A. R. GOMES ET AL.
Copyright © 2011 SciRes. AJAC
315
mine are purchased from Merck, Darmstadt, Germany.
High purity water is pr epared by using Millipo re Milli-Q
plus water purification syste m.
2.2. Procedure
2.2.1. Equipment
The LC system, used for method development and me-
thod validation is Agilent RRLC. The output signal is
monitored and processed using EZ-Chrome elite soft-
ware on Pentium computer (Digital equipment Co).
RRLC is equipped with binary gradient pump, thermos-
tatted auto sampler, thermostatted column compartment,
variable wavelength detector.
2.2.2. Chromatogra phic Conditio ns
The chromatographic column used is Waters Symmetry
RP18 (50 × 4.6 mm) with 3.5 µm particles. The mobile
phase-A contains a 0.05 M of ammonium acetate and 1.0
mL·L–1 diethylamine. Methanol and tetrahydrofuran
(95:5) is used as mobile phase-B. The flow rate of the
mobile phase is 1.5 mL·min–1 with a gra dient p rogra m of
0/2, 2/10, 7/35, 9/45, 10/2 and 12/2 (time (min)/%
B).Th e column temperature is maintained at 30˚C and
the detectio n is monitored at wavelength o f 254 nm. T he
injection volume is 10 µL.
Diluent consists buffer and methanol in the ratio
80:20.
2.2.3. Preparation o f Standard and Sample So l utions:
All the impurities are dissolved in diluents having con-
centr atio n of 0. 1 mg/ mL the n make up to the vo lu me wit h
diluent. A Stock solution of Nizatidine (2000 µg·mL–1) is
prepared by dissolving appropriate amount in the diluent.
Working solution 200 µg·mL–1 is prepared from above
stoc k s olution for a s s ay d eter minati on.
2.3. Method Development and Optimization
The USP [4] method for Nizatidine chromatographic
purity determination has a run time of 70 minutes with
1.0 mL·min –1 flow rate. And the European pharmacopeia
[5] method for Nizatidine chromatographic purity deter-
minat ion has a r un time of 6 0 mi nutes wit h 1.0 mL ·mi n –1
flow rate. The main objective of the present study is to
develop a method having less runtime which can be use
for both c hromatograp hi c pur it y and as s ay d etermination.
To calculate the flow rate we have used the formula in
USP pharmacopeial forum Stimuli article “Transfer of
HPLC Procedures to Suitable Columns of Reduced Di-
mensions and Particle Sizes” [6 ] .
where F, l, and d are the flow rates, the column lengths,
and the column diameters, by this formula a flow rate of
0.2 mL·min–1 was derived from USP method parameters
and by using the USP method details to short length
column and flow specified but when attempted Nizati-
dine peak elute around 32 minutes and impurity-3 (last
eluting impurity) elute around 60 minutes with low re-
sponse.
Flow rate arrived from existing USP method is 0.2
mL·min–1 was derived for 50 × 4.6 mm column in which
a late elution was found. To decrease the run time flow
rate has increased 7.5 times i.e. 1.5 mL·min–1 when ap-
plied in this condition Nizatidine peak elutes around 4.8
minutes and last impurity elutes around 11 minutes with
low response. For decreasing the retention time and to
raise the response of impur i ty-3, 5% tetrahydrofuran
introduced in to the mobile phase-B. In this condition
impurity-3 retention time decreased to 8.2 minutes from
11 minutes and peak responses also enhanced. The typi-
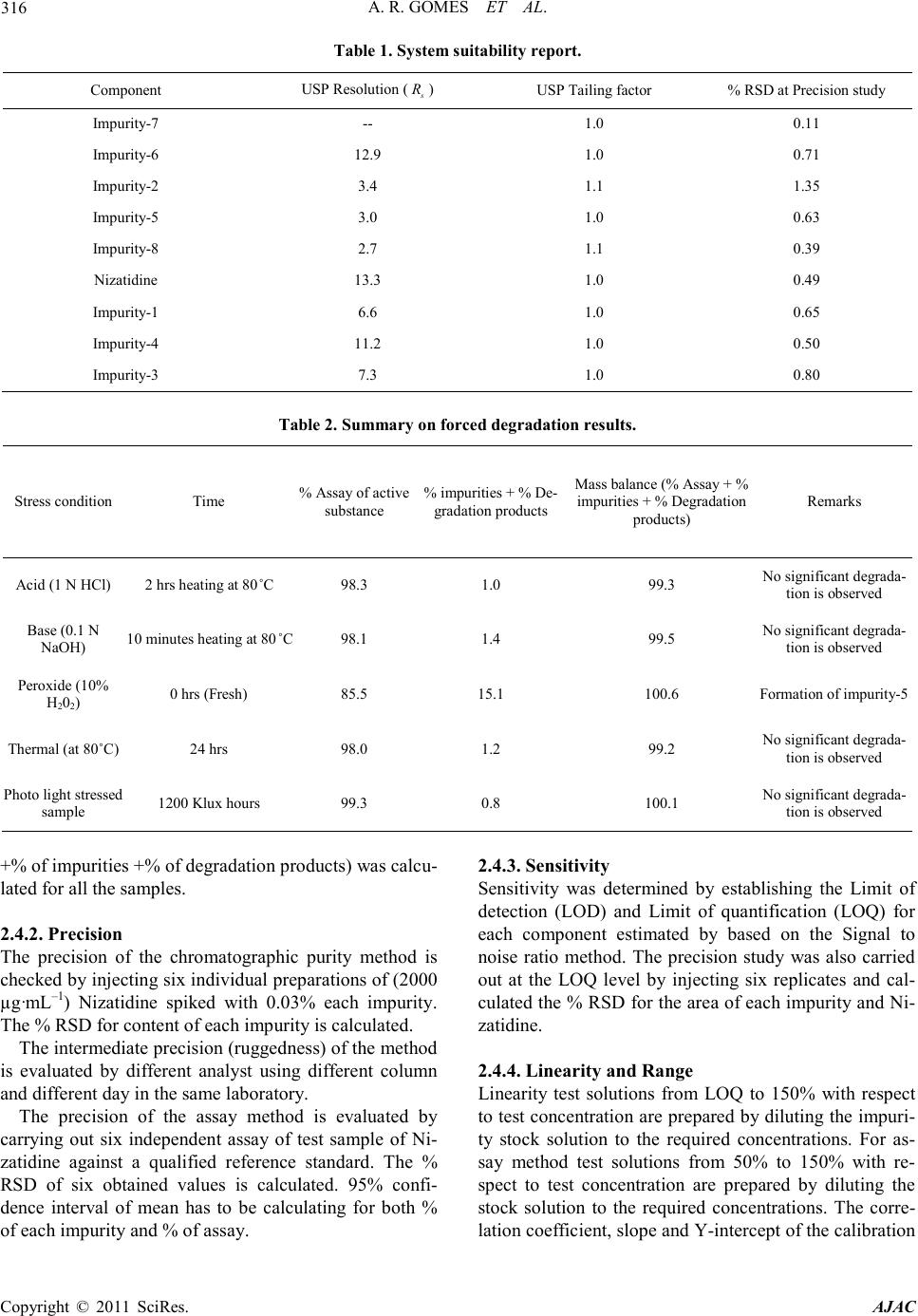
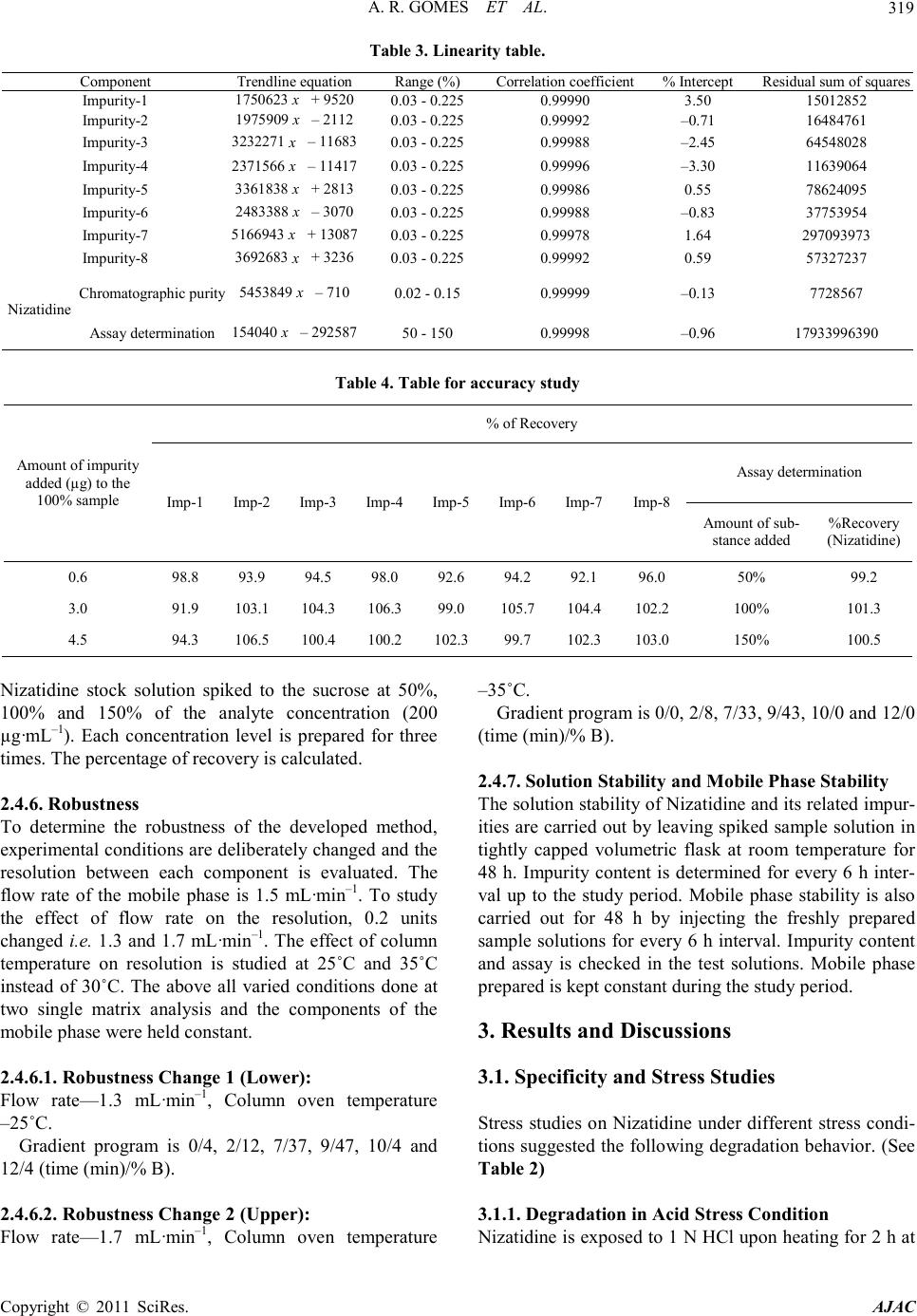
cal retention times of Nizatidine, impurity-1, impurity-2,
i mp urity-3, impurity-4, impurity-5, impurity-6, i mp urity-
7 and impurity-8 were about 4.801, 5.719, 2.595, 8.392,
7.381, 2.930, 2.211, 0.646 and 3.231 minutes respec-
tively meeting the chromatographic system suitability
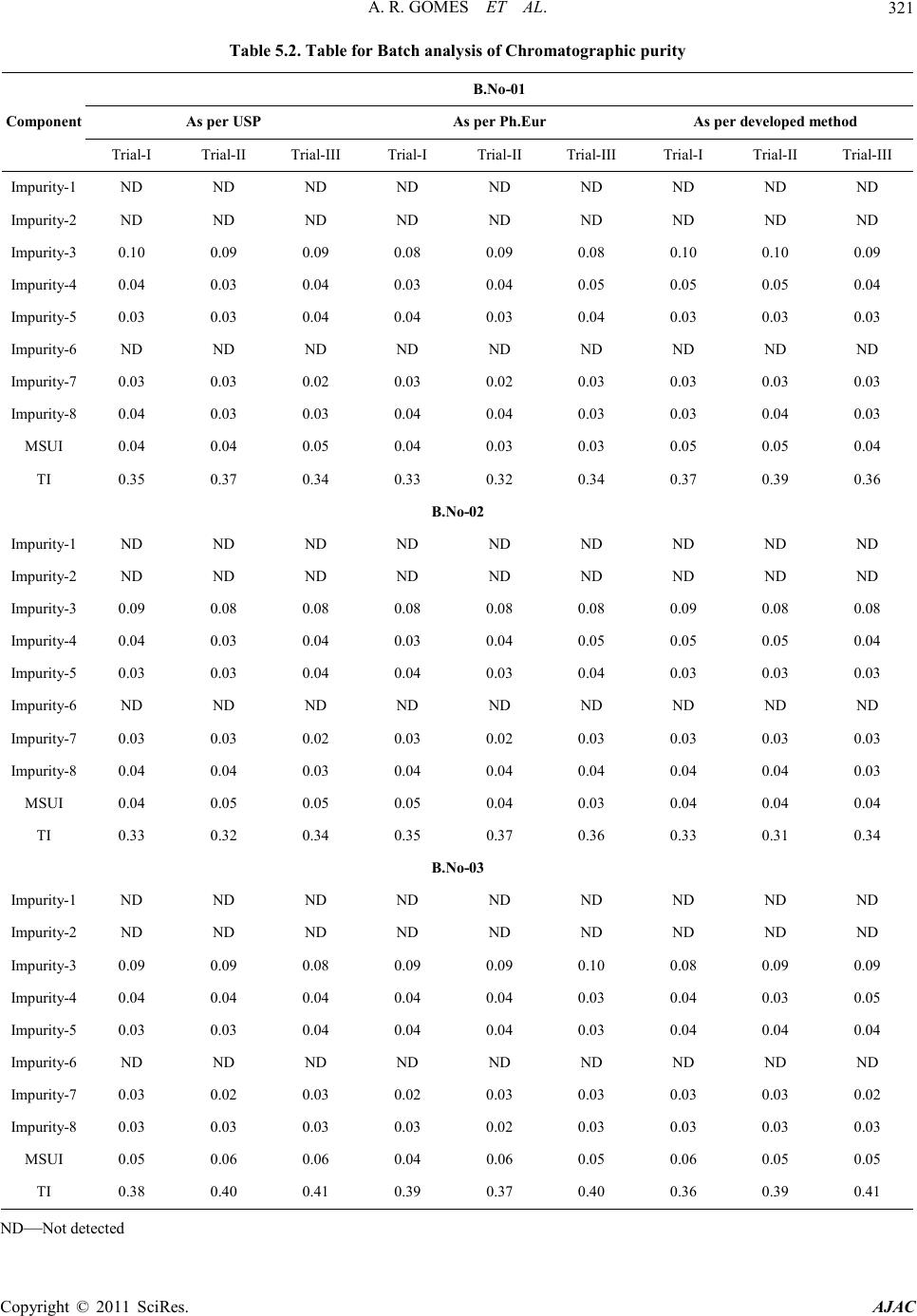
requireme n t s .( See Table 1)
2.4. Analytical Method Validation
The developed chromatographic method is validated for
specificity and stress studies, sensitivity, linearity &
range, precision, accuracy, and robustness and system
suitability for both chromatographic purity and assay
methods [7-15].
2.4.1. Specificity and Stress Studies
Specificity is the ability of the method to measure the
analyte response in the presense of its potential impuri-
ties. The specificity [10,11] of the developed LC method
for Nizatidine was determined in the presence of its im-
purities namely impurity-1, impurity-2, impurity-3, im-
purity-4, impurity-5, impurity-6, impurity-7 and impuri-
ty-8 at a concentration of 30 µg·mL–1. The stress condi-
tions employed for degradation study includes photolytic
(carried out as per ICH Q1B), thermal (80˚C), acid hy-
drolysis (1 N HCl), base hydrolysis (0.1 N NaOH), hy-
drolysis and oxidation (10% H2O2).Peak purity of
stressed samples of Nizatidine was checked by using
2996 Photo diode array detector of Waters (PDA). All
stressed samples of Nizatidine were analysed for an ex-
tended run time of 15 minutes to check the late eluting
degradants.
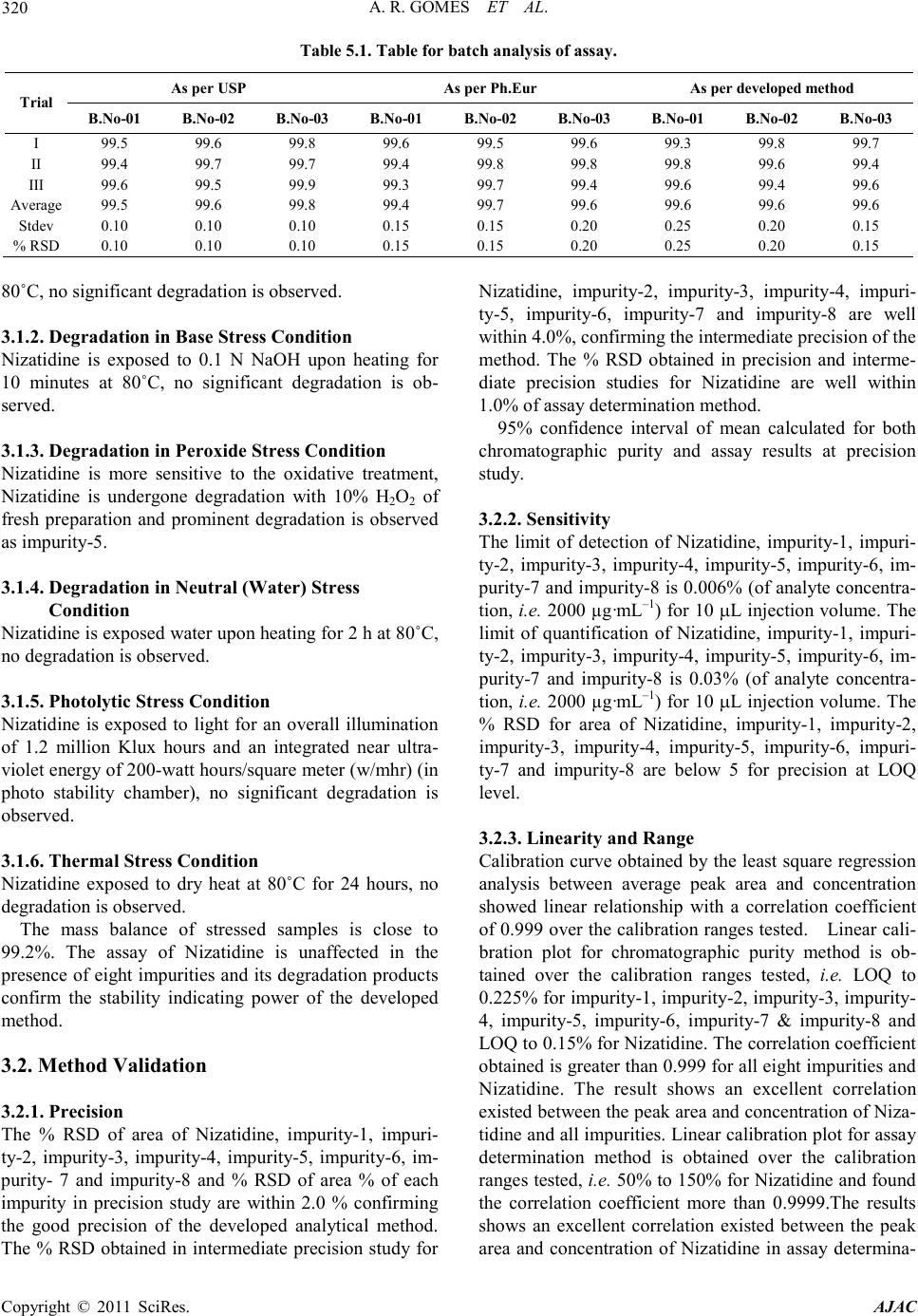
Assay was car ried out fo r stre ss sa mples a gai nst q uali-
fied reference standard and the mass balance (% assay