 Journal of Cancer Therapy, 2011, 2, 181-189 doi:10.4236/jct.2011.22023 Published Online June 2011 (http://www.SciRP.org/journal/jct) Copyright © 2011 SciRes. JCT 181 Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, Senecence Escape, and Cell Polarity Change/Loss Kirsten H. Walen California Department of Public Health, Viral and Rickettsial Disease Laboratory, Richmond, USA. Email: kwalen@dhs.ca.gov Received January 17th, 2011; revised April 11th, 2011; accepted April 18th, 2011. ABSTRACT In cell cultures monola yered cell growth is controlled by c ontact inhibition which again is controlled by the cell polar- ity system by always being positioned in accord with the cytoskeleton axis. Presently, cycling endopolyploid cells (tetraploidy) were shown to undergo perpendicular divisions relative to the cytoskeleton axis which disrupted to some degree contact inhibition in the near-senescent phase of human primary cells. These experiments included genome damage-induced endopolyploidization (TAS-treated) to simulate as a model system the state of in vivo accelerated cell senescence (ACS) which is induced by therapy-associated genomic damage. From ACS delayed tumor re-growth (re- lapse) occurs from “robust” cell propagation, but mechanisms for such cell escape from senescence are unknown. For TAS-treated a karyoplast bud-off process with change to limited mitotic activity occurred in young senescent cultures. In old, deep senescent (5 - 8 weeks) cultures, unexpectedly escape cell-growth showed three dimensional (3-D) tu- mor-like spheres from growths of morphologica lly different cells as compared to th e fibrob lastic phenotyp e. These cells expressed cell polarity change, and very condensed nuclei were variously perpendicularly oriented to what-ever cell polarity was present. These results were discussed in regard to in vivo relapse and, to the importance of cell polarity change in tumorigenesis. Induced senescence as an anti-tumor mechanism in therapy treatment becomes a questionab le procedure from the present experimental results. Keywords: Endo-Division Perpendicularity, Contact Inhibition, Deep-Senescence, Escape-Route, Amorphous Flat Cells 1. Introduction Senescence of cells as an anti-tumor mechanism is con- troversial for both cancer-therapy-induced accelerated senescence (ACS) and telomere associated senescence (TAS) [1-5]. Recent works have shown both in vivo and in vitro reversions to proliferating cells [6-13]. ACS es- cape cells, following a period of time-delay, were shown to re-grow tumors with similar phenotypic tumor cells, but showing increased growth rate and aggressiveness [14-18]. How these sinister tumor cells gain propagating robustness, and how cells escape from the large flat cells which has a wrong nuclear/cytoplasmic ratio for mitotic activity, are unknown. These problems for effective therapy are difficult-to-impossible to assess for in vivo studies. Therefore, in vitro model studies in search of a cellular morphological base seem appropriate because, it can be followed by genetic pathway studies in both in vitro and in vivo. Such in vitro works for TAS has shown that one way of escape from young senescent cells was a karyoplast budding-o ff process [10-13]. These small cells, wrapped in mother-cell membrane grew in cytoplasmic content and entered mitosis, but with very limited pro- pagating capacity (2 - 16 cell stage). Presently, it is un- known if TAS escape cells express differential growth capacity coming from young or old senescent cells. But this possibility has some merit from observation of p53 activation and chromatin-associated DNA damage foci following H2O2 treatment of deep (old) senescent cells [19]. These experiments suggest that both time-delayed ACS- and TAS-associated escape cells have undergone repair of genomic damage during the protracted senes- cence arrest-period. And thus, escape cells from old se-  Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, 182 Senecence Escape, and Cell Polarity Change/Loss nescent cells may show gain in growth capacity (see Discussion). This supposition brings in anoth er relatively new issue in tumorigenesis which also influences growth capacity, namely: change/loss in cell polarity contro l of growth [20, 21]. This change was found to be associated with skewed/perpendicular cell division relative to the cy- toskeleton axis, and is entering human tumorigenesis via worms and Drosophila cancer studies [22,23 ]. Such divi- sion led to cancer cell development, but the mechanism behind perpendicularity is not known. It is however molecularly accepted that cell polarity change occurs in late tumorigenesis [22]. This peculiar positioning of the division plane occurs for the unicellular radiolarian Aulachanta in vegetative reproduction of endopolyploidy to offspring cells with polarity change [24]. Perpendicularity is not known for regular mitotic division, therefore, the idea of it being tied to the endopolyploid irregular division system [25, 26] is being explored in the present study. In human tu- mor progression there is a gradual increase in cells showing morphological, architectural alterations, desc r ib ed as mild, modest and severe dysplasia, which originate from a dedifferentiation process of the original differen- tiated (specialized) mother cell. The famous pathologist von Hansemann, linked such cell alterations to “greater capacity for independent existence” with possible change to “complete spherical cells” [27]. In simple words such cells have lost the original growth controlling cell polar- ity system, and unscheduled proliferation (e.g., telom- erase positive tumor cells) becomes a biological lawless process. The purpose/aim of the present study is to establish a model system for senescence-associated escape cell- events from young and old senescent cell cultures: are the escape routes and the growth capacities the same? But firstly, the presumed perpendicular endopolyploid division in the presenescent phase must be verified and assessed for associations to cell polarity loss/change. Therefore, escape cell growths is not only monitored for extended mitotic capacity, but also for cell morphologi- cal changes of the strongly polarized fibroblastic pheno- type. In order to simulate with greater likeness of the TAS- to the ACS-phase (from therapy-induced genomic dam- age to kill cells), primary diploid cells in the pre-senes- cent phase (3-6 population doublings from senescent flat cell changes) were exposed to genomic damage in addi- tion to that from broken telomeres (TAS-treated). In- crease in cycling endopolyploid cells (from re-replication of G2/G1 arrest escaped cells [28,29]) would precede activation of the senescence-inducing p53/p21/p14 pro- gram which are relatively slow processes in both in vitro and in vivo systems [4,9]. The present data showed karyoplast bud-offs and three dimensional (3-D) tu- mor-like spheres with polarity changed cells, but only in the old, deep senescent cell cultures. These results are discussed regarding: DNA-repair synthesis in flat cells, and how cell polarity change can be augmented by cells going through a senescent phase. 2. Materials and Methods The progressive culturing of diploid cell strains (L645 and WI-38 both from embryonic lungs) to the senescent phase has been described earlier [13,30] which include weekly passages at a 1 to 4 cell dilution factor. This method results in ~50 - 60 population doubling which is the Hayflick-limit [31]. The common Eagles’ basal me- dium supplemented with 10% serum, penicillin/ strepto- mycin and glutamine as cell growth medium was used. Late passage cells, 3 - 5 passages (near-senescent phase) before significant presence of non-proliferation capable large senescent flat cells, were washed 2x in Hank’ bal- anced salt solution. This was followed by a 1x-5-day exposure to medium deficient in amino acid glutamine (TAS-treated) [32,33]. Since 10% serum in such medium revealed continued, but reduced presence of mitotic cells, the amount was reduced to 2% for the present study. Following the five day exposure, complete medium (10%) was added for 1-2 days recovery period before tryp- sin/versene transfer to new flask cultures. These cultures were passaged with diminishing dilution until senescent cells predominated the cell populations. Control cultures (TAS-only) were passaged the same way, and consisted of two cultures each of the respective primary cell strains. The optimum survival for the treated cu ltures to reach 5 - 8 weeks in the senescent phase dependent on high cell density and pH adjustments during maintenance media changes. Low density cultures were therefore, combined by careful trypsin/versene treatment since they did not reveal sigificant re-growth from escape cells. Live cell photography (film) at 100x from well-surviving cultures was done by a VWR Vista Vision inverted, phase con- trast microscope with a Pentax (2x-M) camera attached. For endopolyploid perpendicular divisions pre-senescent cells (both cell strains) were grown on chamber slides for fixation at various harvest days which consisted of sev- eral changes with 3 parts methyl alcohol to one part of acidic acid (3:1 Carnoy’). The slides were air-dried, stained with 2% Giemsa in phosphate buffer solution at pH 6.8 and coverslipped with Coverbond. Photography through a Zeiss Standard microscope with an attached Zeiss camera for the following picture magnifications: 750x and 2475x for Figure 1 were done. Copyright © 2011 SciRes. JCT 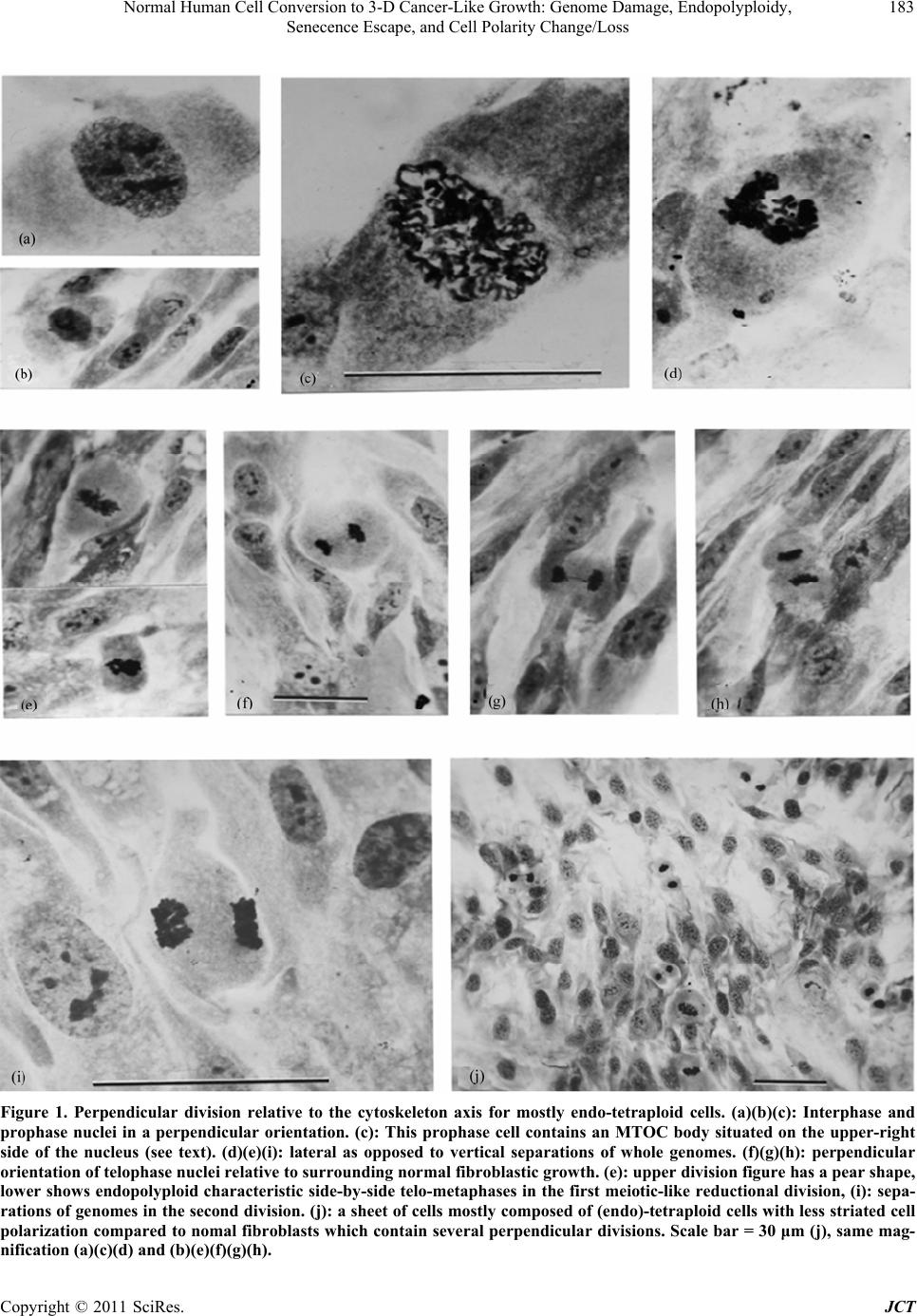 Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, Senecence Escape, and Cell Polarity Change/Loss Copyright © 2011 SciRes. JCT 183 Figure 1. Perpendicular division relative to the cytoskeleton axis for mostly endo-tetraploid cells. (a)(b)(c): Interphase and prophase nuclei in a perpendicular orientation. (c): This prophase cell contains an MTOC body situated on the upper-right side of the nucleus (see text). (d)(e)(i): lateral as opposed to vertical separations of whole genomes. (f)(g)(h): perpendicular orientation of telophase nuclei relative to surrounding normal fibroblastic growth. (e): upper division figure has a pear shape, lower shows endopolyploid characteristic side-by-side telo-metaphases in the first meiotic-like reductional division, (i): sepa- rations of genomes in the second division. (j): a sheet of cells mostly composed of (endo)-tetraploid cells with less striated cell polarization compared to nomal fibroblasts which contain several perpendicular divisions. Scale bar = 30 μm (j), same mag- nification (a)(c)(d) and (b)(e)(f)(g)(h).  Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, 184 Senecence Escape, and Cell Polarity Change/Loss 3. Results 3.1. Pre-Senescence Contact inhibition for normal fibroblast cells prevents cells to grow into multilayered growth and maintains nuclei/mitosis with apical/basal orientation in accord with the cytoskeleton axis (vertical division). Figure 1 shows various key features/consequences of perpendicu- lar endopolyploid divisions starting with interphase and prophase nuclei being perpendicularly oriented to the cell’ longitudinal axis (Figures 1 (a-c)). In Figure 1(c) (prophase) there is a small round body located on the upper-right side of the nucleus from which strands of material is radiating. Presumably, this is a microtubular organizing center (MTOC), but in the wrong po sition for normal mitotic vertical division. Genomic separations with so-called lateral moves for endopolyploid divisions are clearly demonstrated in figures 1DEI. The cells be- come pear-shaped (Figure 1(e)(i)) and in Figure 1(e) the characteristic endopolyploid side-by side star-like telo- metaphases are seen. The endopolyploid second division [13,25,26] is also clearly demonstrated in figure 1I and also perpendicularity of division products in relationship to surrounding fibroblastic cell growth (Figures 1(e)(g) (h)). This confirms the perpendicularity of single endo- polyploid cell division. Lastly Figure 1(j) shows several perpendicular divisions, in a cell sheet mostly composed of (endo)-polyploid cells with less striated polarity as compared to diploid cells. Taken together these few key illustrations undeniably demonstrate perpendicular divi- sions relative to the cytoskeleton axis of endopolyploid genome separations which are derived from re-replica- tion of genome damaged normal, diploid human cells. The five-day exposure of pre-senescent cells to me- dium deficient in glutamine caused a significant slim- ming of the cells and mitotic cells became absent (Figure 2(b) compare Figure 2(a)). In recovery, complete me- dium there was occurrence of synchronized mitotic ac- tivity a few hours later (Figure 2(d)). Figures 2(c) and (h) are growth foci with slight to moderate change in cell polarity axis. Figure 2(h) shows high mitotic activ ity and divisions to four products which are indicative of en- doploid genome reduction divisions [25,26]. The change to the senescent flat cell morphology was associated with presence of large amounts of lysosomes (Figure 2(f)) which is a senescent cell marker [34] and was visibly being extruded into the medium (see insert). The acidity caused considerable cell death such that cultures had to be combined for survival in deep senescencens (see M and M). (These cultures did not show chromocenters and lysosomes were reduced to a ring around the nuclei: Figure 3(a)). Escaped karyoplasts became attached to the cultural surface (Figure 2(g)) for mitotic activity, but as earlier observed the propagation was very limited in these young senescent cultures (not shown). 3.2. Escape Cell Growth from Deep Senescence In the old TAS-treated senescent cell cultures rarer growths as multilayered spheres (3-D) were present. These tumor-like balls were packed with cells (mostly dead) and were both surface attached and free floating in the medium (Figures 3(a-e)). Altogether thirteen varying sized spheres were observed, but the rate of occurrence was likely higher because, detached spheres were no doubt lost through media changes. More recent observa- tions for isolation purposes support this supposition. The cell morphology had chan ged from the striated fibroblas- tic phenotype to tri-quadri-shaped cells, and the nuclei were variously oriented in these cells. The nuclei also showed hyper-chromasia and were very condensed. A beginning focus of such cells appears to be located on top of an amorphous flat cell (Figure 3(a)). These cells showed early-on the ability of growing on top of each other (Figure 3(b)). Around the spheres there are these cells and also cells with less polarity changed morphol- ogy as compared to the fibroblast. 4. Discussion The present bud-off karyoplast mechanism leading to mitotic cells might be one answer to proliferation capable cells before activation of telomerase in tumorigenesis which is a late event [35]. The mechanism achieves es- cape from anti-proliferation proteins in flat cell cyto- plasm [36]. But it is not known to what degree such karyoplasts can rebuild an effective growth controlling cell polarity system. The limited growth from senescence escape cells in young cultures and the apparent mixture of polarized to non-polarized cells around the tumor-like spheres suggest varying ability for different escape cells. But the essential is that change in the cell polarity syste m was associated with 3-D cell growth in the present study. Interestingly, for human genome unstable oral tumors the divisions were oriented in skewed/perpendicular posi- tions relative to the cytoskeleton axis [37], and the ques- tion is: did this happening involve the present sequence of genome damage, cycling endopolyploidy with per- pendicularity, cell senescence, and 3-D escape cell growth with change in the cell polarity axis? The acquired growth characteristic of 3-D which is only known for cancer cells and was presently derived from normal diploid cells is a new finding. That these tumor-like spheres grew surface attached in normal liquid Copyright © 2011 SciRes. JCT 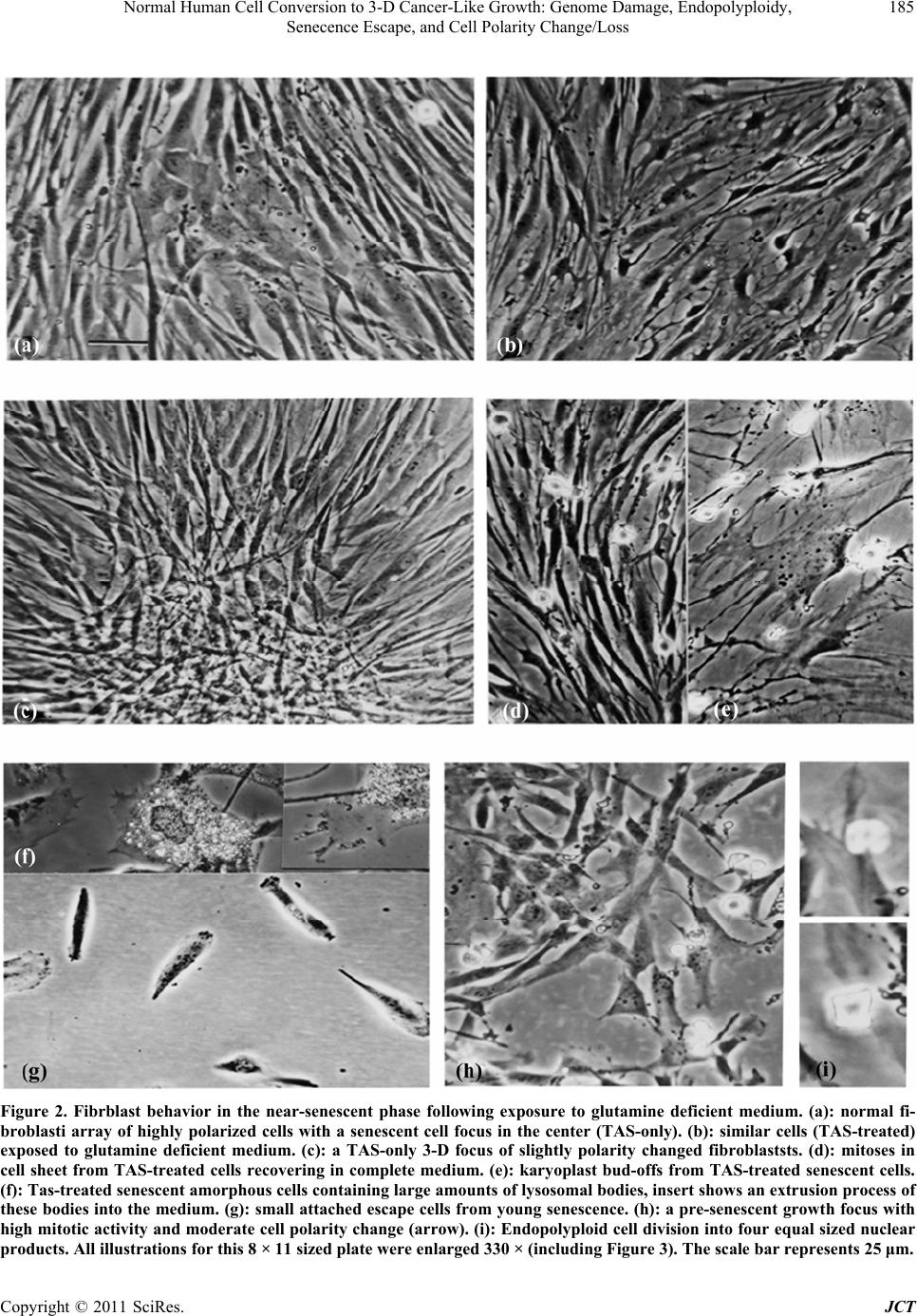 Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, 185 Senecence Escape, and Cell Polarity Change/Loss Figure 2. Fibrblast behavior in the near-senescent phase following exposure to glutamine deficient medium. (a): normal fi- broblasti array of highly polarized cells with a senescent cell focus in the center (TAS-only). (b): similar cells (TAS-treated) exposed to glutamine deficient medium. (c): a TAS-only 3-D focus of slightly polarity changed fibroblaststs. (d): mitoses in cell sheet from TAS-treated cells recovering in complete medium. (e): karyoplast bud-offs from TAS-treated senescent cells. (f): Tas-treated senescent amorphous cells containing large amounts of lysosomal bodies, insert shows an extrusion process of these bodies into the medium. (g): small attached escape cells from young senescence. (h): a pre-senescent growth focus with high mitotic activity and moderate cell polarity change (arrow). (i): Endopolyploid cell division into four equal sized nuclear products. All illustrations for this 8 × 11 sized plate were enlarged 330 × (including Figure 3). The scale bar represents 25 μm. Copyright © 2011 SciRes. JCT 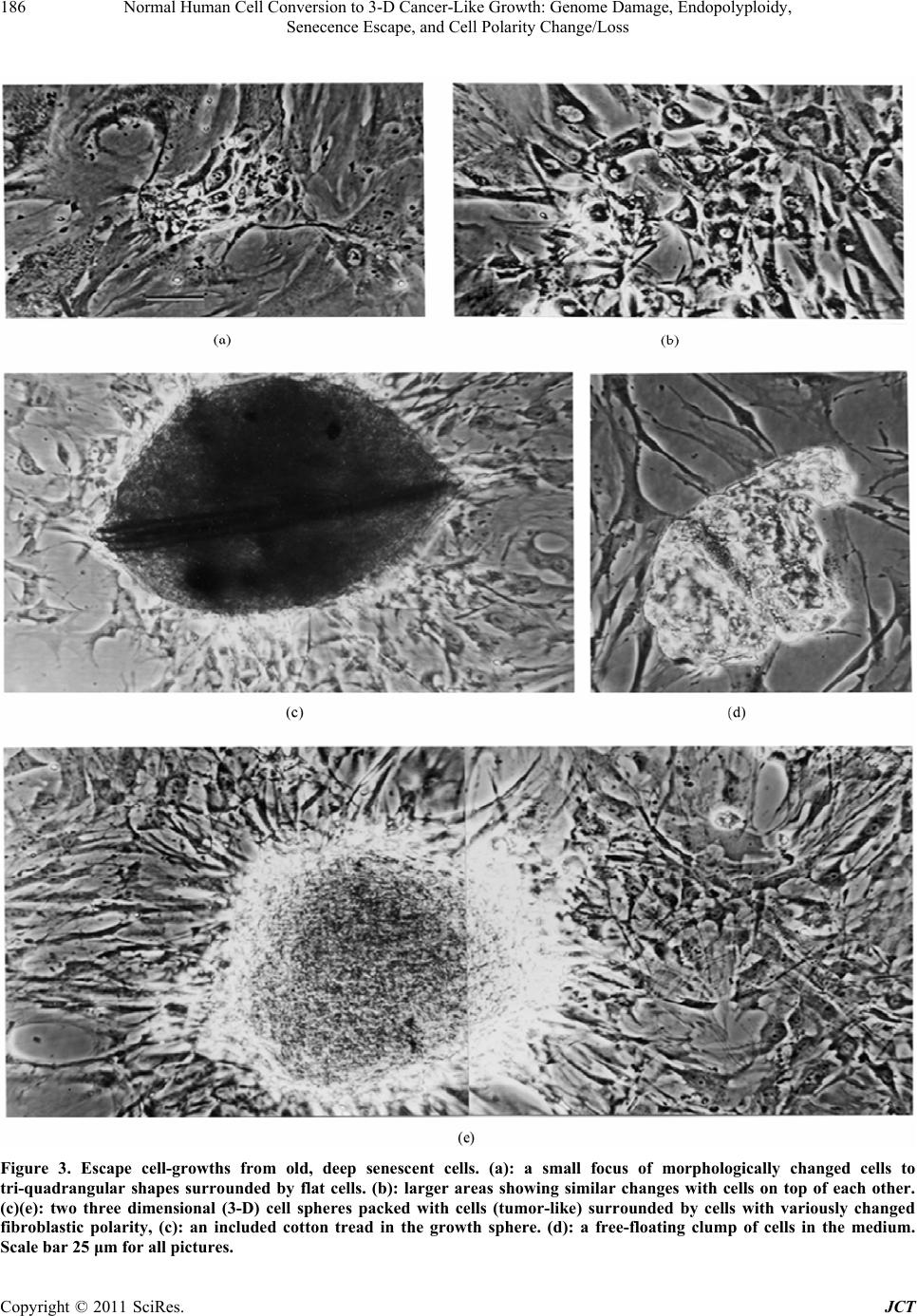 Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, 186 Senecence Escape, and Cell Polarity Change/Loss Figure 3. Escape cell-growths from old, deep senescent cells. (a): a small focus of morphologically changed cells to tri-quadrangular shapes surrounded by flat cells. (b): larger areas showing similar changes with cells on top of each other. (c)(e): two three dimensional (3-D) cell spheres packed with cells (tumor-like) surrounded by cells with variously changed fibroblastic polarity, (c): an included cotton tread in the growth sphere. (d): a free-floating clump of cells in the medium. Scale bar 25 μm for all pictures. Copyright © 2011 SciRes. JCT  Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, Senecence Escape, and Cell Polarity Change/Loss Copyright © 2011 SciRes. JCT 187 growth medium is extraordinary. The questions of how and why this cell growth occurred in deep senescent cul- tures may as already mentioned hinge on observations by Chen and Ozanne [19]. Their experiments showed that deep senescent cells are “active” beyond physiological processes for survival also indicated earlier [38,39]. The speculation was that dysfunctional telomeres that caused TAS can be repaired (gene-conversion recombination?) during the protracted deep senescent arrest-period [19, 40]. This notion implies occurrence of DNA syn thesis in senescent flat cells which was shown to occur by a slow, low rate of H3thymidine nuclear incorporation during a weeks-exposure time to low H3thymidine concentrations [38,41,42]. Synthesis of DNA in senescence is highly controversial [4,9]. Nevertheless, slow repair-synthesis might in general undo senescence arrest-associated ge- nomic damage (TAS-treated and ACS) leading to prefer- ential cell proliferation chance for escape cells from old as compared to young senescent cells. In the old senes- cent cultures a karyoplast bud-off process with attach- ment to free cultural space (Figures 2(e) and (g)) has not yet been observed, but nuclear isolation for cell escape can also be achieved by senescence cell-associated cyto- plasmic compartmentalization [43]. Clearly, this matter, and the difference in growth capacity for early versus late senescent escape cells with possible linkage to DNA- repair synthesis need further clarifications. Similarly, the 3-D growth from TAS-treated and not from TAS-only escape cells is troublesome because, it points to amino acid insufficiency being mutagenic by induction of growth pr omoting m ut a t ions. But interestingly, the present escape cells have also gone through the amorphous flat cell stage which does not express any known type of cell polarity orientations [44]. Their spread-out shapes (Figure 2(e) and (f)) make it difficult to see how escape cells can regain a functional, stabilized, polarity system for growth control by inter- cell-specific contact points (e.g., tight junctions).Thus, both ACS and TAS in pre-neoplasia may actually be promoting phases for augmentation (additional too that from perpendicularity in endopolyploid divisions) of cell polarity change with consequence of increased “inde- pendent existence” [27]. A speculation is that such gai- ned cell polarity change for escape cells which prior to senescence demonstrated mother-cell phenotypic sem- blance, can lead to a discontinuous step/gap in the cellu- lar, architectural, de-differentiation process. Possible examples are changes of adenoma to full-blown carci- noma and nevi to metastatic melanoma [45,46]. Another specific example that of t(14;18)-associated follicular lymphoma which following therape-induced repeated senescence programs, eventually produced escape cells (?) that showed: “histological “upgrade” of aggressive- ness to a so-called “transformed” lymphoma type” [4]. Cell polarity change/loss as a modifier of tumor cell- robustness is consistently not considered in the well- crafted reviews/reports on molecular-ways for improve- ments in cancer therapy. Therefore, the above suggested scenario with involvement of cell polarity change does not speak well for the possibility of cancer therapy by induced senescence [1,4] unless the “late” senescent es- cape route is known and can be controlled. 5. Conclusions The present study followed normal, diploid cells on a pathway of cellular events which was one way to tumor- like 3-D cell growth from morphologically ch anged cells. In fact there are no other routes known in spite of over 100 years search for a beginning and progression to tu- morigenesis. The key issues for the present cellular de- velopments involved: sufficient (?) genomic damage, endopolyploid perpendicular divisions to cell polarity change, disruption of contact inhibition, a protracted se- nescent cell-arrest period, escape cells from senescence, and growth of escape cells to multilayered tumor-like spheres. This is a rather gross sequence of events which however, include several tumorigenesis-associated stages as for instance, hyperplasia before telomerase activation, senescence occurrence in pre-neoplasia, with cell escape into different cancer-cell phenotypes. These similarities suggest a model role for the present observations in ex- plorations of initiation and progression in cancer devel- opment. 6. Acknowledgements I am very grateful for laboratory space, use of equipment and encouragements from Drs. David Schnurr and Sha- ron Messenger and for computer assistance from Mr. Chao Pan, all of the California Department of Public Health, VRDL unit. REFERENCES [1] I. B. Roninson, “Tumor Cell Senescence in Cancer Treatment,” Cancer Research, Vol. 63, 2003, pp. 2705- 2715. [2] J. Campisi, “Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens and Bad Neighbors,” Cell, Vol. 120, No. 4, 2005, pp. 513-522. doi:10.1016/j.cell.2005.02.003 [3] J. Bartkova, Z. Horejsi, K. Koed, A. Krammer, F. Tort, K. Zieger, P. Guldberg, M. Sehested, J. M. Nesland, C. Lu- kas, T. Orntoft, J. Lukas and J. Bartek, “DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis,” Nature, Vol. 434, No. 7035, 2005, pp. 864-870. doi:10.1038/nature03482  Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, 188 Senecence Escape, and Cell Polarity Change/Loss [4] C. A. Schmitt, “Cellular Senescence and Cancer Treat- ment,” Annals of Hematology, Vol. 1775, 2007, pp. 5-20. [5] D. M. Feldser and C. W. Gr eider, “Short T elome res Li mit Tumor Progression by Inducing Senescence,” Cancer Cell, Vol. 11, No. 5, 2007, pp. 461-469. doi:10.1016/j.ccr.2007.02.026 [6] R. S. Roberson, S. J. Kussick, E. Vallieres, S.-Y. J. Chen and D. Y. Wu, “Escape from Therapy-Induced Acceler- ated Cellular Senescence in P53-Null Lung Cancer Cells and in Human Lung Cancers,” Cancer Research, Vol. 65, 2005, pp. 2795-2803. doi:10.1158/0008-5472.CAN-04-1270 [7] L. W. Elmore, X. Di, C. Dumur, S. E. Holt, D. A. Ge- wirtz, “Evasion of a Single-Step, Chemotherapy-Induced Senescence in Breast Cancer Cells: Implications for Treatment Response,” Clinical Cancer Research, Vol. 11, No. 7, 2005, pp. 2637-2643. doi:10.1158/1078-0432.CCR-04-1462 [8] M. Sabisz and A. Skladanowski, “Cancer Stem Cells and Escape from Drug-Induced Premature Senescence in Human Lung Tumor Cells. Implications for Drug Resis- tance and In Vitro Drug Screening Models,” Cell Cycle, Vol. 8, No. 19, 2009, pp. 3208-3217. doi:10.4161/cc.8.19.9758 [9] C. M. Beausejour, A. Krtolica, F. Galimi, M. Narita, S. W. Lowe, P. Yaswen and J. Campisi, “Reversal of Human Senescence: Roles of P16 and P53 Pathways,” EMBO Journal, Vol. 22, No. 16, 2003, pp. 4212-4222. doi:10.1093/emboj/cdg417 [10] K. H. Walen, “Spontaneous Cell Transformation: Karyo- plasts Derived from Multinucleated Cells Produce New Cell Growth in Senescent Human Epithelial Cell Cul- tures.” In Vitro Cellular & Developmental Biology, Vol. 40, No. 5-6, 2004, pp. 150-158. doi:10.1290/1543-706X(2004)40<150:SCTKDF>2.0.CO; 2 [11] K. H. Walen, “Budded Karyoplasts from Multinucleated Fibroblast Cells Contain Centrosomes and Change Their Morphology to Mitotic Cells,” Cell Biology International, Vol. 29, No. 12, 2005, pp. 1057-1065. doi:10.1016/j.cellbi.2005.10.016 [12] K. H. Walen, “Human Diploid Fibroblast Cells in Senes- cence: Cycling Through Polyploidy to Mitotic Cells,” In Vitro Cellular & Developmental Biology, Vol. 42, No. 7, 2006, pp. 216-224. doi:10.1290/0603019.1 [13] K. H. Walen, “Genetic Stability of Senescence Reverted Cells: Genome Reduction Division of Polypolid Cells, Aneuploidy and Neoplasia,” Cell Cycle , Vol. 7, 2008, pp. 1623-1629. doi:10.4161/cc.7.11.5964 [14] M. Braig, S. Lee, C. Loddenkemper, C. Rudolph, A. H. Petrs, B. Schleglberger, H. Stein,B. Dorken, T. Jenuwein and C. A. Schmitt, “Oncogen-Induced Senescence as an Initial Barrier in Lymphoma Development,” Nature, Vol. 436, 2005, pp. 660-665. doi:10.1038/nature03841 [15] P.-E. Puig, M.-N. Guilly, A. Bouchot, N. Droin, D. Cath- elin, F. Boyer, L. Favier, F. Ghiringhelli, G. Kroemer, E. Solari, F. Martin and B. Chauffert, “Tumor Cells can Es- cape DNA-Damaging Cisplatin Through DNA Endore- duplication and Reversible Polyploidy,” Cell Biology In- ternational, Vol. 32, No. 9, 2008, pp. 1031-1043. doi:10.1016/j.cellbi.2008.04.021 [16] D. N. Wheatley, “Groving Evidence of the Re-population of Regressed Tumors by the Division of Giant Cells,” Cell Biology International, Vol. 32, No. 9, 2008, pp. 1029-1030. doi:10.1016/j.cellbi.2008.06.001 [17] M. V. Blagosklonny, “Cancer Stem Cell and Cancer Stem- loids.” Ca ncer Biology and Therapy, Vol. 6, No. 11, 2007, pp. 1684-1690. doi:10.4161/cbt.6.11.5167 [18] C. A. Schmitt, J. S. Fridman, M. Yang, S. Lee. E. Baranov, R. M. Hoffman and S. W. Lowe, “A Senescence Program Controlled by P53 and P16Ink4a Contributes to the Outcome of Cancer Therapy,” Cell, Vol. 109, No. 3, 2002, pp. 335-346. doi:10.1016/S0092-8674(02)00734-1 [19] J. H. Chen and S. E. Ozanne, “Deep Senescent Human Fibroblasts Show Diminished DNA Damage Foci but Retain Checkpoint Capacity to Oxidative Stress,” FEBS Letters, Vol. 580, No. 28, 2006, pp. 6669-6673. doi:10.1016/j.febslet.2006.11.023 [20] E. M. Munrov, “PAR Proteins and the Cytoskeleton: A Marrige of Equals,” Current Opinion in Cell Biology, Vol. 18, No. 1, 2006, pp. 86-94. doi:10.1016/j.ceb.2005.12.007 [21] S. Yoshida and D. Pellman, “Plugging the GAP between Cell Polarity and Cell Cycle,” EMBO Reports, Vol. 9, No. 1, 2008, pp. 39-41. doi:10.1038/sj.embor.7401142 [22] A. Wodarz and I. Nathke, “Cell Polarity in Development and Cancer,” Nature Cell Biology, Vol. 9, No. 9, pp. 1016-1024. doi:10.1038/ncb433 [23] P. Gonczy, “Mechanisms of Asymmetric Cell Division: Flies and Worms Pave the Way,” Nature Review, Vol. 9, No. 5, 2008, pp. 355-366. doi:10.1038/nrm2388 [24] K. G. Grell and A. Ruthmann, “Uber Die Karyologie des Radiolars Aulachantha scolymantha und die Feinstruktur Seiner Chromosomen,” Chromosoma, Vol. 15, 1964, pp. 185-211. doi:10.1007/BF00285729 [25] K. H. Walen, “Bipolar Genome Reductional Division of Human Near-Senescent, Polyploid Fibroblast Cells,” Cancer Genet Cytogenet, Vol. 173, 2007, pp.43-50. doi:10.1016/j.cancergencyto.2006.09.013 [26] K. H. Walen, “Origin of Dipochromosomal Polyploidy in Near-Senescent Fibroblast Cultures: Heterochromatin, Telomeres and Chromosomal Instability,” Cell Biology International, Vol. 31, 2007, pp. 1447-1455. doi:10.1016/j.cellbi.2007.06.015 [27] L. P. Bignold, B. L. D. Coghlan and H. P. A. Jersmann, “David von Hansemann: Contributions to Oncology: Context, Comment, and Translations,” Birkhauser Verlag, Basel, Switzerland, 2007. [28] D. J. Galgoczy and D. P. Toczyski, “Checkpoint Adapta- tion Precedes Spontaneous and Damage-Induced Ge- nomic Instability in Yeast,” Molecular and Cellular Bi- ology, Vol. 21, No. 5, 2001, pp. 1710-1718. doi:10.1128/MCB.21.5.1710-1718.2001 Copyright © 2011 SciRes. JCT  Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, Senecence Escape, and Cell Polarity Change/Loss Copyright © 2011 SciRes. JCT 189 [29] D. Brito and C. L. Rieder, “Mitotic Slippage in Humans Occurs Via Cyclin B Destruction in the Presence of an Active Checkpoint,” Current Biology, Vol. 16, No. 12, 2006, pp. 1194-2001. doi:10.1016/j.cub.2006.04.043 [30] K. H. Walen, “Spindle Apparatus Uncoupling in Endo-Tetraploid Asymmetric Division of Stem and Non-Stem Cells,” Cell Cycle, Vol. 8, 2009, pp. 3234- 3237. doi:10.4161/cc.8.19.9570 [31] L. Hayflick and P. S. Moorhead, “The Serial Cultivation of Human Diploid Cell Strains,” Experimental Cell Re- search, Vol. 25, 1961, pp. 585-621. doi:10.1016/0014-4827(61)90192-6 [32] J. J. Freed and S. A. Schatz, “Chromosome Aberrations in Cultured Cells Deprived of Single Essential Amino Acid,” Experimental Cell Research, Vol. 55, 1969, pp. 393-409. doi:10.1016/0014-4827(69)90574-6 [33] R. Phillip, E. Campbell and D. N. Wheatley, “Arginine Deprivation, Growth Inhibition and Tumor Cell Death: 2. Enzymatic Degradation of Arginine in Normal and Ma- lignant Cell Cultures,” British Journal of Cancer, Vol. 88, 2003, pp.613-623. doi:10.1038/sj.bjc.6600681 [34] B. Y. Lee, J. A. Han, J. S. Im, A. Morrone, K. Johung, E. C. Goodwin. W. Kleijer, D. DiMaio and E. S. Hwang, “Se- nescence-Associated Beta-Galactosidase is Lysosomal Beta-Galactosidase,” Aging Cell, Vol. 5, 2006, pp. 187-19 5. doi:10.1111/j.1474-9726.2006.00199.x [35] D. M. Feldser, J. A. Hackett and C. W. Greider, “Te- lomere Dysfunction and Initiation of genome Instability,” Nature Reviews Cancer, Vol. 53, 2003, pp. 623-627. [36] J. R. Smith and O. Pereira-Smith, “Replicative Senes- cence: Implications for IN Vivo Aging and Tumor Sup- pression.” Scienc e, Vol. 273, No. 5271, 1996, pp. 63-67. doi:10.1126/science.273.5271.63 [37] W. S. Saunders, M. Shuster, X. Huang, B. Gharaibe, A. H. Enyenihi, I. Petersen and S. M. Gollin, “Chromosomal Instability and Cytoskeletal Defects in Oral Cancer,” Proceedings of the National Academy of Sciences, Vol. 97, No. 1, 2000, pp. 303-308. doi:10.1073/pnas.97.1.303 [38] V. Gire and D. Wynford-Thomas, “Reinitiation of DNA Synthesis and Cell Division in Senescent Human Fibro- blasts by Microinjection of Anti-P53 Antibodies,” Mo- lecular and Cellular Biology, Vol. 18, 1998, pp. 1611- 1621. [39] A. M. Dirac and R. Bernards, “Reversal of Senescence in Mouse Fibroblasts through Lentiviral Suppression of P53,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 11731-11734. doi:10.1074/jbc.C300023200 [40] D. M. Baird, “Telomeres II,” Experimental Gerontology, Vol. 43, 2008, pp. 15-19. [41] V. J. Cristofalo and B. B. Sharf, “Cellular Senescence and DNA Synthesis. Thymidine Incorporation as a Measure of Population Age in Human Diploid Cells,” Experimen- tal Cell Research, Vol. 76, 1973, pp. 419-427. doi:10.1016/0014-4827(73)90394-7 [42] T. Matsumura, Z. Zerrudo and L. Hayflick, “Senescent Human Diploid Cells in Culture: Survival, DNA Synthe- sis and Morphology,” Journal of Gerontology, Vol. 34, 1979, pp. 328-335. [43] K. H. Walen, “Mitosis is not the Only Distributor of Mu- tated Cells: Non-Mitotic Endopolyploid Cells produce Reproductive Genome Reduced Cells,” Cell Biology In- ternational, Vol. 34, 2010, pp. 867-872. doi:10.1042/CBI20090502 [44] K. Nishio, A. Inoue, S. Qiao, H. Kondo and A. Mimura, “Senescence and Cytoskeleton: Overproduction of Vimentin Induces Senescent-Like Morphology in Human Fibroblasts,” Histochemistry and Cell Biology, Vol. 116, No. 4, 2001, pp. 321-327. doi:10.1007/s004180100325 [45] N. E. Sharpless and R. A. DePinho, “Crime and Punish- ment,” Nature, Vol. 436, 2005, pp. 636-637. doi:10.1038/436636a [46] C. Michaloglou, L. C. W. Vredeveld, M. S. Soengas, C. Denotells, T. Kuilman, C. M. A. M. Van der Horst, D. M. Majoor, J. W. Shay and W. J. Mooi, “BRAF-E600-Asso- ciated Senescence-Like Cell Cycle Arrest of Human Naevi,” Nature, Vol. 436, pp. 720-724. doi:10.1038/nature03890
|