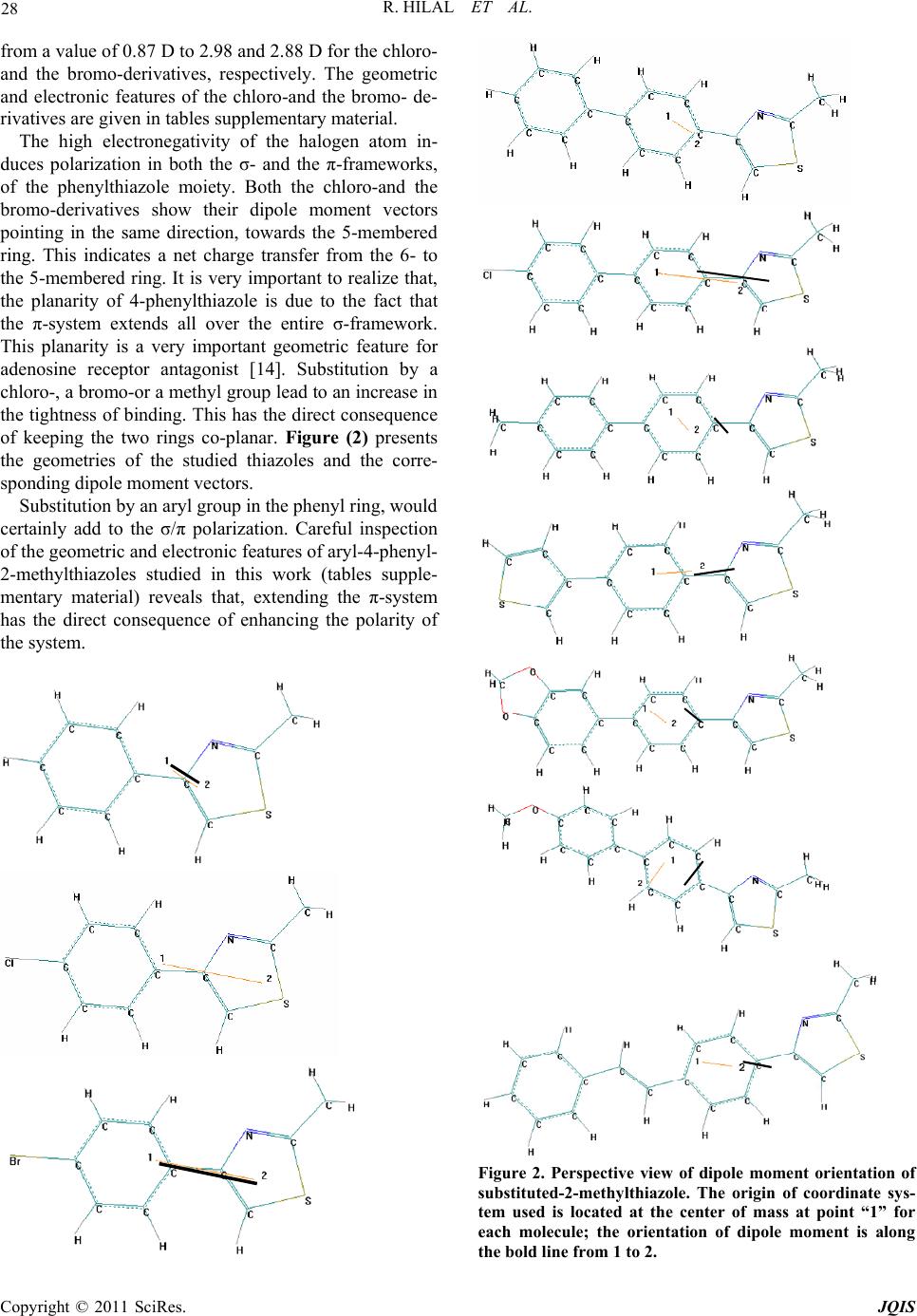
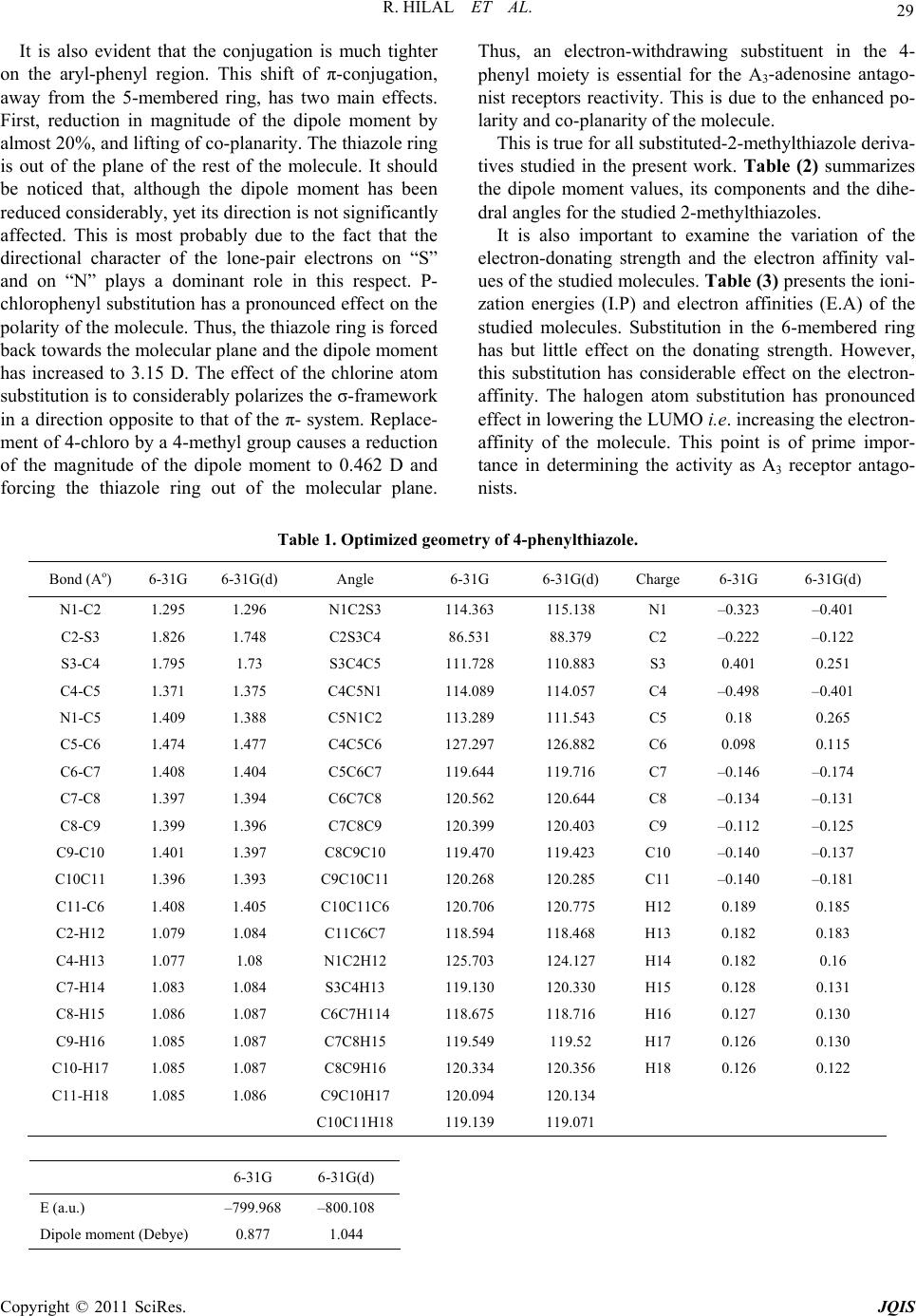
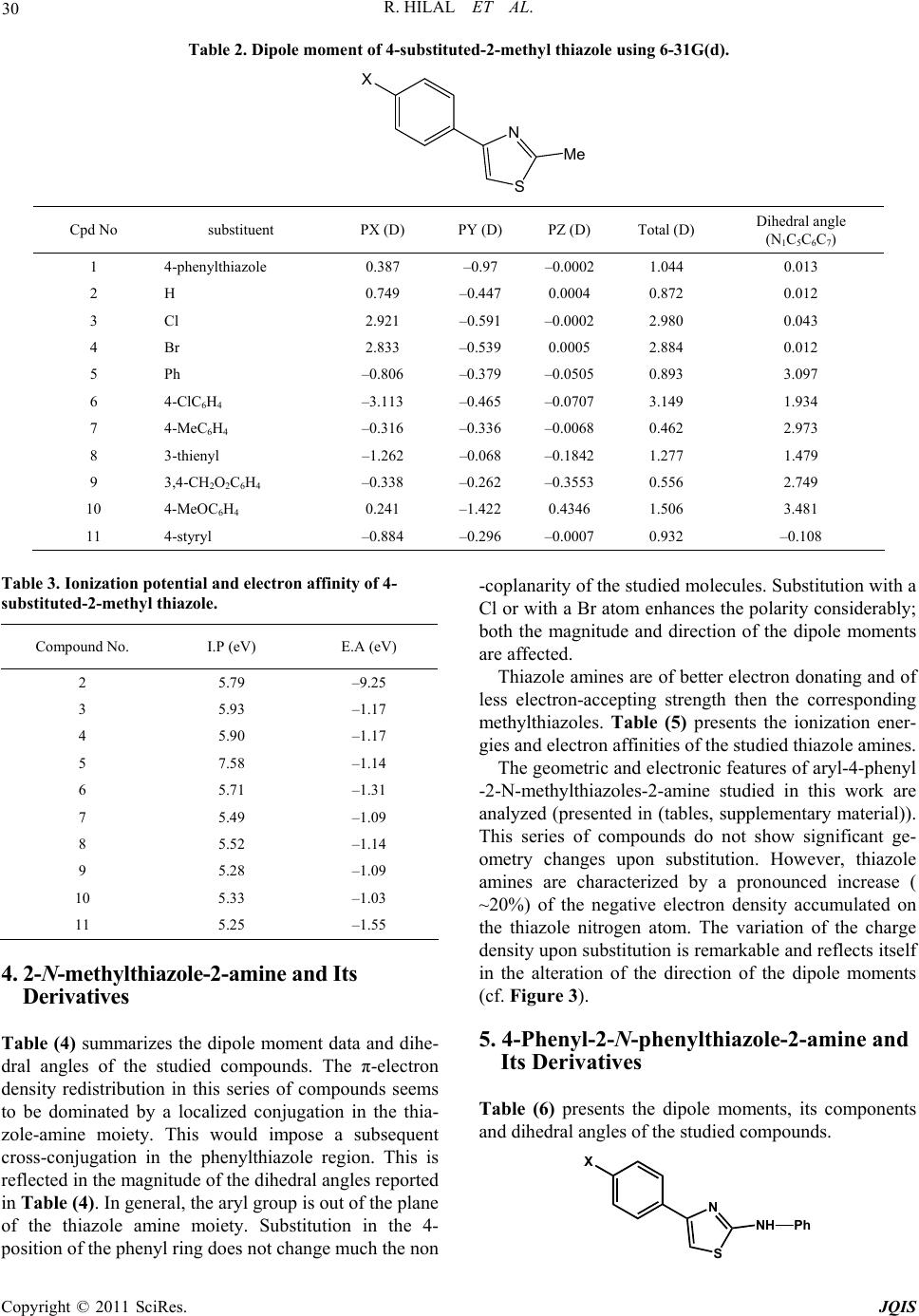
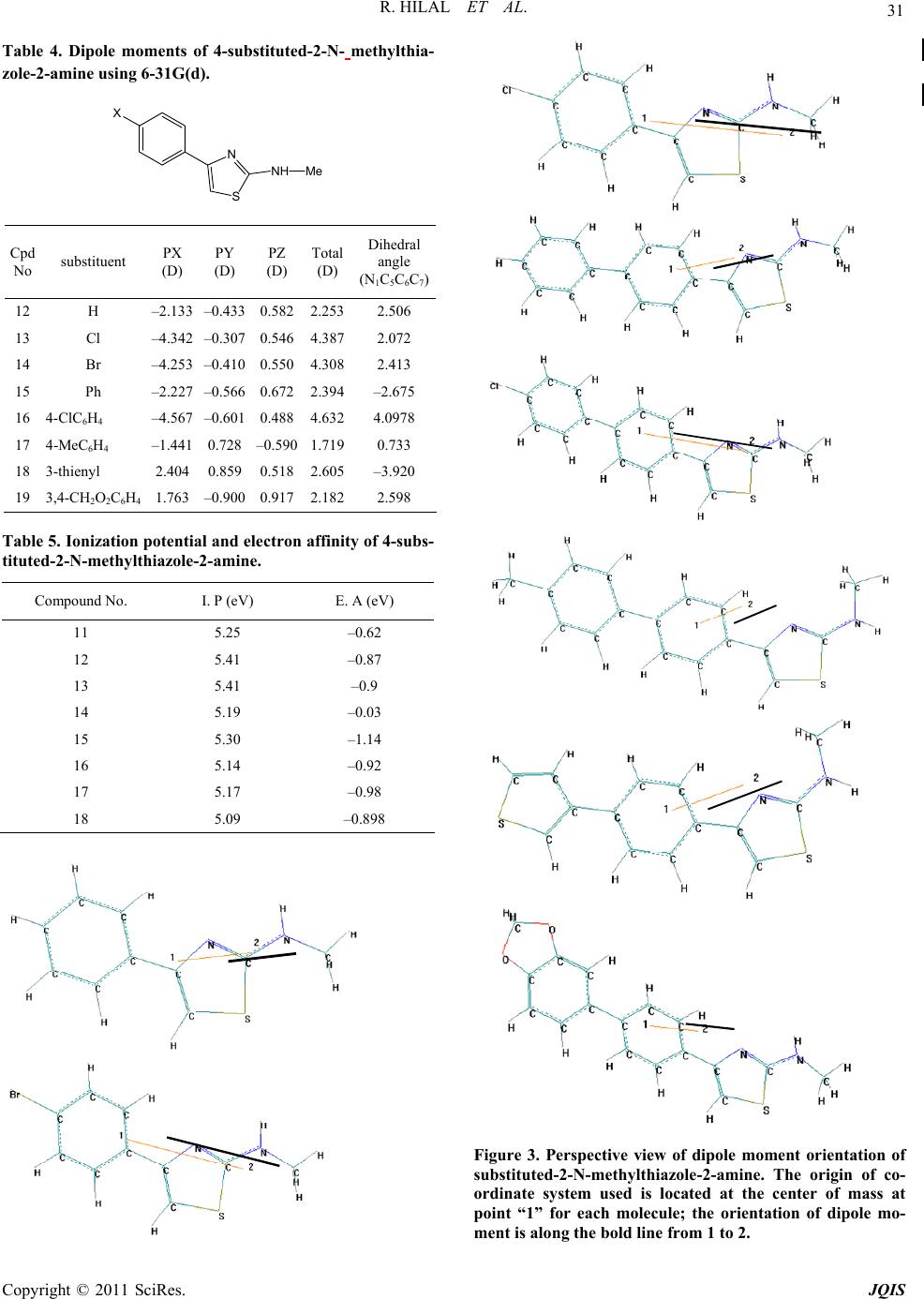
R. HILAL ET AL.
Copyright © 2011 SciRes. JQIS
33
vestigated. The energies of the HOMO's of the studied
series of thiazoles are around 5 eV. This low ionization
potential indicates a high tendency toward electron dona-
tion. Furthermore, aryl-2-methylthiazoles are character-
ized by low lying LUMO's, indicating considerable elec-
tron-affinity. This is of prime importance in enhancing
the activity towards A3-receptor sites.
The present analysis of the structure and electronic
properties of phenylthiazoles focuses on four main
structural features. The first feature is the co-planarity of
molecules. The second is the polarity, as indicated by the
dipole moments and their directions. The third feature is
the net negative charge on the thiazole “N” atom. This
charge enhances H-bond formation; a property of prime
importance for binding to A3-receptor sites. Finally, the
fourth feature is the electron donating and accepting ten-
dency of the studied thiazoles. A substituent that forces
co-planarity increases the dipole moment and the charge
accumulated on the thiazole “N” atom, and lowers the
LUMO, would certainly enhance the activity as potent and
selective adenosine A3-receptor antagonists.
6. References
[1] S. El-Tahar, K. M. El-Sawy and R. Hilal, “Electronic
Structure of Some Adenosine Receptor Antagonists: V.
QSAR Investigation ,”Journal of Chemical Information
And Computer Science, Vol. 42, No. 2, 2002, pp. 386-392.
doi:10.1021 /ci010307x
[2] Y.-C. Kim and K. A. Jacobson, “Derivatives of the Tri-
zoloquinazoline Adenosine Antagonist (CGS15943) are
Selective for the Human A(3) Receptor Subtype,” Jour-
nal Medicinal Chemistry, Vol. 39, No. 21, 1996, pp.
4142-4148.doi:10.1021/jm960482i
[3] P. G. Baraldi, B. Cacciari, R. Romagonoli, G. Spalluts,
K.-N. Klotz, E. Leung, S. Gessi, S. Merighi and P. A.
Porea, “Pyrazolo [4,3-e]-1, 2, 4-Triazolo[1,5-c]-Pyrimi-
dine Dervatives as Highly Potent and Selective Human
A(3) Adenosine Peceptor Antagonists,” Journal Medici-
nal Chemistry, Vol. 42, No. 22, 1999, pp, 4473-4478.
[4] K.-Y. Jung, S.-K. Kim, Z.-G. Gao, A. S. Gross, N. Mel-
man, K. A. Jacobson and Y-C. Kim, “Structure-Activity
Relationships of Thiazole and Thiadiazole Derivatives as
Potent and Selective Human Adenosine A(3) Receptor
Antagonists,” Bioorganic and Medicinal Chemistry, Vol.
12, No. 3, 2004, pp. 613-623.
doi:10.1016/j.bmc.2003.10.041
[5] J. E. V. Muijlwijk-Kooezen, H. Timmerman, R. C.
Vollinga, J. F. Kunzel, M. de Goote, S. Visser and A. P.
Ijzernan, “Thiazole and Thiadiazole Analogues as a
Novel Class of Adenosine Receptor Antagonists,” Jour-
nal Medicinal Chemistry, Vol. 44, No. 5, 2001, pp.
749-762.doi:10.1021/jm0003945
[6] N. J. Press, J. R. Fozard, D. Beer, R. Heng, F. Padova, P.
Tranter, A. Trifilieff, C. Walker and T. H. Keller, “New
Highly Potent and Selective Adenosine A3 Receptor An-
tagonists,” Abstracts of Pape rs of the American Chemical
Society, Vol. 224, 2002,1-2, MEDI 419.
[7] K. M. Dawood and M. M. El-Deftar, “Microwave-Assisted
Synthesis of 2-Substituted 4-Biarylyl-1, 3-thiazoles by
Carbon-Carbon Cross-Coupling in Water,” Synthesis, Vol.
6, 2010, pp. 1030-1038.doi:10.1055/s-0029-1218662
[8] F. Bellina, A. Carpita and R. Rossi, “Palladium Catalysts for
the Suzuki Cross-Coupling Reaction: An Overview of Re-
cent Advances,” Synthesis, Vol. 15, 2004, pp. 2419-2440.
[9] L. Botella and C. Nájera, “A Convenient Oxime-Carbap-
alladacycle-Catalyzed Suzuki Cross-Coupling of Aryl
Chlorides in Water,” Angewandte Chemie International
Edition, Vol. 41, No. 1, 2002, pp. 179-181.
doi:10.1002/1521-3773(20020104)41:1<179::AID-ANIE
179>3.0.CO;2-O
[10] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E.
Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Jr.
Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M.
Millam, S. S. Iyengar, J. Tomasi, V. Barone, B.
Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A.
Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota,
R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.
Honda, O. Kitao, H. Nakai, M. Klene, Li, J. E. Knox, H.
P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J.
Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J.
Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala,
K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg,
V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C.
Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K.
Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G.
Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu,
A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J.
Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A.
Nanayakkara, M. Challacombe, P. M. W. Gill, B. G.
Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A.
Pople, “Gaussian,” Revision C.02, Gaussian, Inc.,
Wallingford, CT, 2004.
[11] P. Hohenberg and W. Kohn, “Inhomogenous Electron
Gas,” Physical Review, Vol. 136, No. 3, 1964, pp. 864-871.
doi:10.1103/PhysRev.136.B864
[12] A. D. J. Becke, “Density-Functional Thermochemistry. 3.
The Role of Exact Exchange,” The Journal of Chemical
Physics, Vol. 98, No. 7, 1993, pp. 5648-5652.
doi:10.1063/1.464913
[13] W. Y. Lee and R. G. Parr, “Development of The Colle-
Salvetti correlation-energy Formula Into A Functional of
The Electron-Density,” Physical Review, Vol. 37, No. 2,
1988, pp, 785-789.doi:10.1103/PhysRevB.37.785
[14] K. A. Jacobson, P. J. M. Van Galen and M. Williams,
“Adenosine Receptors-pharmacology, Structure- Activ-
ity-Relationships, and Therapeutic Potential,” Journal
Medicinal Chemistry, Vol. 35, No. 3, 1992, pp. 407-422.
doi:10.1021/jm00081a001