Journal of Materials Science and Chemical Engineering
Vol.03 No.07(2015), Article ID:57688,6 pages
10.4236/msce.2015.37013
Effect of Melt Mixing Time in Internal Mixer on Mechanical Properties and Crystallization Behavior of Glycidyl Methacrylate Grafted Poly (Lactic Acid)
Nguyen Chi Thanh1, Chaiwat Ruksakulpiwat2, Yupaporn Ruksakulpiwat1*
1School of Polymer Engineering, Institute of Engineering, Suranaree University of Technology, Nakhon Ratchasima, Thailand
2Department of Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, Thailand
Email: *yupa@sut.ac.th


Received 30 March 2015; accepted 24 June 2015; published 1 July 2015

ABSTRACT
Glycidyl methacrylate (GMA) was grafted onto poly (lactic acid) (PLA) by melt mixing in internal mixer using dicumyl peroxide (DCP) as an initiator. The results from proton nuclear magnetic resonance (1H-NMR) and Fourier transform infrared (FTIR) spectroscopy indicated that the grafting reaction of GMA onto PLA took place successfully. The impact strength of PLA-g-GMA was significantly higher than that of pure PLA. The crystallinity of PLA, obtained from differential scanning calorimetry (DSC), decreased after grafting. In order to obtain the optimal mixing conditions, the mixing time was varied into 7, 10 and 14 min. The optimum mixing time of 10 min was found to give the optimum mechanical properties of glycidyl methacrylate grafted poly (lactic acid) (PLA-g- GMA). However, the mixing time played no important role in impact behavior of PLA-g-GMA. In addition, the highest crystallinity was obtained with the PLA-g-GMA prepared with the mixing time of 7 min.
Keywords:
PLA, Grafting, PLA-g-GMA, Mixing Time, Crystallization

1. Introduction
Due to the environmental issue related to non-degradable plastic wastes, the research and development of biodegradable polymers have recently taken considerable attention. Poly (lactic acid) (PLA), a synthetic aliphatic polyester derived from biomasses, is an environmentally friendly polymer and has been emerging as an alternative to conventional petroleum-based polymeric materials because of its renewability, biodegradability and greenhouse gas neutrality. Moreover, PLA also possesses good mechanical properties and can be easily processed [1]. However, PLA is still more expensive than many petroleum-derived commodity plastics. It has some poor physical properties such as slow crystallization rate and high brittleness, which hinder PLA to be used for various end-use applications [2].
The reactive grafting of glycidyl methacrylate (GMA) onto non-polar polyolefin such as polypropylene (PP) backbones via reactive extrusion was carried out successfully [3] [4]. Kuk Young Cho et al. investigated the melt grafting of GMA onto high-density polyethylene (HDPE) in the presence of free radical initiators in the batch mixer [5]. The results showed that the grafting reaction was significantly influenced by reaction time. It is one of the important variables for the preparation of functional polymer.
Recently, GMA has been increasingly used as a grafting monomer because of its dual functionality, which consists of epoxy and acrylic groups. It is believed that the epoxy group of GMA can react with many other groups, such as hydroxyl and carboxyl groups. Meanwhile, acrylic groups show the capability of free-radical grafting of GMA onto the polymer chain [6].
Even the radical grafting of GMA onto polyolefin in melt mixing process has been investigated extensively, there have been very few studies focusing on the grafting of GMA onto PLA and effect of reaction time on physical properties of prepared PLA-g-GMA. Therefore, in this study, we report on the GMA grafting onto PLA with different mixing times using dicumyl peroxide (DCP) as an initiator in the internal mixer.
2. Experimental
2.1. Materials
Commercial PLA (PLA 4043D) was purchased from Nature Works LLC. Dicumyl peroxide (DCP, 99%) and glycidyl methacrylate (GMA, 99%), dichlorobenzene (DCB), chloroform (HPLC grade) and methanol were supplied from Sigma-Aldrich. Before being used for grafting, GMA was purified by basic alumina pack column to remove inhibitor and stored in the refrigerator at about 5˚C in order to ensure that GMA is not decomposed.
2.2. Preparation of Glycidyl Methacrylate Grafted Poly (Lactic Acid)
The grafting of GMA onto PLA chain was carried out by an internal mixer (Hakke Rheomix, 3000p). Before being used, PLA pellet was dried at 70˚C for 2 hours. The grafting reaction was carried out under constant temperature of 170˚C. The mixing time was varied in the range of 7, 10 and 14 min. The mixing speed was held at 60 rpm. The GMA content of 10 wt% of the PLA weight was used. Before being used for grafting, GMA was stored in the refrigerator at 5˚C. Concentration of DCP used was 0.2 wt% of the PLA weight. The grafting procedure is described as follows:
First, determined amounts of PLA and DCP were premixed by hand in a small plastic bag before being charged into the mixing chamber. After the addition of the mixture, the mixing chamber was closed immediately in order to minimize the loss of GMA due to vaporization. After two minutes of mixing, GMA was added into chamber.
2.3. Purification of Glycidyl Methacrylate Grafted Poly (Lactic Acid)
In order to remove unreacted GMA and possible homopolymer of GMA, which can be generated during the grafting reaction, after completion of mixing, the obtained samples were dissolved in DCB (5 wt% solution) at 120˚C. After that, the solution was poured into a large excess of methanol to precipitate PLA-g-GMA. The obtained white PLA-g-GMA precipitate was washed several times with methanol to remove completely the residual GMA and homopolymer of GMA. After purification process, the pure white PLA-g-GMA was dried in a vacuum oven at 70˚C for 24 h.
2.4. Characterization of Glycidyl Methacrylate Grafted Poly (Lactic Acid)
2.4.1. NMR Analysis
1H-NMR spectra of PLA, PLA-g-GMA and GMA monomer were recorded on a Varian model Inova 300 NMR spectrophotometer at 25˚C and 300 MHz using CDCl3 as a solvent and tetramethylsilane (TMS) as an internal standard.
2.4.2. FTIR
The measurements of structural characterization of PLA and PLA-g-GMA were conducted by Fourier transform infrared spectroscopy Bruker Tensor 27 using attenuated total reflectance (ATR) equipped with platinum diamond crystal (TYPE A225/QL). Spectra were obtained at 4 cm−1 resolution and 32 scans in the wavenumber range from 4000 to 400 cm−1. All samples were dried in a vacuum oven at 60˚C for 24 h before testing.
2.4.3. Mechanical Properties
Tensile properties of all samples were measured according to ASTM D638 using an Instron Universal Testing Machine (UTM 5565) with a load cell of 5 kN. The unnotched Izod impact strengths of pure PLA and PLA-g- GMA were obtained by using Instron CEAST 9050 Pendulum Impact System according to ASTM D256.
2.4.4. Crystallization Behavior
The nonisothermal crystallization behavior of pure PLA and PLA-g-GMA was investigated with a differential scanning calorimetry (DSC) (model: DSC204F1 Phoenix). The samples were first heated to 200˚C with the rate of 5˚C/min and kept at 200˚C for 5 min. After that, they were cooled to 40˚C with the rate of 5˚C/min. Then they were heated again to 200˚C with the rate of 5˚C/min. The degree of crystallinity (%Xc) of pure PLA and PLA-g- GMA was examined by Equation (1) [7]:
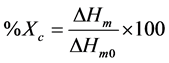 (1)
(1)
where, ΔHm is the measured melting enthalpy (in J∙g−1) from the second heating scan and ΔHm0 is the theoretical melting enthalpy of completely crystalline PLA (93.7 J∙g−1).
3. Results and Discussion
The FTIR spectra of pure PLA and PLA-g-GMA are shown in Figure 1. The spectrum of pure PLA indicated the peaks at 3000 - 2940 cm−1, 1761 cm−1 and 1190?1090 cm−1, which are associated with CH3 stretching, C=O stretching and the O?C=O stretching, respectively [8]. The peaks at 910 cm−1, 1150 cm−1, 1761 cm−1 and 3000 cm−1 were observed in the spectrum of PLA-g-GMA. Compared to the spectrum of pure PLA, a small new peak appeared at 910 cm−1 in the spectrum of PLA-g-GMA was observed, which is attributed to the asymmetric stretching of the epoxy group. This evidence demonstrates that the GMA was successfully grafted onto the PLA chain. Furthermore, as can be seen from Figure 1, there was no significant difference in FTIR spectra of PLA-g- GMA when the mixing time increased from 7 to 14 min. The NMR analysis was also used to confirm the result above. As seen in Figure 2, the 1H-NMR spectra of PLA and PLA-g-GMA showed two peaks at 5.2 and 1.6 ppm, which represent methylene protons and methyl protons of the PLA chain, respectively. However, compared to the spectrum of pure PLA, the new and weak peaks appeared at 0.9 - 4.3 ppm in the spectrum of PLA- g-GMA were observed. These peaks represent the protons 1 - 7 in the chemical structure of GMA as shown in Figure 2. Moreover, from the 1H-NMR spectrum of GMA, it can be observed that the chemical shifts of all peaks, which are associated with the GMA constitutional unit of proton CH, CH2 and CH3, are different from those in the spectrum of PLA-g-GMA. This observation demonstrates that the new multipeak belong to GMA
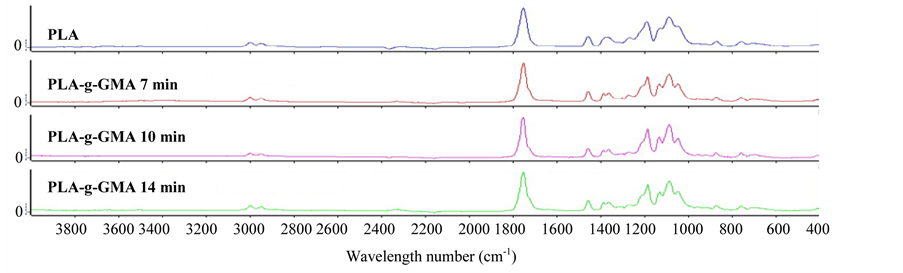
Figure 1. FTIR spectra of pure PLA and PLA-g-GMA with different mixing times.
grafted on PLA chain (not unreacted GMA). These results again confirmed that the GMA was successfully grafted onto the PLA chain. Similar result was also reported [9].
Effect of mixing time on mechanical properties of PLA-g-GMA was shown in Figure 3 and Figure 4. From Figure 3, PLA-g-GMA showed ductile fracture with necking and cold drawing in stress-strain curve. In contrast, brittle fracture of pure PLA was demonstrated. The elongation at break of PLA-g-GMA was considerably higher than that of PLA. From this result, it can be thought that the re-orientation of molecular chains of PLA-g-GMA and cold crystallization can occur under tensile force.
These phenomena did not occur in the case of pure PLA, which indicated the difference in crystallization behavior between pure PLA and PLA-g-GMA under applied tensile force. However, the tensile strength at break of PLA-g-GMA significantly decreased compared to that of neat PLA. This decrease is attributed to the decrease of crystallinity of the PLA, which is due to the higher hindrance of PLA crystallization by the increase of chain structural irregularity caused by grafting reaction. Moreover, Figure 3 also shows that the elongation at break of PLA-g-GMA increased slightly with the increase of mixing time from 7 to 10 min. This is possibly attributed to the increase of GMA grafting yield as mixing time increased from 7 to 10 min. On the other hand, it significantly decreased when the mixing time reached 14 min. It has been known that PLA is highly sensitive to heat, therefore, if the mixing time was kept too long, the degradation of PLA chain can occur easily. Furthermore, the GMA grafting yield had tendency to drop at certain mixing time as observed in another research [5] where
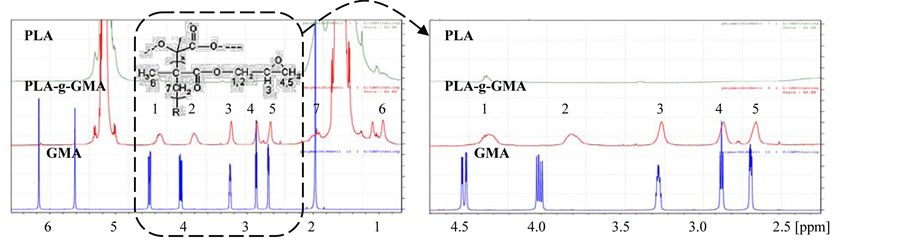
Figure 2. 1H-NMR spectra of pure PLA and PLA-g-GMA.
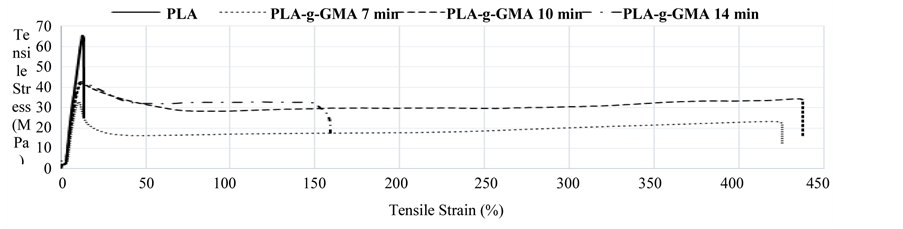
Figure 3. Effect of mixing time on tensile properties of PLA-g-GMA.
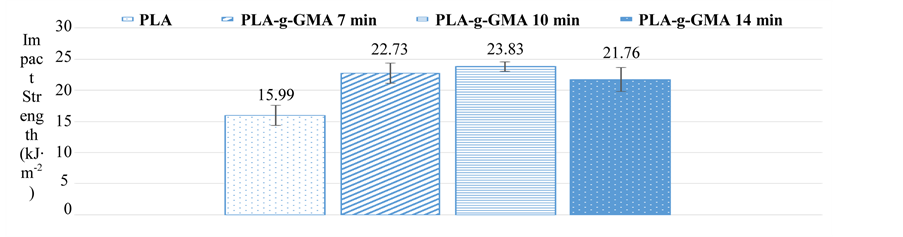
Figure 4. Effect of mixing time on impact strength of PLA-g-GMA.
GMA was grafted on HDPE. The reason for this may be attributed to depolymerization of GMA which has taken place as the mixing time is too long.
Moreover, as observed in Figure 4 that the impact strength of PLA-g-GMA was considerably higher than that of pure PLA. However, it was not significantly influenced by the mixing time. The impact strength of PLA-g- GMA increased slightly as mixing time increased from 7 to 10 min. Conversely, it decreased as mixing time increased from 10 to 14 min. This decrease is possibly due to the decrease of GMA grafting yield and degradation of PLA chains as the mixing time was kept too long. The results above indicate that the mixing time of 10 min was optimal for the mechanical properties of PLA-g-GMA.
Figure 5 shows the effect of mixing time on crystallization behavior of PLA-g-GMA. As seen from Figure 5 that the glass transition temperature of pure PLA was higher than that of PLA-g-GMA, which indicates the higher flexibility of PLA-g-GMA compared to pure PLA. Moreover, the cold crystallization temperature of PLA-g-GMA was lower than that of pure PLA. However, as observed in Figure 5 that the cold crystallization temperature of PLA-g-GMA was slightly shifted to higher value as mixing time increased from 7 to 10 min. This indicated that the recrystallization behavior occurred at higher temperature as mixing time increased from 7 to 10 min. This is possibly thought that the GMA grafting degree increased when mixing time increased from 7 to 10 min, which caused the decrease in chain mobility of PLA. As a result, the cold crystallization temperature increased. After that, with the further increase of mixing time, however, the cold crystallization temperature was shifted to lower value. This is due to the depolymerization of GMA occurring as mixing time was kept too long, which caused the increase of chain mobility.
The double melting peaks located at lower and higher temperatures were observed in Figure 5 for both pure PLA and PLA-g-GMA, which related to the less organized crystals and well-organized crystals, respectively [10]. However, the melting temperature of PLA-g-GMA was found to be lower than that of pure PLA. This implied that the formation of lamella was hindered by GMA grafted on PLA chains, which led to less perfect crystals of PLA-g-GMA [10]. Furthermore, it also indicated that GMA, on the one hand, acted as nucleation agent for the crystallization of PLA but reduced the chain mobility on the other hand.
As a result, the crystallinity of PLA-g-GMA as shown in Table 1 decreased compared to that of pure PLA. Moreover, it can be clearly seen from Figure 5 that the melting temperature of PLA-g-GMA did not change significantly with respect to the increase of mixing time from 7 to 10 min. However, when the mixing time increased from 10 to 14 min, the area of melting peak located at lower temperature became larger. It indicates that
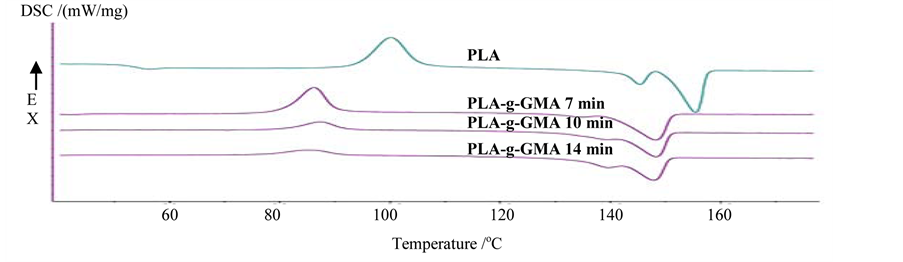
Figure 5. Effect of mixing time on crystallization behavior of PLA-g-GMA under second heating scan.

Table 1. Crystallinity of PLA and PLA-g-GMA at different mixing times.
more less perfect crystals were formed. As seen in Table 1, the highest crystallinity was obtained with the PLA- g-GMA prepared with the mixing time of 7 min.
4. Conclusion
The glycidyl methacrylate was successfully grafted onto poly (lactic acid) by internal mixer using dicumyl peroxide as an initiator. The mechanical properties and crystallization behavior of PLA-g-GMA were found to be considerably different from those of pure PLA. Moreover, these properties were slightly affected by melt mixing time. The mixing time of 10 min was found to be optimal for the grafting reaction under other certain conditions.
Acknowledgements
The authors would like to express gratitude to the SUT-PhD Scholarship for Asian Countries Program of Suranaree University of Technology, Thailand for the financial support and analytical instruments.
Cite this paper
Nguyen Chi Thanh,Chaiwat Ruksakulpiwat,Yupaporn Ruksakulpiwat, (2015) Effect of Melt Mixing Time in Internal Mixer on Mechanical Properties and Crystallization Behavior of Glycidyl Methacrylate Grafted Poly (Lactic Acid). Journal of Materials Science and Chemical Engineering,03,102-107. doi: 10.4236/msce.2015.37013
References
- 1. Garlotta, D., et al. (2003) Mechanical and Thermal Properties of Starch-Filled Poly (D, L-Lactic Acid)/Poly (Hydroxy Ester Ether) Biodegradable Blends. Journal of Applied Polymer Science, 88, 1775-1786. http://dx.doi.org/10.1002/app.11736
- 2. Rudnik, E. (2010) Compostable Polymer Materials. Elsevier.
- 3. Pesetskii, S. and Makarenko, O. (2002) Grafting of Glycidyl Methacrylate to Polypropylene in an Extruder, Initiated with Organic Peroxides. Russian Journal of Applied Chemistry, 75, 629-635. http://dx.doi.org/10.1023/A:1019529400627
- 4. Burton, E.L., et al. (2010) Reactive Grafting of Glycidyl Methacrylate onto Polypropylene. Journal of Applied Polymer Science, 117, 2707-2714. http://dx.doi.org/10.1002/app.31085
- 5. Cho, K.Y., et al. (2008) Grafting of Glycidyl Methacrylate onto High-Density Polyethylene with Reaction Time in the Batch Mixer. Journal of Applied Polymer Science, 108, 1093-1099. http://dx.doi.org/10.1002/app.27715
- 6. Xu, T., Tang, Z. and Zhu, J. (2012) Synthesis of Polylactide-Graft-Glycidyl Methacrylate Graft Copolymer and Its Application as a Coupling Agent in Polylactide/Bamboo Flour Biocomposites. Journal of Applied Polymer Science, 125, E622-E627. http://dx.doi.org/10.1002/app.36808
- 7. Huang, J., et al. (1998) Crystallization and Microstructure of Poly (l-lactide-co-meso-lactide) Copolymers. Macromolecules, 31, 2593-2599. http://dx.doi.org/10.1021/ma9714629
- 8. Orozco, V.H., et al. (2009) Preparation and Characterization of Poly (Lactic Acid)-g-Maleic Anhydride + Starch Blends. Macromolecular Symposia, 277, 69-80. http://dx.doi.org/10.1002/masy.200950309
- 9. Liu, J., Jiang, H. and Chen, L. (2012) Grafting of Glycidyl Methacrylate onto Poly (Lactide) and Properties of PLA/ Starch Blends Compatibilized by the Grafted Copolymer. Journal of Polymers and the Environment, 20, 810-816. http://dx.doi.org/10.1007/s10924-012-0438-1
- 10. Chen, R.-Y., et al. (2014) Poly(Lactic Acid)/Poly(Butylene Succi-nate)/Calcium Sulfate Whiskers Biodegradable Blends Prepared by Vane Extruder: Analysis of Mechanical Properties, Morphology, and Crystallization Behavior. Polymer Testing, 34, 1-9. http://dx.doi.org/10.1016/j.polymertesting.2013.12.009
NOTES

*Corresponding author.