion Caused by 6-Hydroxydopamine
All procedures described in this study were approved by the BUAP Animal Care Committee and met governmental guidelines (Mexican Council for Animal Care, Norma.
Oficial Mexicana NOM-062-ZOO-1999). Male Wistar rats (from Bioterio Claude Bernard, BUAP) weighed 250 - 350 g at the beginning of the experiment. Animals were housed in pairs in ventilated sound-attenuated rooms under a 12 h light:dark cycle at an ambient temperature with free access to water and food. The animals were deeply anaesthetized with choral hydrate (350 mg/kg ip) and received an injection of 2 µL 6-hydroxydopamine (6-OHDA) over a period of 10 min (8 µg/µL dissolved in 0.1% ascorbic acid) into the left substantia nigra pars compacta (SNc), according to the atlas of Paxinos and Watson 1998 [30] (stereotaxic coordinates; A = 4.0 mm anterior to bregma, L = 1.3 mm side lateral to the midline, V = 6.5 below the skull) to produce a unilateral dopamine depletion.
Two weeks after the lesion caused by the 6-OHDA the rats were evaluated with methamphetamine (METH, 7 mg/kg sc) starting 10 minutes after injection. To measure turning behavior the rats were placed in a plastic chamber and the number of complete (360˚) turns, made over a period of 10 minutes, were counted. The mean number of ipsilateral rotations per minute was measured. This model is the one of the most reliable rodent models for the assessment of anti-Parkinsonian activity of the news drugs [31]. Only animals exhibiting at least 10 ipsilateral rotations/minute were used in subsequent experiments.
Ten days after the first evaluation of the turning behavior, the animals were administrated with (E)-3-ethyl- 1-propyl-8-styrylxanthine (A15Et, 11a) or (E)-3-butyl-1- propyl-8-styrylxanthine (A15Bu, 11c) at dose of 1 mg/kg sc or with vehicle (mineral oil + DMSO, 0.1 ml/100 g sc), and 10 min later the animals received METH (7 mg/kg sc) and a second turning behavior was evaluated over a period of 80 min.
2.4. Statistical Analysis
The results were expressed as the mean ± SEM. The statistical analysis for turning behavior was done using t of student, with p < 0.05 consider significant.
3. Results
3.1. Chemistry
The synthesis of the compounds 11a-d was accomplished following the general pathway [25,32,33] depicted in the Scheme 1, optimizing all involved reactions.
The alkylation of the N atom at position 3 in the 6- aminouracil (5) was highly selective using the known “one pot” procedure [32]: trisilylation of 5 by means of hexamethyldisilazane (HMDS), followed by a typical N-alkylation using an alkyl iodide. We have obtained excellent yields using small quantities of HMDS. The identification of the 3-substituted 6-aminouracil was carried out through the analysis of the chemical shift of the signal for NH2 protons in the 1H-NMR spectrum: in the 6-amino-1-propyluracil the NH2 protons are shifted downfield with respect of those in 6-aminouracil; in the 6-amino-3-propyluracil the NH2 protons are not influenced [34]. The UV spectrum showed the classical absorption of a b-amino a,b-unsaturated carbonyl (lmax 261 nm).
In the preparation of the 6-amino-5-nitroso-3-propyluracil (7) better results were obtained when only 2 eq of sodium nitrite were used, working at room temperature. The yellow compound 7 (lmax 320 nm) was isolated in 85% yield. In the 13C-NMR spectrum the signal for C-5 appears at 140.3 ppm, down-shifted in comparison to that in the starting material (74.6 ppm), caused by the electron-withdrawing effect of the nitroso group. In the IR spectra the characteristic band for the N=O group is shown at 1510 cm−1.
The nitroso compound 6 was chemoselectively reduced by sodium dithionite in alkaline solution to the corresponding 5,6-diamino-3-propyluracil (8). It was easily isolated and was used for the next step without further purification because of its instability to light, oxygen and acidic medium. The chemoselective reduction was carried out in the range from 40˚C to 80˚C; we found that at lower temperatures the reaction needs a huge excess of reducing agent, causing difficulties in the purification of the diamino compound 8. For this reaction, we additionally examined the influence of the concentration of ammonia. All concentrations used (24%, 12%, 6% and 3%) led successfully to the diamino compound 8 in about 70% yield.
The condensation of the diamino compound 8 with cinnamic acid to form (E)-6-amino-3-propyl-5-styrylcarboxamidouracil (9) was achieved using N-ethyl-N’-[3- (dimethylamino)propyl]carbodiimide (EDC) as the coupling agent. The configuration of the double bond in the styryl moiety was corroborated by its 1H-NMR spectrum: the doublet signal at 6.85 ppm, assigned to vinyl proton, has a typical Jtrans = 16 Hz. The alkylation at N1 of the uracil derivative 9 could easily be performed under mild conditions. A DMF solution of 9, treated with K2CO3 and an alkyl bromide or iodide, yielded the 1-substituted (E)-6-amino-3-propyl-5-styrylcarboxamidouracil 10a-d in good yields. UV absorptions are in the range of 278 - 280 nm. No alkylation at position 7 was observed.
Finally, an alkaline treatment promoted the cyclization
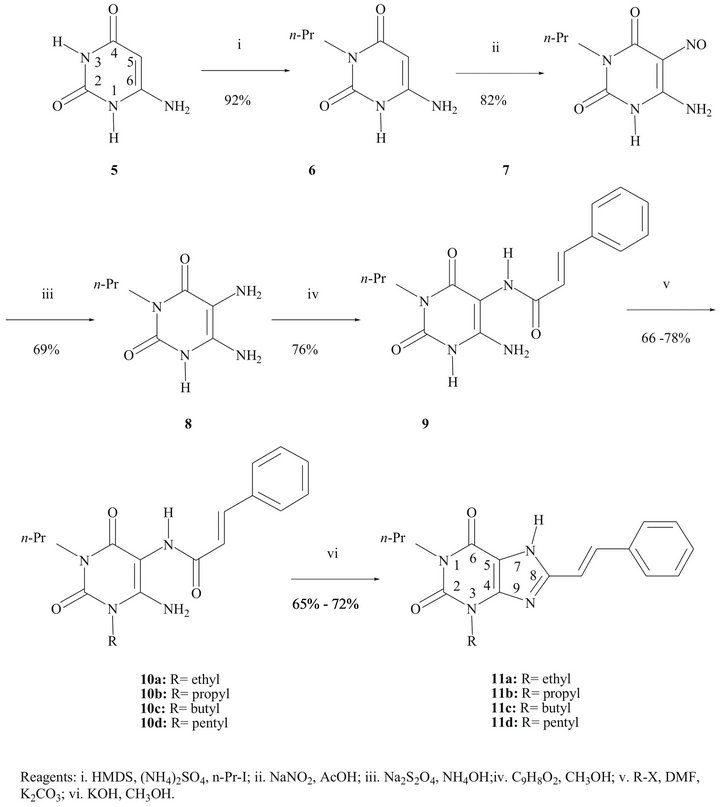
Scheme 1. Synthetic pathway for the styrylxanthines 11a-d.
between the amino and carboxamido groups to provide compounds 11a-d, in around 70% yield. The UV spectra showed bands in the range lmax = 342 - 347 nm, characteristic for 8-styrylxanthines.
3.2. Binding Studies
The results of binding studies showed that all the 1,3- disubstituted 8-styrylxanthines synthetized have affinity to both adenosinergic receptors. Table 1 shows the different affinities of each compound analyzed; it is worth to notice that gradual increase in the length of the carbon chain of the alkyl group at the N atom at position 3 increases the affinity towards A1 receptors. However, opposite effect is observed for affinity toward A2A recaptors.
3.3. Behavioral Test Method
In these conditions the animals showed a clear increase in the turning behavior 14.21 ± 3.206 minutes after METH injection. The difference was stable over a period of 80 minutes with a significant decrease of 1.938 ± 0.6003 minutes respect to group that only received to vehicle (mineral oil + DMSO; 1:1). Animals that only received vehicle did not show deficit in movement initiation or speed of turning (Figure 2).
Based on binding studies we evaluated the effect on turning behavior of (E)-3-ethyl-1-propyl-8-styrylxanthine (A15Et, 11a), compound that showed highest affinity towards A2A receptor and also (E)-3-butyl-1-propyl-8- styrylxanthine (Al5Bu, 11c) with approximately half affinity to A2A receptor (see Table 1).
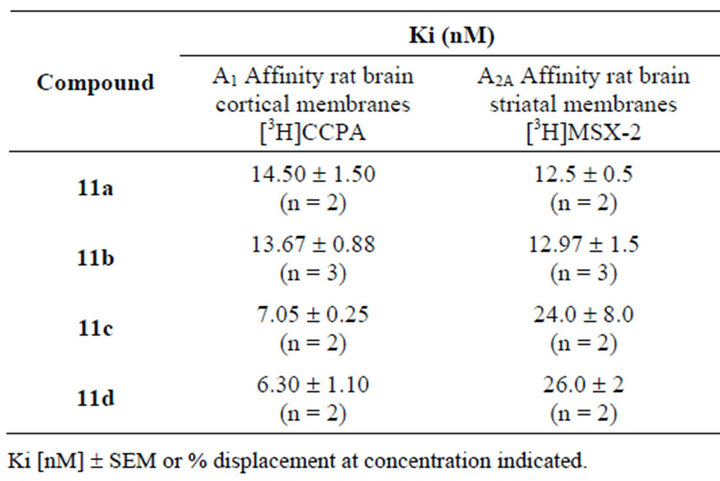
Table 1. Adenosine receptor affinities of 3-substituted (E)-1- propyl-8-styrylxanthines.
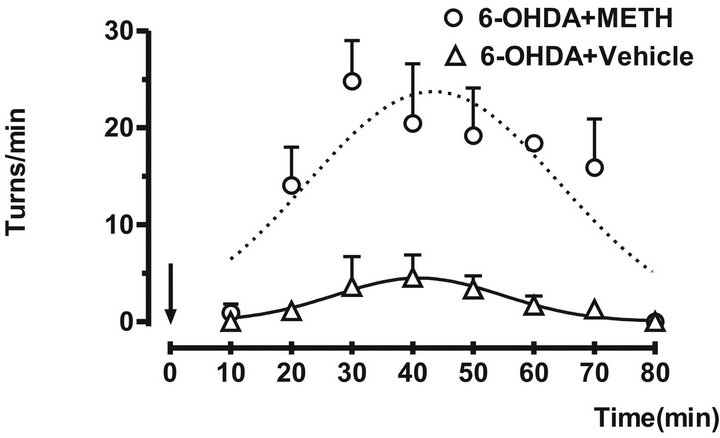
Figure 2. The ipsilateral turn behavior were induced by administration of METH (7 mg/kg sc) in rats with 6-OHDA. The turns/min were counted over 10 minutes. The arrow indicates the administration time. Each point represents the mean number of ipsilateral turns each 10 min ± SEM. Vehicle (Δ) and 6-OHDA (○).
The A15Et decreased the turning behavior and was administrated as a dose of 1 mg/Kg, 30 minutes before the administration of METH (7 mg/kg sc). This significant difference was maintained over a period of 50 minutes respect to the vehicle group (Figure 3(a)).
The Al5Bu was administrated 30 min before the METH (7 mg/kg sc), decreased significantly the turning behavior and this difference was maintained over a period of 50 - 70 min respect to the vehicle group (Figure 3(b)).
4. Discussion
The administration of the A15Et (11a) or Al5Bu (11c) effectively reduces the ipsilateral turning behavior caused by METH in rats with unilaterally lesions caused by 6-OHDA. These compounds can exert significant functional effects on motor behavior. Also we have found that rotational asymmetry in rats with unilateral lesions caused by 6-OHDA can be counteract by administration of compounds A15Et and A15Bu (Figures 3(a) and (b)). These effects were more potent and with more prolonged duration with A15Et, this effect was expected in order
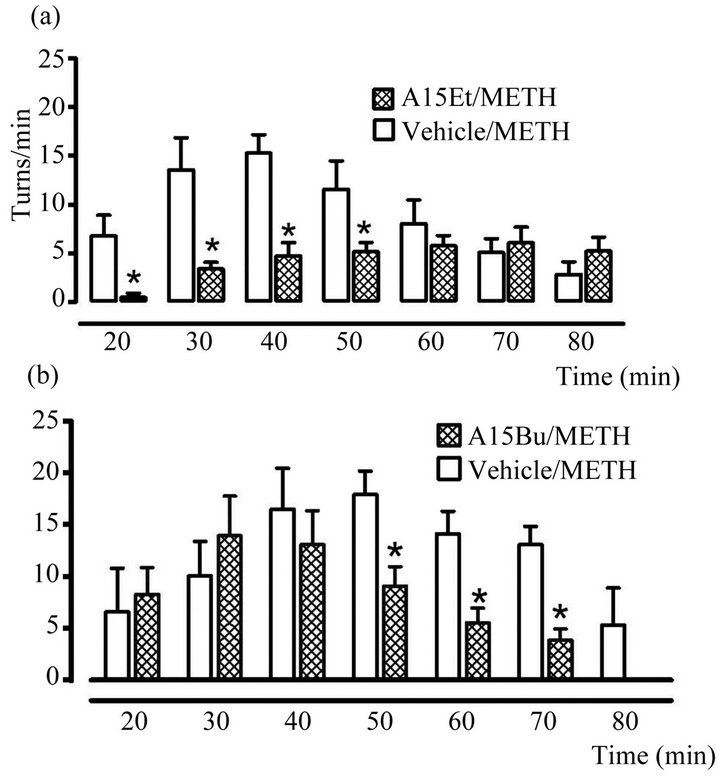
Figure 3. Effect of (a) A15Et and (b) A15Bu on number of ipsilateral turns in rats with unilateral lesions caused by 6-OHDA. Both novel xanthines were administrated at dose of 1 mg/kg sc 10 min before the administration of METH (7 mg/kg sc). Open bars represent ipslateral turns in the vehicle group and hatched bars show the turns with derivates of xanthines. The data were given as the mean ± SEM. The statistical analysis was made using t of student test. *p < 0.05 vs Vehicle/METH group.
that A15Et showed highest affinity to A2A receptor and this effect may be related to modulation of A2A receptor over D2 receptor. Recently it has been reported that other compounds possessing the property of adenosine A2A- receptor antagonism [35,36], improved the motor effect in an experimental model of Parkinson’s disease [37]. Because adenosine A2A receptors have a profound influence on motor functions via the modulation of basal ganglia output pathways [37], blockade of this modulatory function by an A2A antagonist could repair striatopallidal abnormal-neuronal activities caused by striatal dopamine depletion in the Parkinsonian state [37].
5. Conclusion
In summary, we describe the synthesis of the 1,3-disubstituted 8-styrylxanthines 11a-d with optimized conditions and excellent yield. The results of the present study provide evidence that all compounds synthetized have affinity towards A1 and A2A receptors but compound A15Et showed the highest affinity towards A2A receptor. The administration of the A15Et (11a) or Al5Bu (11c) effectively reduces the ipsilateral turning behavior caused by METH in rats with unilaterally lesions caused by 6-OHDA. Based on these results we propose to A15Et as a potential compound to treat some symptoms of Parkinson’s disease.
6. Acknowledgements
The authors are grateful to Prof. C. E. Müller, Universität Bonn, for radioligand binding essays. The authors thank CONACyT-México for financial support (50682) and (CB2011-169023 give to D.L.).
REFERENCES
- R. A. Olsson and J. D. Pearson, “Cardiovascular Purinocaptors,” Physiological Reviews, Vol. 70, No. 3, 1990, pp. 761-845.
- C. Londos, D. M. Cooper and J. Wolff, “Subclasses of External Adenosine Receptors,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 77, No. 5, 1980, pp. 2551-2554. doi:10.1073/pnas.77.5.2551
- B. B. Fredholm, M. P. Abbracchio, G. Burnstock, J. W. Daly, T. K. Harden, K. A. Jacobson, P. Leff and M. Williams, “Nomenclature and Classification of Purinoceptors,” Pharmacological Reviews, Vol. 46, No. 2, 1994, pp. 143-156.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, “International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update,” Pharmacological Reviews, Vol. 63, No. 1, 2011, pp. 1-34. doi:10.1124/pr.110.003285
- D. L. Rosin, A. Robeva, R. L. Woodard, P. G. Guyenet and J. Linden, “Immunohistochemical Localization of Adenosine Receptors in the Rat Nervous System,” The Journal of Comparative Neurology, Vol. 401, No. 2, 1998, pp. 163-186. doi:10.1002/(SICI)1096-9861(19981116)401:2<163::AID-CNE2>3.0.CO;2-D
- P. Svenningsson, C. Le Moine, B. Kull, R. Sunahara, B. Bloch and B. B. Fredholm, “Cellular Expression of Adenosine A2A Receptor Messenger RNA in the Rat Nervous System with Special Reference To Dopamine Innervated Areas,” Neuroscience, Vol. 80, No. 4, 1997, pp. 1171- 1185. doi:10.1016/S0306-4522(97)00180-2
- J. Shimada and F. Suzuki, “Medicinal Chemistry of Adenosine Receptors in the Brain and Periphery,” In: H. Hiroshi, P. J. Richardson and P. Jenner, Eds., Adenosine Receptors and Parkinson’s Disease, Academic Press, New York, 2000, pp. 31-50. doi:10.1016/B978-012400405-4/50004-6
- S Ferré, G. von Euler, B. Johansson, B. B. Fredholm and K. Fuxe, “Stimulation of High-Affinity Adenosine A2 Receptors Decreases the Affinity of Dopamine D2 Receptors in Rat Striatal Membranes,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 88, No. 16, 1991, pp. 7238-7241. doi:10.1073/pnas.88.16.7238
- J. D. Peterson, J. A. Goldberg and D. J. Surmier, “Adenosine A2a Receptor Antagonists Attenuate Striatal Adaptations Following Dopamine Depletion,” Neurobiology of Disease, Vol. 45, No. 1, 2012, pp. 401-416. doi:10.1016/j.nbd.2011.08.030
- P. Jenner, “Istradefylline, a Novel Adenosine A2A Receptor Antagonist, for the Treatment of Parkinson’s Disease,” Expert Opinion on Investigational Drugs, Vol. 14, No. 6, 2005, pp. 729-738. doi:10.1517/13543784.14.6.729
- M. T. Armentero, A. Pinna, S. Ferré, J. L. Lanciego, C. E. Müller and R. Franco, “Past, Present and Future of A2A Adenosine Receptor Antagonists in Therapy of Parkinson’s Disease,” Pharmacology and Therapeutics, Vol. 132, No. 3, 2011, pp. 280-299. doi:10.1016/j.pharmthera.2011.07.004
- N. Szabó, Z. T. Kincses and L. Vécsei, “Novel Therapy in Parkinson’s Disease: Adenosine A2A Receptor Antagonists,” Expert Opinion on Drug Metabolism and Toxicology, Vol. 7, No. 4, 2011, pp. 441-455. doi:10.1517/17425255.2011.557066
- N. Castagnoli Jr., J. P. Petzer, S. Steyn, K. Castagnoli, J. F. Chen, M. A. Schwarzschild and C. J. Van der Schyf, “Monoamine Oxidase B Inhibition and Neuroprotection: Studies on Selective Adenosine A2A Receptor Antagonists,” Neurology, Vol 61, No. 11, 2003, pp. S62-S68. doi:10.1212/01.WNL.0000095215.97585.59
- P. Popoli, C. Frank, M. T. Tebano, R. L. Potenza, A. Pintor, M. R. Domenici, V. Nazzicone, A. Pèzzola and R. Reggio, “Modulation of Glutamate Release and Excitotoxicity by Adenosine A2A Receptors,” Neurology, Vol. 61, No. 11, 2003, pp. S69-S71. doi:10.1212/01.WNL.0000095216.89483.A2
- J. P. Petzer, S. Steyn, K. P. Castagnoli, J. F. Chen, M. A. Schwarzchild, C. J. Van der Schyf and N. Castagnoli, “Inhibition of Monoamine Oxidase B by Selective Adenosine A2A Receptor Antagonists,” Bioorganic & Medicinal Chemistry, Vol. 11, No. 7, 2003, pp. 1299-1310. doi:10.1016/S0968-0896(02)00648-X
- P. Hickey and M. Stacy, “Adenosine A2A Antagonists in Parkinson’s Disease: What’s Next?” Current Neurology and Neuroscience Reports, Vol. 12, No. 4, 2012, pp. 376- 385. doi:10.1007/s11910-012-0279-2
- A. H. Schapira, E. Bezard, J. Brotchie, F. Calon, G. L. Collingridge, B. Ferrer, B. Hengerer, E. Hirsch, P. Jenner, N. Le Novère, J. A. Obeso, M. A. Schwarzschild, U. Spampinato and G. Davidai, “Novel Pharmacological Targets for the Treatment of Parkinson’s Disease,z” Nature Reviews, Drug Discovery, Vol. 5, No. 10, 2006, pp. 845- 854. doi:10.1038/nrd2087
- J. Bove, J. Serrats, G. Mengod, R. Cortés, E. Tolosa and C. Marin, “Neuroprotection Induced by Adenosine A2A Antagonist CSC in the 6-OHDA Rat Model of Parkinsonism: Effect on the Activity of Striatal Output Pathways,” Experimental Brain Research, Vol 165, No. 3, 2005, pp. 362-374. doi:10.1007/s00221-005-2302-1
- W. Bara-Jiminez, A. Sherzai, T. Dimitrova, A. Favit, F. Bibbiani, M. Gillespie, M. J. Morris, M. M. Mouradian and T. N. Chase, “Adenosine A2A Receptor Antagonist Treatment of Parkinson’s Disease,” Neurology, Vol. 61, No. 3, 2003, pp. 293-296. doi:10.1212/01.WNL.0000073136.00548.D4
- N. Vlok, S. F. Malan, N. Castagnoli Jr., J. J. Bergh and J. P. Petzer, “Inhibition of Monoamine Oxidase B by Analogues of the Adenosine A2A Receptor Antagonist (E)-8- (3-chlorostyryl)caffeine (CSC),” Bioorganic & Medicinal Chemistry, Vol. 14, No. 10, 2006, pp. 3512-3521. doi:10.1016/j.bmc.2006.01.011
- M. B. Youdim and P. F. Riederer, “A Review of the Mechanisms and Role of Monoamine Oxidase Inhibitors in Parkinson’s Disease, ” Neurology, Vol. 63, No. 7, 2004, pp. S32-S35. doi:10.1212/WNL.63.7_suppl_2.S32
- M. A. Schwarszchild, J. F. Chen and A. Ascherio, “Caffeinated Clues and the Promise of Adenosine A2A Antagonists in PD,” Neurology, Vol. 58, No. 8, 2002, pp. 1154-1160. doi:10.1212/WNL.58.8.1154
- A. Muñoz-Zurita, J. Sandoval-Ramírez, L. E. EspinozaMoreno, C. Parra-Cid, L. A. Juárez-Leyva, D. LimónPérez de León and G. Flores, “ Synthesis and Evaluation of Ansiolytic Effect of a New Derivate Pirimide [1,6-a] Pirimidinic,” Revista de la Sociedad Química de México, Vol. 47, No. 4, 2003, pp. 298-302.
- C. B. Vu, D. Pan, B. Pen, G. Kumaravel, G. Smits, X. Jin, D. Phadke, T. Engber, C. Huang, J. Reilly, S. Tam, D. Grant, G. Hetu and R. C. Petter, “Novel Diamino Derivates of [1,2,4]Triazolo[1,5-a][1,3,5]triazine as Potent and Selective Adenosine A2a Receptor Antagonists,” Journal of Medicinal Chemistry, Vol. 48, No. 6, 2005, pp. 2009-2018. doi:10.1021/jm0498396
- S. Massip, J. Guillon, D. Bertarelli, J. J. Bosc, J. M. Léger, S. Lacher, C. Bontemps, T. Dupont, C. E. Mulle and C. Jarry, “Synthesis and Preliminary Evaluation of New 1- and 3-[1-(2-Hydroxy-3-phenoxypropyl)]xanthines from 2-Amino-2-oxazolines as Potential A1 and A2A Adenosine Receptor Antagonists,” Bioorganic & Medicinal Chemistry, Vol. 14, No. 8, 2006, pp. 2697-2719. doi:10.1016/j.bmc.2005.11.050
- K. A. Jacobson, C. Gallo-Rodriguez, N. Melman, B. Fischer, M. Maillard, A. van Bergen, P. J. van Galen and Y. Karton, “Structure-Activity Relationships of 8-Styrylxanthines as A2-Selective Adenosine Antagonists,” Journal of Medicinal Chemistry, Vol. 36, No. 10, 1993, pp. 1333- 1342. doi:10.1021/jm00062a005
- M. Williams, “Purinergic Pharmaceuticals for the 1990,” Nucleosides and Nucleotides, Vol. 10, No. 5, 1991, pp. 1087-1099. doi:10.1080/07328319108047246
- C. E. Müller, J. Maurinsh and R. Sauer, “Binding of [3H]MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine) to Rat Striatal Membranes—A New, Selective Antagonist Radioligand for A(2A) Adenosine Receptors,” European Journal of Pharmaceutical Sciences, Vol. 10, No. 4, 2000, pp. 259-265. doi:10.1016/S0928-0987(00)00064-6
- A. Galvan, B. Floran, D. Erlij, J. Aceves, “Intrapallidal Dopamine Restores Motor Deficits Induced by 6-Hydroxydopamine in the Rat,” Journal of Neural Transmission, Vol. 108, No. 2, 2001, pp. 153-166. doi:10.1007/s007020170085
- G. Paxinos and C. Watson, “The Rat Brain in Stereotaxic Coordinates,” Academic Press, San Diego, 1998.
- U. Ungerstedt and G. W. Arbuthnott, “Quantitative Recording of Rotational Behavior in Rats after 6-HydroxyDopamine Lesions of the Nigrostriatal Dopamine System,” Brian Research, Vol. 24, No. 3, 1970, pp. 485-493. doi:10.1016/0006-8993(70)90187-3
- C. E. Müller, “Synthesis of 3-Substituted 6-aminouracils,” Tetrahedron Letters, Vol. 32, No. 45, 1991, pp. 6539- 6540. doi:10.1016/0040-4039(91)80214-Q
- C. E. Müller, “A2A Adenosine Receptor Antagonists— Future Drugs for Parkinson’s Disease?” Drugs of the Future, Vol. 25, No. 10, 2000, p. 1043. doi:10.1358/dof.2000.025.10.858696
- C. E. Müller and J. Sandoval-Ramírez, “A New Versatile Synthesis of Xantines with Variable Substituents in the 1-, 3-, 7- and 8-Positions,” Synthesis, Vol. 1995, No. 10, 1995, pp. 1295-1299. doi:10.1055/s-1995-4082
- M. Ochi, K. Koga, M. Kurokawa, H. Kase, J. Nakamura and Y. Kuwana, “Systemic Administration of Adenosine A2A Receptor Antagonist Reverses Increased GABA Release in the Globus Pallidus of Unilateral 6-Hydroxydopamine-Lesioned Rats: A Microdialysis Study,” Neuroscience, Vol. 100, No. 1, 2000, pp. 53-62. doi:10.1016/S0306-4522(00)00250-5
- H. Kase, “New Aspects of Physiological and Pathophysiological Functions of Adenosine A2A Receptor in Basal Ganglia,” Bioscience, Biotechnology and Biochemistry, Vol. 65, No. 7, 2001, pp. 1447, 1457.
- F. Pedata, A. M. Pugliese, A. Melani and M. Gianfriddo, “A2A Receptors in the Neuroprotection of Dopaminergic Neurons,” Neurology, Vol. 61, No. 11, 2003, pp. S49- S50. doi:10.1212/01.WNL.0000095212.19029.04
NOTES
*Corresponding author.