Journal of Cancer Therapy
Vol.2 No.2(2011), Article ID:5711,15 pages DOI:10.4236/jct.2011.22013
The Opioid Growth Factor Inhibits Established Ovarian Cancer in Nude Mice and Can Be Combined with Taxol or Cisplatin to Enhance Growth Inhibition
![]()
Department of Neural and Behavioral Sciences, College of Medicine, the Pennsylvania State University, Hershey, USA.
Email: isz1@psu.edu
Received March 15th, 2011; revised April 12th, 2011; accepted April 19th, 2011.
Keywords: Cell Proliferation, [Met5]-Enkephalin, Opioid Growth Factor, OGFr, Ovarian Cancer
ABSTRACT
Ovarian cancer is the 5th leading cause of cancer-related mortality in women. Seventy-five percent of ovarian cancer patients present in advanced stages, and receive cytoreductive surgery and adjuvant chemotherapy. However, within 2 years 65% of these patients relapse and thereafter only receive palliative care. Novel therapies based on the biology of these cancers are urgently needed. The opioid growth factor (OGF)-OGF receptor (OGFr) axis is an endogenous opioid system known to inhibit proliferation of human ovarian cancer cells in tissue culture, but does not affect cell survival. The present study determined whether OGF in combination with standard of care chemotherapy, provides an inhibitory effect on the growth of human ovarian cancer cells in vitro. In addition, this investigation assessed whether OGF biotherapy, alone or in combination with taxol or cisplatin, inhibits tumor growth in mice with xenografts of ovarian cancer. The combination of OGF (10–6 M) with taxol (10–9 M or 10–10 M) or cisplatin (0.01 mg/ml or 0.001 mg/ml) markedly reduced cell number and DNA synthesis in vitro to a greater extent than individual compounds. OGF, but not taxol or cisplatin, altered growth in an opioid receptor mediated and reversible manner. Female nu/nu mice inoculated subcutaneously with SKOV-3 cells, and treated with OGF (10 mg/kg) for 5 weeks commencing at the time tumors became measurable, had tumor volumes and weight that were reduced by up to 50% from animals receiving saline. The combination of OGF with taxol (3 mg/kg, weekly) or cisplatin (4 mg/kg, weekly for 2 weeks) for 37 days reduced tumor volumes and weight in contrast to mice receiving individual agents alone. Moreover, OGF treatment in mice receiving cisplatin provided protection against the weight loss associated with cisplatin alone. All treatments suppressed DNA synthesis and angiogenesis, whereas exposure to taxol or cisplatin, but not OGF, induced apoptosis. Additive inhibitory effects on DNA synthesis and angiogenesis were recorded in animals treated with both OGF and taxol, or OGF and cisplatin, in comparison to individual compounds alone. OGF and OGFr were detected in tumor tissue; however OGFr expression was reduced 51% - 81% by OGF treatment. This preclinical evidence demonstrates that OGF biotherapy markedly inhibits ovarian tumorigenesis in a non-toxic manner, and can be combined with taxol or cisplatin to provide an enhanced therapeutic benefit.
1. Introduction
Ovarian cancer is the leading cause of death from gynecological malignancies [1], resulting in an estimated 140,200 deaths worldwide annually [2]. Approximately 90% of ovarian cancers are epithelial in origin [3], and the most common presentation (75%) is in the advanced stages (stage III/IV). Cytoreductive surgery and adjuvant chemotherapy with taxol and a platinum containing compound serve as the standard of care [4]. Although initial clinical response is excellent [4], 65% of patients relapse within 2 years and thereafter only receive palliative care [1]. Major improvements in the treatment of ovarian cancer patients will require novel therapies that capitalize on biological pathways [1].
Dysregulation of the cell cycle is an integral component of ovarian cancer [5]. One native biological regulator of cell replication in normal cells and a variety of cancers, including ovarian cancer, is the opioid growth factor (OGF) and its receptor, OGFr [6-11]. OGF is a constitutively active native opioid peptide, chemically known as [Met5]-enkephalin, that is autocrine produced and secreted, and interacts with OGFr to delay the G1/S interface of the cell cycle without affecting cell survival by modulating cyclin-dependent kinase inhibitory (CKI) pathways [11-15]. Although OGFr pharmacologically resembles classical opioid receptors (recognizes opioids, naloxone (Nal) reversibility, stereospecificity), it shares no sequence homology, has a different cellular localization (detected in the outer nuclear envelope, nucleus, and perinuclear cytoplasm [6,16-19]), and undergoes trafficking into the nucleus in a process that requires nuclear localization signals and transport by karyopherin b and Ran [17,20].
The OGF-OGFr axis has been shown to be present and involved in the regulation of human ovarian cancer cell proliferation in a tissue culture model [11,21]. OGFr RNA, protein, and binding activity have been documented in ovarian cancer cells in vitro [11], and OGF has been detected by radioimmunoassay in surgical samples taken from human ovarian neoplasms [22]. Moreover, an increase in OGF-OGFr activity in human ovarian cancer cells in tissue culture by the addition of exogenous OGF has been shown to markedly suppress cell proliferation in a non-toxic manner by targeting the CKI pathways [11, 21]. The present investigation explores whether OGF biotherapy 1) modulates human ovarian tumorigenesis in mice with established subcutaneous xenografts, and 2) can be combined with standard of care chemotherapies (taxol, cisplatin) to elicit an additive inhibitory effect on cell proliferation and tumorigenesis. The results demonstrate that OGF inhibits ovarian cancer in vivo, and can be combined with taxol or cisplatin for improved efficacy. Moreover, our data suggest that the toxic effects associated with chemotherapeutic agents utilized in the treatment of ovarian cancer can be alleviated by combining these drugs with OGF. Thus, the use of both chemotherapy and OGF biotherapy may provide a novel strategy in the treatment of this deadly disease.
2. Materials and Methods
2.1. Cell Culture
The human ovarian cancer cell line SKOV-3 [23], obtained from the American Type Culture Collection (Manassas, VA), was grown in a humidified atmosphere of 5% CO2/95% air at 37˚C in RPMI medium supplemented with 1.2% sodium bicarbonate, 10% fetal calf serum, and antibiotics (5000 units/ml penicillin, 5 µg/ml streptomycin, and 10 mg/ml neomycin).
2.2. Growth Assays
Cells were plated and counted 24 h later (time 0) to determine seeding efficiency. Compounds or vehicle were added at time 0; media and compounds were replaced daily unless otherwise indicated. Taxol was dissolved in DMSO (10–2 M) and further diluted in sterile water; all other compounds were prepared in sterile water, and dilutions represent final concentrations. An equivalent volume of vehicle was added to control (Co) wells. Cells were harvested at designated times, stained with trypan blue, and counted with a hemacytometer. At least 2 aliquots/well and 2 wells/treatment/time point were sampled.
2.3. Animals, Tumor Cell Implantation, and Tumor Growth
Four week-old athymic nu/nu female mice, purchased from The Charles River Laboratory (Wilmington, MA), were housed in pathogen-free isolator ventilated cages in a controlled-temperature room (22˚C - 25˚C) with a 12 - 12 h light/dark cycle (lights on 0700 - 1900). Sterile rodent diet (Harlan Teklad, Fredrick, MD) and water were available ad libitum. All procedures were approved by the IACUC Committee of the Pennsylvania State University College of Medicine, and conformed to the guidelines established by the NIH. Following a 48 h acclimation period, unanaesthetized mice were injected subcutaneously (s.c.) with SKOV-3 cells (4 × 106/mouse) into the right scapula region. Mice were weighed 3x/week, observed daily for initial appearance of tumors, and tumors were measured 3 times/week using vernier calipers. Volume was calculated using the formula l × w2 × π/6 where length (l) is the longest dimension, and the width (w) is the dimension perpendicular to the length [24].
2.4. Drug Treatment
Beginning on the day tumors became visible (day 0), six groups of mice (n = 12) were randomly assigned to receive intraperitoneal (i.p.) injections of OGF (10 mg/kg, daily), taxol (3 mg/kg, days 0, 7, 14, 21, 28, and 35), cisplatin (4 mg/kg, days 0 and 7), OGF (10 mg/kg, daily) and taxol (3 mg/kg, days 0, 7, 14, 21, 28, and 35), OGF (10 mg/kg, daily) and cisplatin (4 mg/kg, days 0 and 7), or an equivalent volume of saline (daily). These dosages and regimens were selected based on published reports [25-29]. To ensure that all mice received an equivalent number of injections, mice not assigned to receive treatment on a given day were injected with saline. In groups receiving combined therapy, OGF was administered first. Taxol was dissolved in DMSO (10–2 M) and further diluted in saline, while OGF and cisplatin were dissolved in saline; all drugs were prepared weekly.
2.5. Termination Day Measurements
According to the IACUC guidelines, the study was terminated when tumors became ulcerated or grew to 2 cm in diameter. All mice were euthanized by an overdose of sodium pentobarbital (100 mg/kg) and cervical dislocation 37 days following initiation of treatments. For examination of DNA synthesis rates in tumors, a subset of mice from each group were injected i.p. twice with 100 mg/kg BrdU (Sigma-Aldrich, St. Louis, MO) at 6 and 3 h prior to euthanasia. Tumors and spleens were removed and weighed, and the lymph nodes, liver, and spleen examined for metastases. Tumor tissues were assessed for expression of OGF and OGFr, cell survival, angiogenesis, and DNA synthesis.
2.6. Semiquantitative Immunohistochemistry
Immunohistochemistry was utilized to evaluate the presence and relative levels of OGF and OGFr in tumor tissue following published procedures [11]. Tumors were excised, frozen in chilled isopentane, sectioned at 10 μm, fixed, permeabilized, and stained with antibodies to OGF and OGFr that were generated in our laboratory [30]. Images were taken at the same exposure time with care not to photobleach the preparations. A random sample of at least 10 fields/section, 2 sections/tumor, and 2 tumors/group were evaluated. Controls were incubated with secondary antibodies only.
2.7. Protein Isolation and Western Blotting
Expression of OGFr was evaluated in tumors by Western blotting following published procedures [11]. Briefly, tissue was homogenized in RIPA buffer containing a cocktail of protease and phosphatase inhibitors (Roche, Indianapolis, IN). Protein (60 μg) was subjected to 15% SDS-PAGE, transferred to nitrocellulose, and probed with antibodies to OGFr (1:200). Optical densities were normalized to β-actin (1:5000, Sigma-Aldrich), and the percent change in expression was calculated by dividing the normalized values of experimental samples to that of saline controls. Means and SE were determined from 2 independent experiments.
2.8. OGFr Binding Assays
Tumors were assayed for OGFr binding using custom synthesized [3H]-[Met5]-enkephalin (Perkin Elmer, Waltham, MA; 52.7 Ci/mmol) following published procedures [11,30,31]. Saturation binding isotherms were generated using GraphPad Prism software (La Jolla, CA), and independent assays were performed at least 3 times.
2.9. Mechanism of Growth Inhibition: DNA Synthesis, Angiogenesis, Apoptosis and Necrosis
Cells were assayed for DNA synthesis, necrosis, and apoptosis, whereas tumor tissue was evaluated for DNA synthesis, apoptosis, and angiogenesis. To measure DNA synthesis, cells were treated with 30 μM BrdU for 3 h prior to fixation, while tumors from mice receiving BrdU on the day of sacrifice were fixed in formalin overnight, processed in paraffin, and sectioned at 10 μm. Preparations were processed with antibodies to BrdU (1:200, Invitrogen, Carlsbad, CA) [11,31,32] to assess DNA synthesis, stained with Hematoxylin/Eosin [33,34] to evaluate endothelial cell-lined vessels containing red blood cells, or processed for TUNEL according to the manufacturer’s instruction to measure apoptosis (Trevigen, Gaithersburg, MD). For cells in tissue culture, the proportion of BrdU or TUNEL positive cells was determined for at least 500 cells on 2 coverslips/treatment group. For tumors, the proportion of BrdU positive cells, number of TUNEL positive cells, and blood vessel density were determined from at least 10 random fields around the periphery of each tumor, with at least 2 sections/tumor, and 2 tumors/treatment group evaluated. BrdU and TUNEL positive cells were counted in a 0.003 mm2 area, while blood vessel density was determined in a 0.16 mm2 area.
2.10. Chemicals
OGF and Nal were obtained from Sigma-Aldrich. Cisplatin was purchased from Alexis Biochemicals (Lausen, Switzerland), and taxol was obtained from Toronto Research Chemicals (North York, ON).
2.11. Statistics
All data were analyzed using one way analysis of variance (ANOVA), with subsequent comparisons made using Newman-Keuls tests (Graph Pad Prism Software). In some cases, data were evaluated with unpaired t-tests; p values less than 0.05 were considered significant.
3. Results
3.1. Combination of OGF with Taxol or Cisplatin Provides an Additive Inhibitory Effect on Cell Number: In Vitro Studies
To establish the efficacy of the combination of OGF with taxol on the growth of human ovarian cancer cells, SKOV-3 cultures were treated with OGF (10–6 M, a dosage known to inhibit cell proliferation [11,21]), taxol (10–9 M or 10–10 M, dosages selected because preliminary experiments revealed no logarithmic growth at higher concentrations), taxol and OGF, or an equivalent volume of sterile water and monitored for 120 h. Relative to sterile water treated controls, cell number was reduced in cultures exposed to OGF (17% - 36%), taxol at 10–9 and 10–10 M (26% - 47%), and OGF and taxol at 10–9 and 10–10 M (24% - 61%) (Figure 1(a)). In cultures receiving both OGF and taxol (at either concentration), cell number was reduced 16% - 48% from 72 h to 120 h compared to cells exposed to OGF alone. In comparison to cells treated with taxol alone (at either concentration), cell number in cultures treated with both OGF and taxol (at either concentration) was reduced 15% - 28% at 96 h and 120 h (Figure 1(a)). At all time points evaluated, an equivalent number of cells were noted in cultures receiving both OGF and taxol (10–10 M) compared to those receiving the higher concentration of taxol (10–9 M) alone (Figure 1(a)).
To establish the efficacy of the combination of OGF with cisplatin on the growth of human ovarian cancer cells, SKOV-3 cultures were treated for 120 h with OGF (10–6 M), cisplatin (0.01 μg/ml or 0.001 μg/ml; dosages selected because preliminary experiments revealed no logarithmic growth at higher concentrations), OGF and cisplatin, or an equivalent volume of sterile water. Relative to sterile water treated controls, cell number was reduced by OGF (25% - 37%), cisplatin at either concentration (18% - 52%), or both OGF and cisplatin (at either concentration) (24% - 60%) (Figure 1(b)). In cultures receiving a combination of OGF and cisplatin (at either concentration), cell number was reduced 24% - 46% compared to cells exposed to OGF alone, and decreased 15% - 30% relative to cells exposed to cisplatin (at either concentration) alone (Figure 1(b)). At all time points evaluated, an equivalent number of cells were noted in cultures receiving OGF and cisplatin (0.001 μg/ml) compared to those receiving the higher concentration of cisplatin (0.01 μg/ml) alone (Figure 1(b)).
3.2. Opioid Receptor Mediated Effects of OGF, but Not Taxol or Cisplatin
To determine whether the effects of OGF, taxol, or cisplatin, were mediated by opioid receptors, cultures were exposed to the short acting opioid receptor antagonist Nal. Cells were treated for 96 h with OGF (10–6 M), Nal (10–6 M), taxol (10–9 M or 10–10 M), cisplatin (0.01 μg/ml or 0.001 μg/ml), OGF and Nal, taxol and Nal, cisplatin and Nal, or an equivalent volume of sterile water. Relative to control levels at 96 h, addition of OGF, taxol, or cisplatin inhibited cell number by 26% - 49% (Figures 1(c) and (d)). Addition of Nal, at a concentration that alone had no effect on cell number, completely blocked the growth inhibitory effects of OGF, but had no influence on the inhibitory action of taxol or cisplatin (Figures 1(c) and (d)).
3.3. Reversibility of the Inhibitory Effects of OGF, but Not Taxol or Cisplatin
To establish whether the inhibitory effects of OGF, taxol, or cisplatin on cell number could be reversed by withdrawing cells from drug exposure, cultures of SKOV-3 cells were exposed for 72 h to OGF (10–6 M), taxol (10–9 M or 10–10 M), cisplatin (0.01 μg/ml or 0.001 μg/ml), or an equivalent volume of sterile water. At 72 h, one-half of the groups stopped receiving drug treatment, and media was replaced without compounds (i.e. OGF-reversal, taxol-reversal, cisplatin-reversal); the remaining cultures continued to receive media and compounds. At 96 and 120 h, the OGF-reversal group had 40% and 42% more cells, respectively, than in the group continuing to receive OGF, and at 120 h had a comparable number of cells as sterile water treated cultures (Figures 1(e)-(h)). However, the taxol and cisplatin reversal groups did not differ from cultures continuing to be treated with taxol or cisplatin, respectively, and remained significantly decreased from sterile water controls (Figures 1(e)-(h)).
3.4. Mechanism of Enhanced Growth Inhibition In Vitro
To evaluate the mechanism(s) by which treatment with both OGF and taxol, or a combination of OGF and cisplatin, decrease SKOV-3 cell number, DNA synthesis and cell survival were evaluated. Compared to the BrdU labeling index of sterile water treated cells (28.5% + 2.1%), cells treated with OGF (10–6 M), taxol (10–9 M or 10–10 M), or cisplatin (0.01 μg/ml or 0.001 μg/ml) had 31% - 46% less cells incorporating BrdU (Figures 2(a) and (b)). Cultures receiving both OGF and taxol (10–10 M), or OGF in combination with taxol (10–9 M), had 45% and 60%, respectively, fewer cells labeled with BrdU relative to cells exposed to OGF, and 56% and 60%, respectively, less cells labeled with BrdU relative to cultures treated with taxol alone (Figure 2(a)). Cultures receiving OGF and cisplatin (0.01 μg/ml) had decreased labeling indexes of 47% and 43% relative to cultures receiving either OGF or cisplatin (0.01 μg/ml), respectively (Figure 2(b)). However, comparable BrdU labeling indexes were noted in cultures exposed to OGF and cisplatin (0.001 μg/ml) in contrast to cells receiving OGF or cisplatin (0.001 μg/ml) alone (Figure 2(b)).
Examination of apoptosis or necrosis in SKOV-3 cells treated with OGF (10–6 M), taxol (10–9 M or 10–10 M), cisplatin (0.01 μg/ml or 0.001 μg/ml), OGF and taxol, or OGF and cisplatin, revealed less than 0.1% positive cells for apoptosis and necrosis, and these data
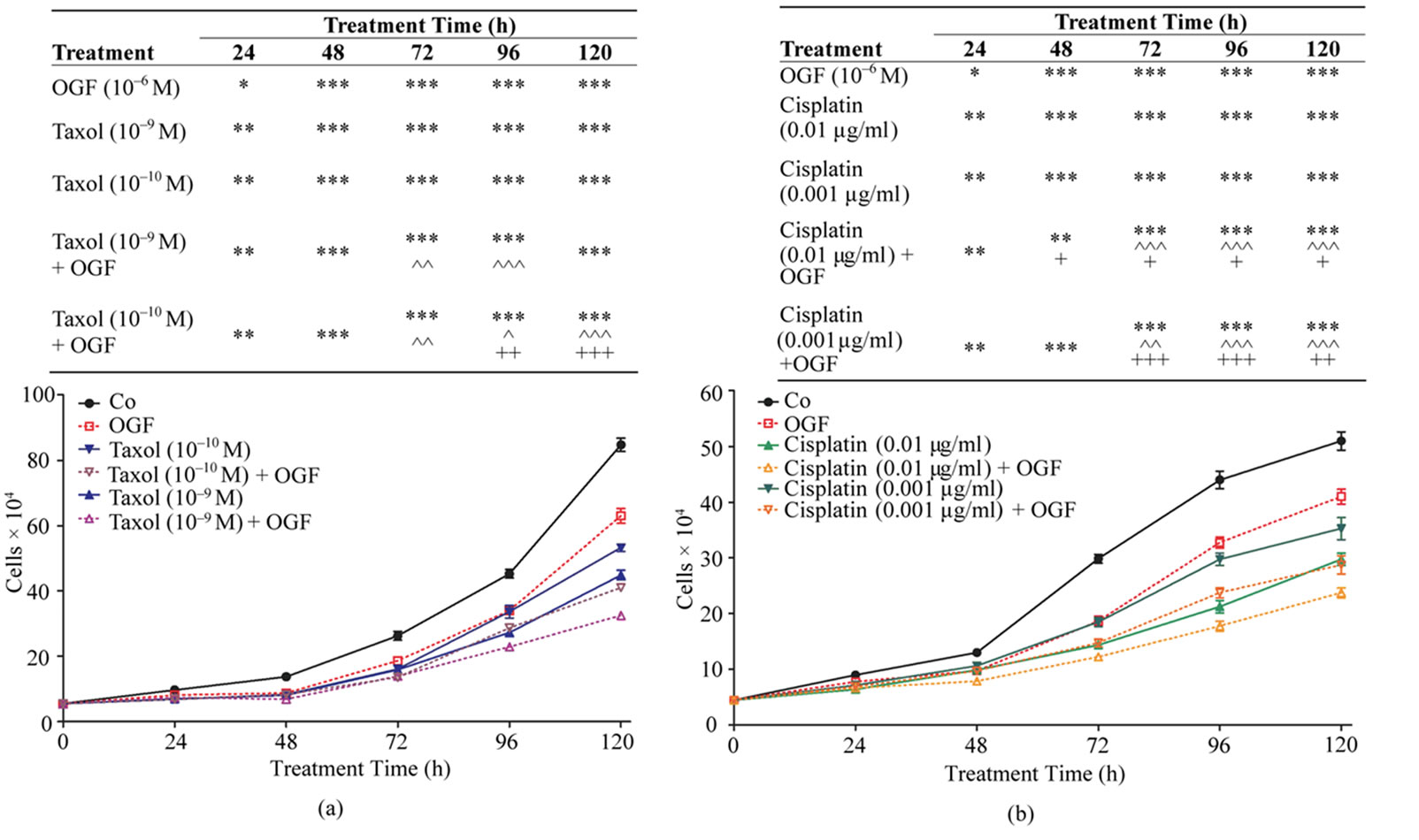
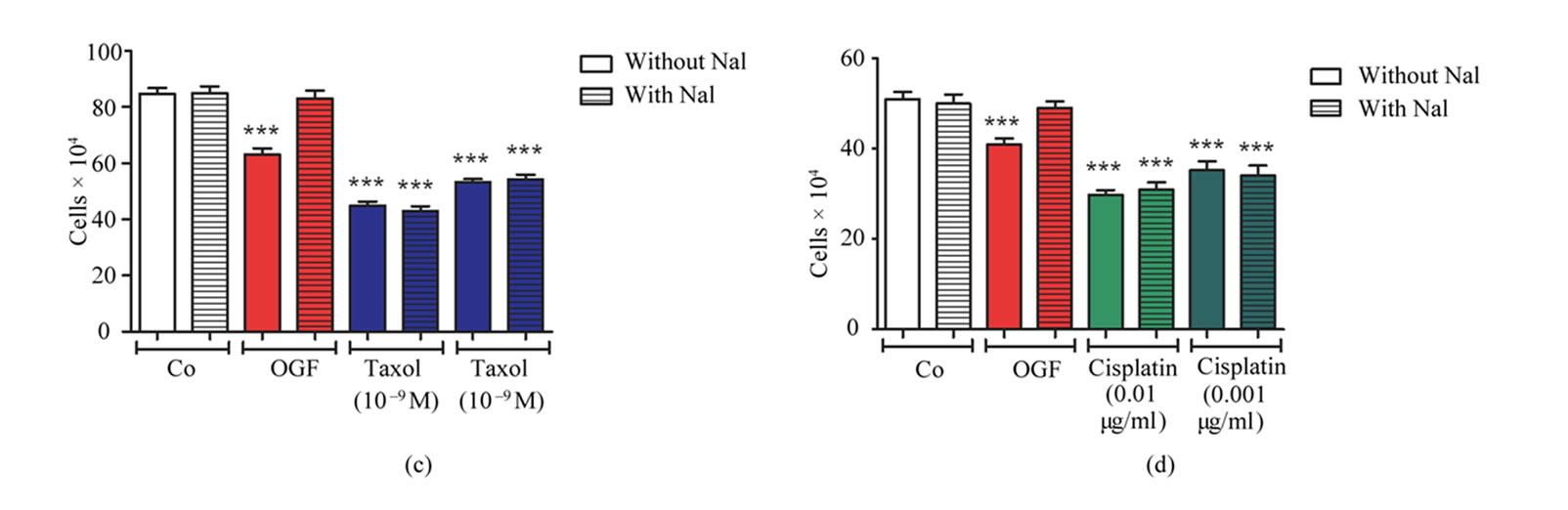
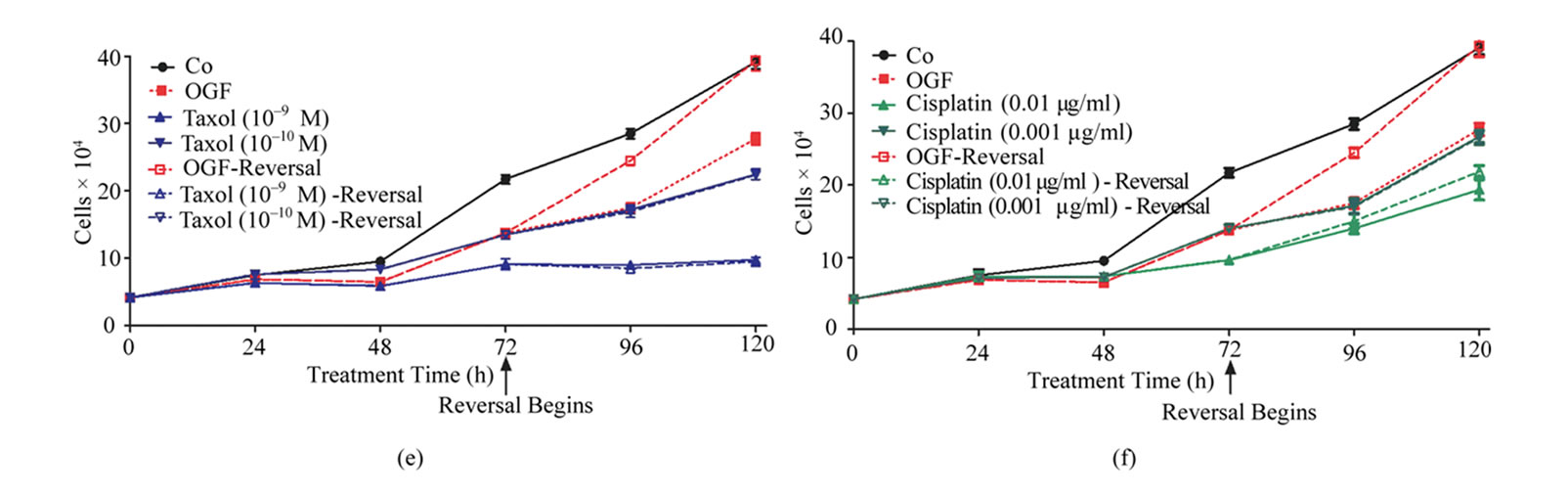
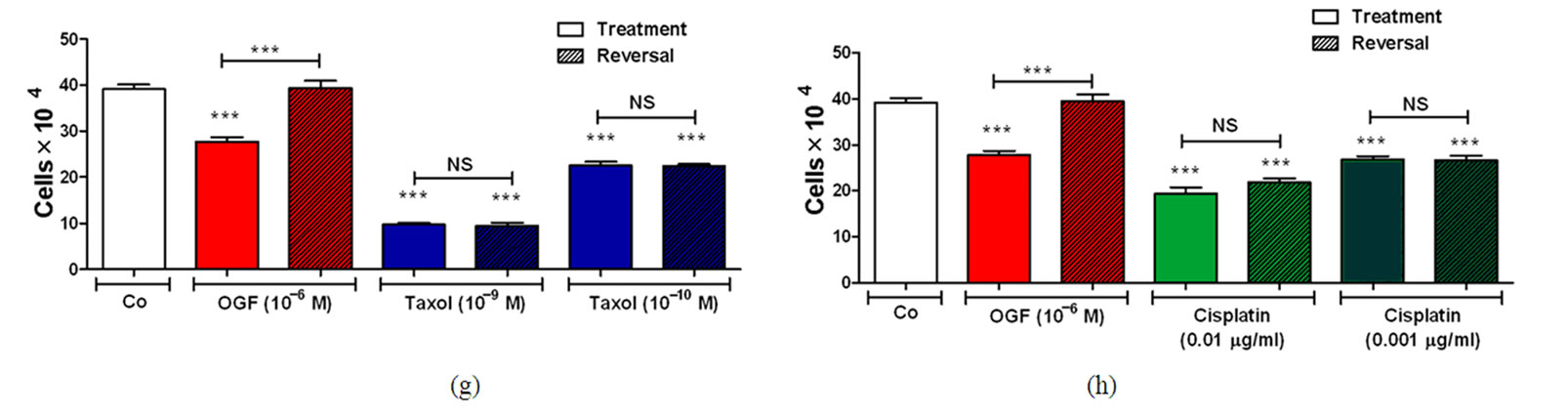
Figure 1. Effects of OGF in combination with taxol or cisplatin on the growth of SKOV-3 human ovarian cancer cells. Cells were treated with OGF (10–6 M), taxol (10–9 M or 10–10 M), cisplatin (0.01 mg/ml or 0.001 mg/ml), OGF and taxol, OGF and cisplatin, or an equivalent volume of sterile water (Co). Media and compounds were replaced daily unless otherwise indicated. (a) and (b) Growth of cells subjected to OGF alone or in combination with taxol (a) or cisplatin (b) over a 120 h period. (c) and (d) Opioid receptor mediation of the growth inhibitory effects of OGF, taxol, or cisplatin. Cell number following 96 h of treatment with OGF, taxol, cisplatin, the opioid antagonist Nal (10–6 M), or the combination of Nal with these compounds. (e) and (f) Growth of SKOV-3 cells in reversibility experiments with OGF, taxol, or cisplatin for 72 h; at 72 h, a subset of cultures continued to receive treatments for an additional 48 h, while the other cultures were administered sterile water for 48 h (Reversal). (g) and (h) Cell number at 120 h in cultures from the reversibility experiments. Values represent mean ± SE cell counts for at least 2 aliquots/well and 2 wells/treatment/timepoint. Significantly different from Co at *p < 0.05, **p < 0.01, and ***p < 0.001, from OGF at ^p < 0.05, ^^p < 0.01, and ^^^p < 0.001, and from taxol or cisplatin at +p < 0.05, ++p < 0.01, and +++p < 0.001. NS = not significant.
 (a)
(a)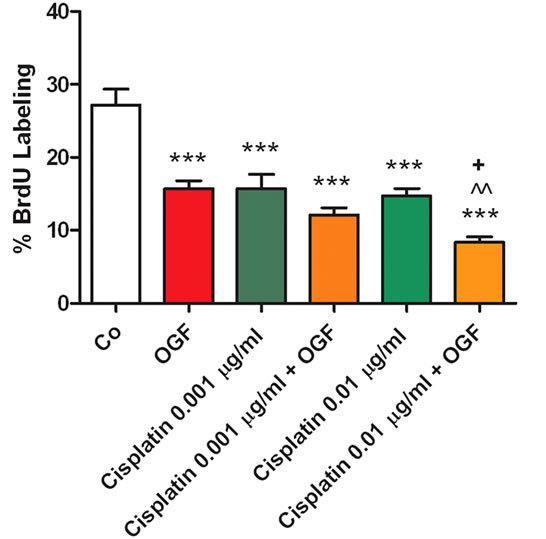 (b)
(b)
Figure 2. Mechanism of enhanced growth inhibition in vitro by both OGF and taxol, or a combination of OGF and cisplatin. (a) and (b) Evaluation of DNA synthesis (% BrdU incorporation) in SKOV-3 cells treated with OGF, taxol, cisplatin, OGF and taxol, OGF and cisplatin, or an equivalent volume of sterile water (Co) for 120 h. Media and compounds were replaced daily. Cells were pulsed with BrdU (30 mM) for 3 h prior to fixation and processed for BrdU immunoreactivity. Values represent %BrdU incorporation (means ± SE) determined for at least 500 cells on 10 fields/coverslip and 2 coverslips/treatment group. Significantly different from Co at ***p < 0.001, from OGF at ^^p < 0.01 and ^^^p < 0.001, and from taxol or cisplatin at +p < 0.01 and +++p < 0.001.
were comparable to values obtained with cells subjected to sterile water (data not shown).
3.5. OGF Inhibits Established Ovarian Cancer, and Can Be Combined with Taxol or Cisplatin for an Additive Inhibitory Effect on Tumor Progression
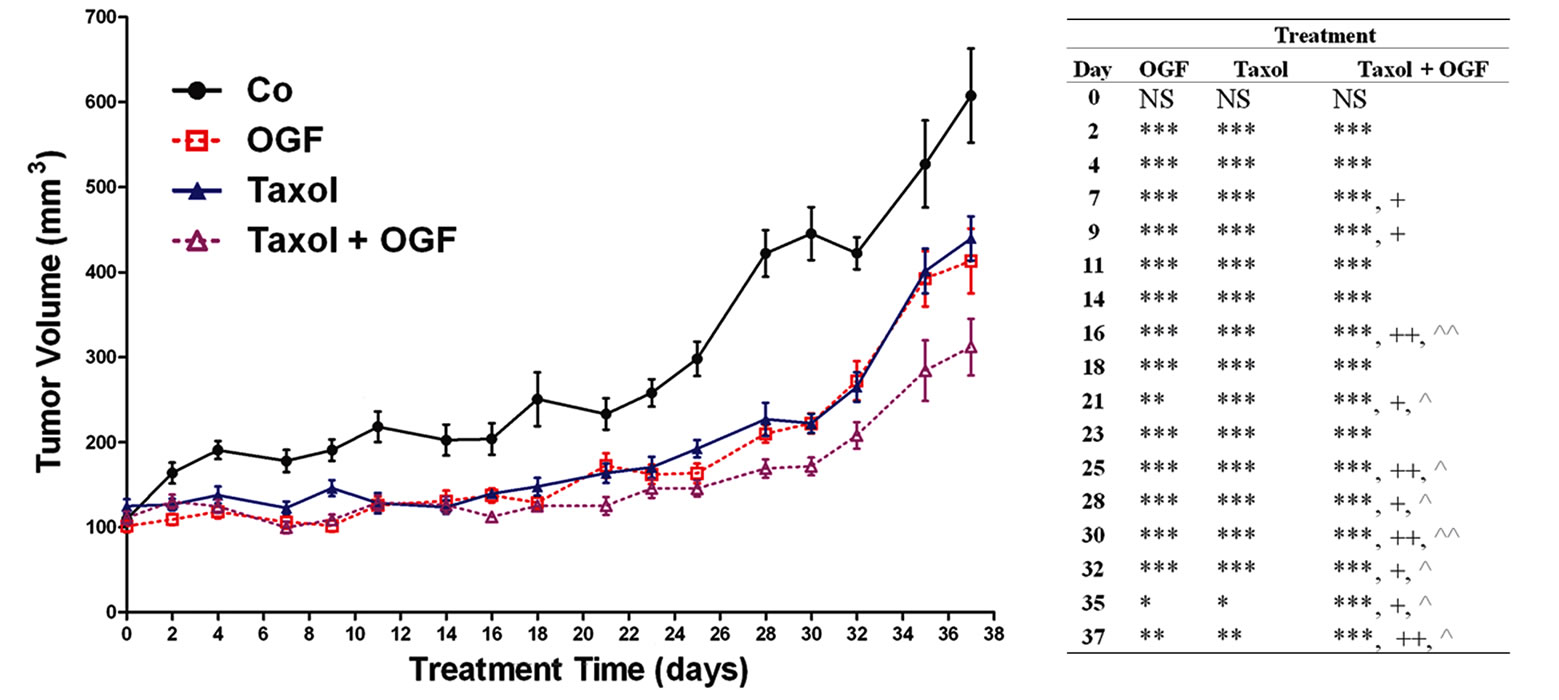
Beginning 2 days after initiation of treatments and persisting throughout the study, tumor volumes in mice with established s.c. ovarian xenografts were reduced by treatment with OGF (26% - 50%), taxol (22% - 50%), cisplatin (31% - 58%), both OGF and taxol (21% - 62%), and a combination of OGF and cisplatin (30% - 70%) compared to control animals receiving saline (Figures 3(a) and (b)). In mice receiving OGF in combination with taxol, tumor volumes were reduced 11% - 28% from
 (a)
(a)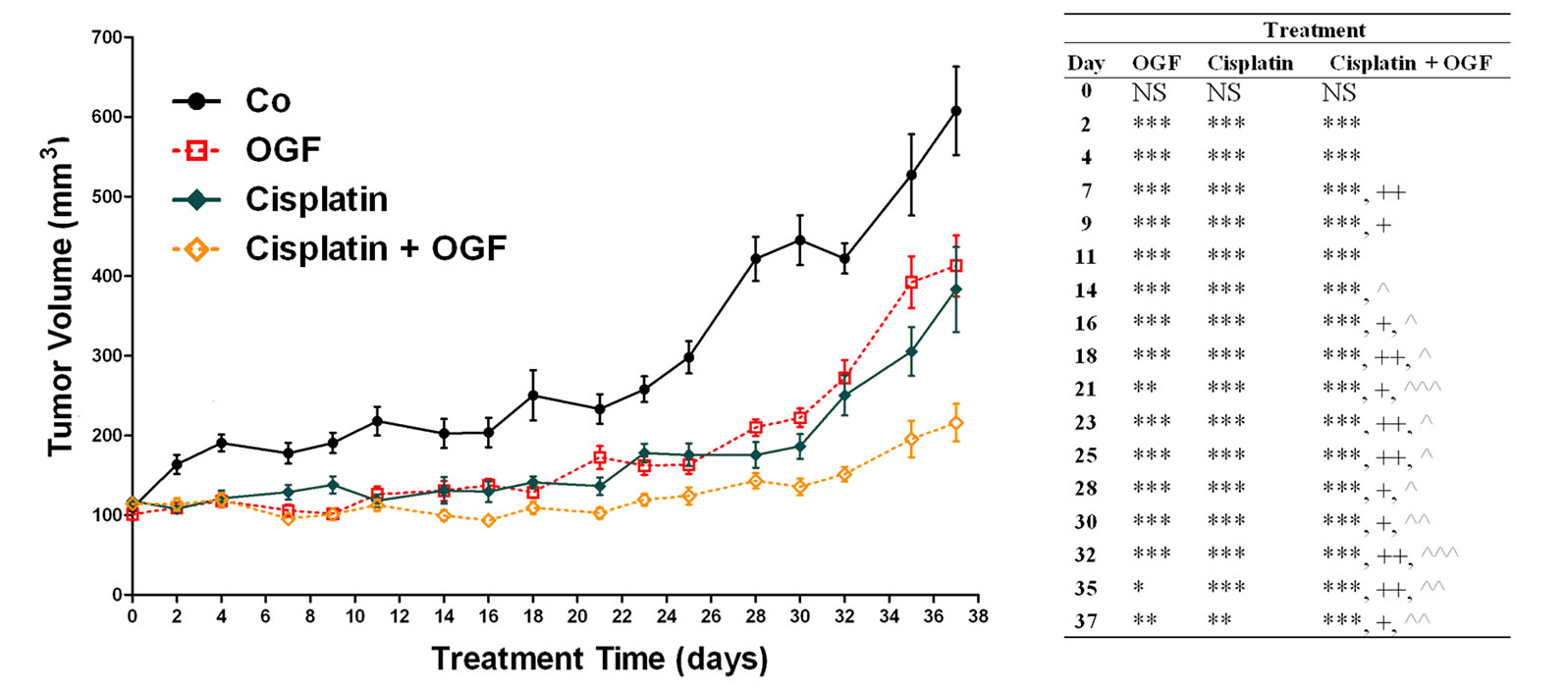 (b)
(b)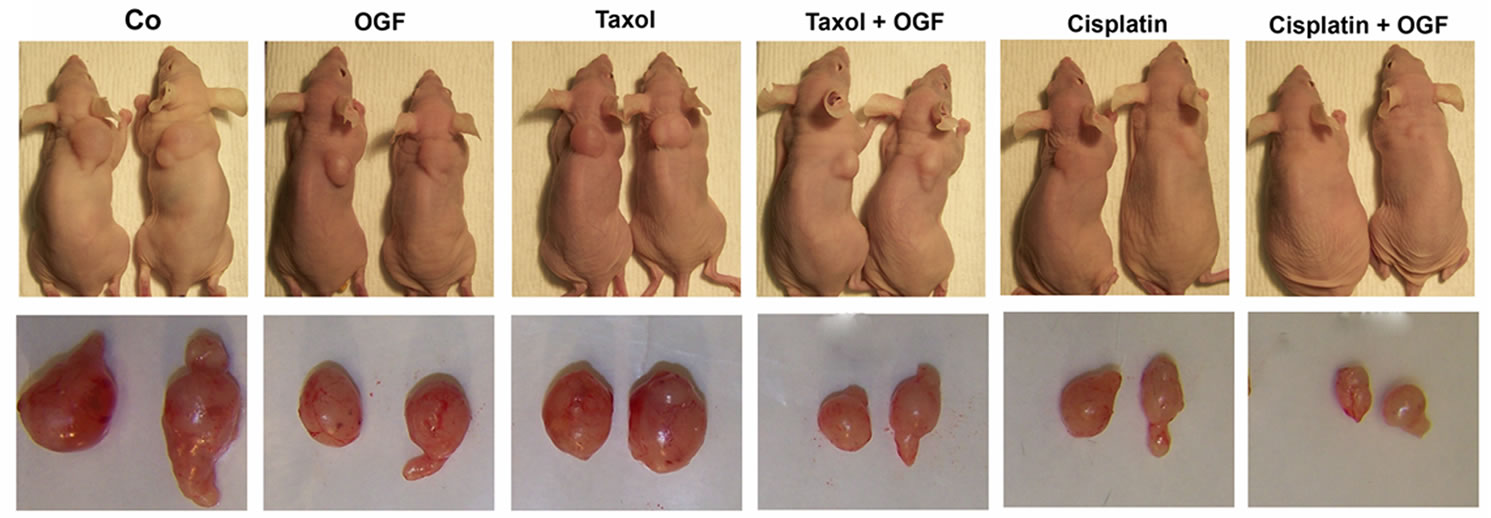 (c)
(c)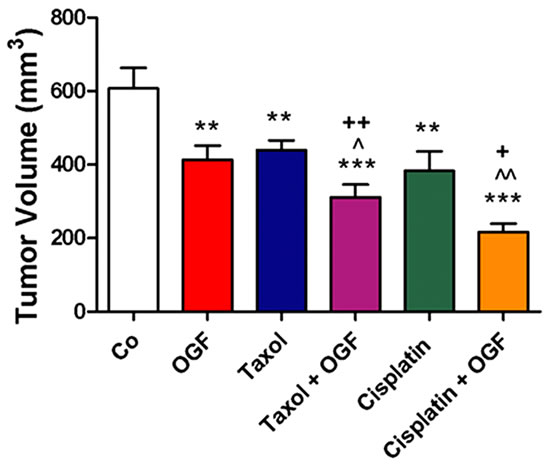 (d)
(d)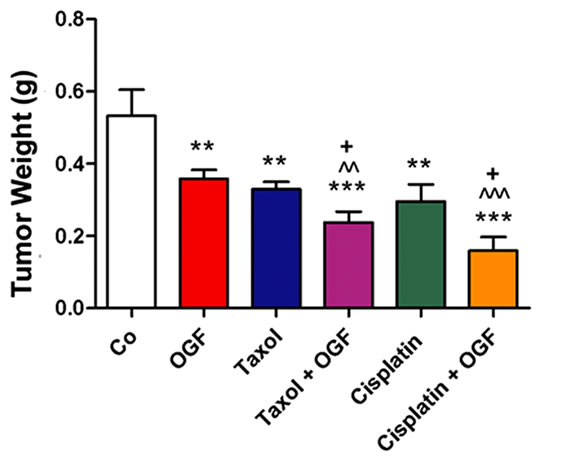 (e)
(e)
Figure 3. Growth of subcutaneous xenografts with SKOV-3 cells in mice treated with OGF, taxol, cisplatin, or OGF in combination with taxol or cisplatin chemotherapy. When tumors became visible (day 0), animals were injected with either OGF (10 mg/kg, daily), taxol (3 mg/kg, days 0, 7, 14, 21, 28, 35), cisplatin (4 mg/kg, days 0, 7), taxol and OGF, cisplatin and OGF, or an equivalent volume of saline (daily). (a) and (b) Tumor volumes were assessed 3x/week. Data for the saline and OGF groups in (a) and (b) are from the same mice. (c) Representative images of tumors before and after removal from mice following 37 days of treatment. (d) Terminal tumor volume (mm3). (e) Terminal tumor weight (g). Values represent means + SE for 12 mice/group. Significantly different from saline at *p < 0.05, **p < 0.01, and ***p < 0.001, from OGF at ^p < 0.05, ^^p < 0.01, and ^^^p < 0.001, and from taxol or cisplatin at +p < 0.05 and ++p < 0.01.
mice treated with OGF alone beginning on day 16 and continuing through the remainder of the experiment. Moreover, mice treated with OGF and taxol had tumor volumes that were decreased 19% - 21% from mice receiving taxol alone beginning on day 7 (Figure 3(a)). Mice administered OGF and cisplatin had tumor volumes that were reduced 15% - 50% from mice treated with OGF alone beginning on day 14, and decreased 19% - 44% in comparison to mice treated with cisplatin alone beginning on day 7 (Figure 3(b)).
On the day of termination (day 37), mice from all treatment groups displayed a visible reduction in tumor size (Figure 3(c)) compared to controls subjected to saline, with decreases in both tumor volume (28% - 64%, Figure 3(d)) and tumor weight (32% - 70%, Figure 3(e)) recorded. Relative to tumor bearing mice treated with either OGF or taxol alone, animals exposed to the combination of OGF and taxol had reductions in tumor volumes (24% and 29%, respectively) and tumor weights (34% and 28%, respectively) (Figures 3(d) and (e)). Similarly, relative to mice treated with either OGF or cisplatin, OGF and cisplatin in combination depressed tumor volumes (48% and 44%, respectively) and tumor weights (56% and 46%, respectively) (Figures 3(d) and (e)).
3.6. Body Mass and Gross Observations
Although all mice weighed approximately 18 to 20 g at the beginning of the experiment (Figure 4), mice receiving cisplatin had a 14% - 21% reduction in body weight compared to saline administered controls beginning on day 9 and extending through day 16 of the study. Mice receiving the combination of OGF and cisplatin also were reduced from saline treated controls in body weight (11% - 13%) from days 9 through 14 of the study. However, mice exposed to both OGF and cisplatin weighed 7% - 12% more than mice treated with cisplatin alone. Body weights of mice administered OGF, taxol, or the combination of OGF and taxol, were comparable to saline controls throughout the study.
Terminal spleen weights were similar in all groups of mice (data not shown), and behavioral abnormalities were not evident. Metastasis or lesions were not noted in mice from any group.
3.7. Mechanism of Enhanced Tumor Growth Inhibition In Vivo
Examination of apoptosis by TUNEL assay revealed similar levels of programmed cell death in tumors taken from mice treated with either OGF or saline (Figure 5(a)). Mice treated with taxol or cisplatin, either alone or in combination with OGF, had 97% - 122% and 239% - 273%, respectively, more apoptotic cells compared to saline administered controls (Figure 5(a)). With respect to cells in tumors undergoing DNA synthesis, a reduction of 34% - 51% was noted in all treatment groups compared to saline controls (Figure 5(b)). The BrdU labeling index in mice receiving the combination of OGF and taxol, or both OGF and cisplatin, was reduced 26% compared to mice treated with taxol or cisplatin alone (Figure 5(b)). Levels of DNA synthesis in tumors were similar in mice treated with OGF, the combination of
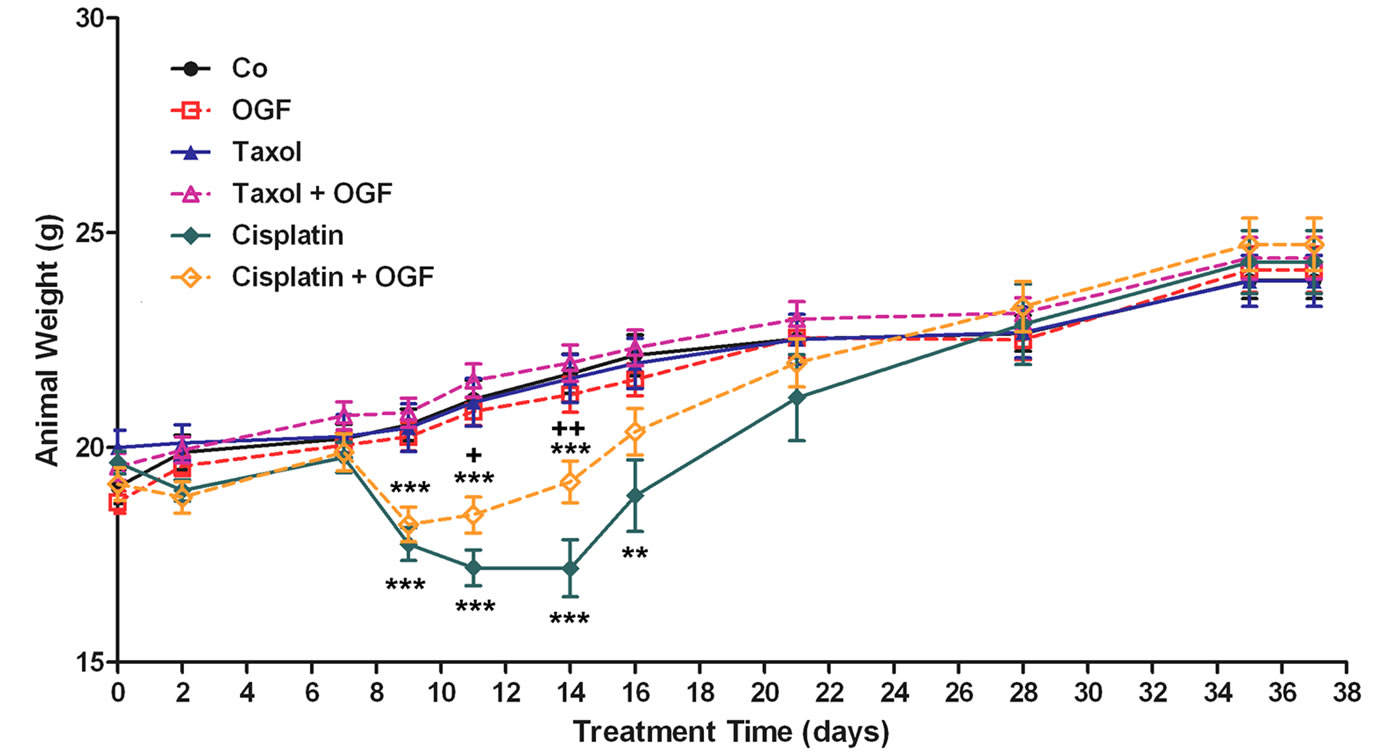
Figure 4. Body weight of mice with subcutaneous xenografts of SKOV-3 cells treated with OGF, taxol, cisplatin, or OGF in combination with taxol or cisplatin, commencing when tumors were visible. Values represent means + SE body weight for 12 mice/group. Significantly different from saline at **p < 0.01 and ***p < 0.001, and from cisplatin at +p < 0.05 and ++p < 0.01.
 (a)
(a) (b)
(b)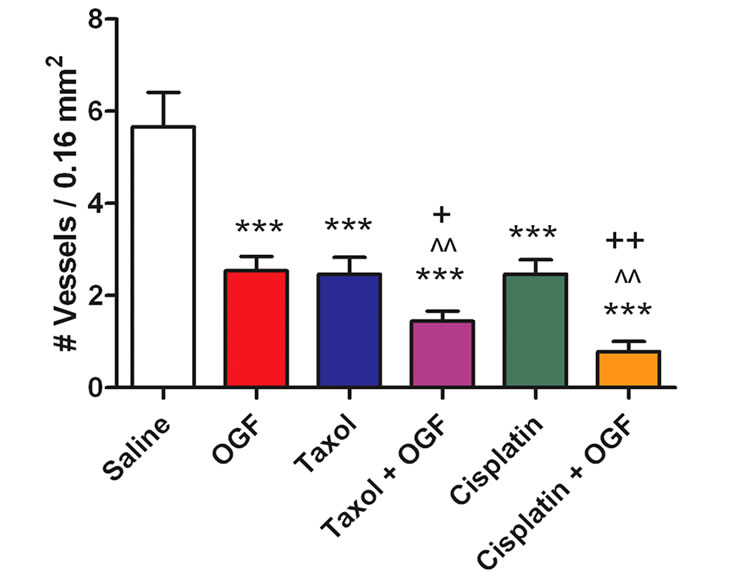 (c)
(c)
Figure 5. Mechanism of tumor growth inhibition by treatment with OGF, taxol, cisplatin, both OGF and taxol, or OGF in combination with cisplatin: effects on apoptosis, DNA synthesis, and angiogenesis. Treatments were initiated when tumors were visible (day 0) and tumor tissue was assessed 37 days later. (a) Number of apoptotic cells per 0.003 mm2, as measured by TUNEL assay. (b) % BrdU labeling. (c) Number of blood vessels per 0.16 mm2, as assessed by Hematoxylin/Eosin staining to identify endothelial cell-lined blood vessels. Values represent means ± SE determined from at least 10 random fields from the periphery of 2 tumor sections/mouse and 2 mice/group. Significantly different from saline at ***p < 0.001, from OGF at ^^p < 0.01 or ^^^p < 0.001, and from taxol or cisplatin by +p < 0.05 or ++p < 0.01.
OGF and taxol, or the combination of OGF and cisplatin (Figure 5(b)). With respect to the density of blood vessels in tumors, blood vessel density was reduced 55% - 86% in all treatment groups relative to animals exposed to saline (Figure 5(c)). Blood vessel density was decreased approximately 43% in mice treated with the combination of OGF and taxol, and decreased 69% in mice treated with the combination of OGF and cisplatin, compared to mice administered individual treatments (Figure 5(c)).
3.8. Presence and Expression of OGF and OGFr in Xenografts
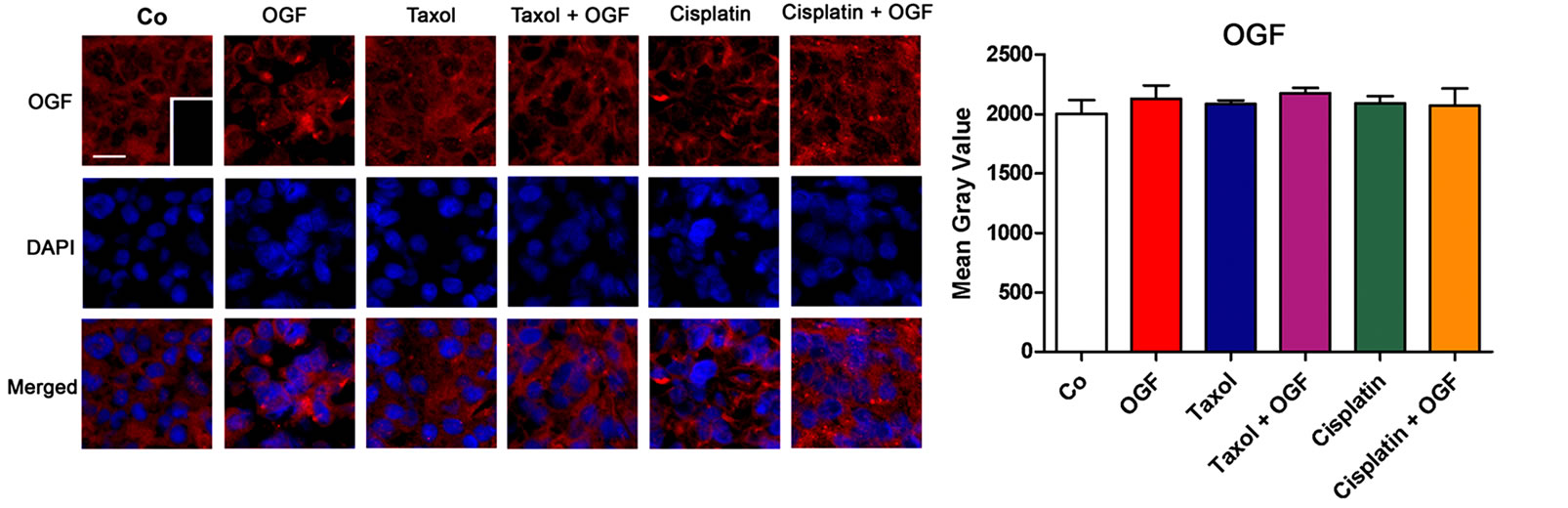
To evaluate the distribution and relative expression of OGF in xenografts, semiquantitative immunohistochemistry was performed. OGF was visible in the cytoplasm and a speckling of immunoreactivity often was noted in cell nuclei (Figure 6(a)). Tumors processed with only secondary antibody showed no staining (Figure 6(a) inset). OGF distribution and immunofluorescence (mean gray value) did not differ between any group (Figure 6(b)).
To examine OGFr distribution and relative expression, immunohistochemistry, Western blotting, and receptor binding assays were performed on xenografts. The cellular location of OGFr was similar in all groups, with immunoreactivity detected in the cytoplasm and nucleus (Figure 6(c)). Tumors processed with only the secondary antibody showed no staining (Figure 6(c, insert)). Relative to saline administered controls, OGFr expression in mice treated with OGF, both OGF and taxol, or OGF combined with cisplatin was decreased 47% - 51% using semiquantitative immunohistochemistry (Figure 6(d)). Further evaluation of OGFr expression using Western blotting, showed that mice treated with OGF had an 81% reduction in OGFr expression in their tumors compared to saline administered controls (Figures 6(e) and (f)).
Receptor binding assays indicated specific and saturable binding for OGFr in tumors of all groups, with a one site model of binding recorded (Figure 6(g)). Binding capacity (Bmax) values were markedly reduced (44% - 51%) in mice treated with OGF, both OGF and taxol or OGF in combination with cisplatin, compared to control animals receiving saline (Figure 6(g)). However, binding affinity (Kd) for OGFr did not differ among treatment groups and ranged from 2.0 nM to 7.5 nM (data not shown).
4. Discussion
The present study demonstrates for the first time that OGF biotherapy, alone or in combination with taxol or cisplatin chemotherapies, has a potent inhibitory effect on the proliferation of a human ovarian cancer cell line, SKOV-3. The repressive effects of OGF, taxol, or cisplatin alone on cell number were consonant with previous reports with respect to a variety of ovarian cancer cells in vitro [11,35,36]. We now have discovered that the anti-proliferative effect of the combination of OGF with taxol or cisplatin was greater than that of the individual drugs, indicating an additive action of these biotherapeutic and chemotherapeutic agents. Indeed, cell number was comparable between cultures receiving taxol or cisplatin alone and cultures receiving OGF in combination with a 10-fold lower concentration of these agents. Two major differences in the effects of OGF on cell growth were noted in contrast to taxol or cisplatin. First, the inhibition of cell number by OGF, but not taxol or cisplatin, was mediated by opioid receptors, with the opioid antagonist naloxone neutralizing the repercussions of OGF in the absence of a growth effect of naloxone alone. Second, the suppressive effect of OGF on cell proliferation was reversed when media was replaced without drug, and cell number returned to normal. In contrast, a discontinuation of taxol or cisplatin treatment did not change the growth characteristics of SKOV-3 cells, with these cultures resembling those that continued to receive taxol or cisplatin. Moreover, the number of ovarian cancer cells in cultures undergoing reversal from OGF was significantly greater than in the taxol or cisplatin reversal groups. These results show that the combination of two treatment modalities with differing mechanisms act in a synergistic fashion to impede the growth of human ovarian cancer cells in tissue culture.
The results of this study make the seminal observation that daily exposure to OGF markedly impedes the progression of human ovarian tumorigenesis. This was evident from our data demonstrating that even 5 weeks after initiating peptide treatment both tumor volume and tumor weight were reduced one-third from tumor-bearing mice injected with saline. Moreover, the magnitude of effects of OGF on tumorigenicity was similar to that of standard of care agents: taxol and cisplatin. We also discovered that the combination of OGF with either taxol or cisplatin had greater anti-tumor activity than individual agents alone. For example, the additive inhibitory effects of OGF and cisplatin on tumor weight were almost 2-fold greater compared to either agent alone. Tolerance in suppressing tumor growth, at least within the 5-week period of our observation, was not observed in any of the treatment groups. These results with a xenograft model in mice support and extend our findings from the tissue culture model. Additionally, our data validate not only that OGF has a potent antitumor effect on ovarian carcinogenesis, but that a combination of biotherapy with
 (a)
(a)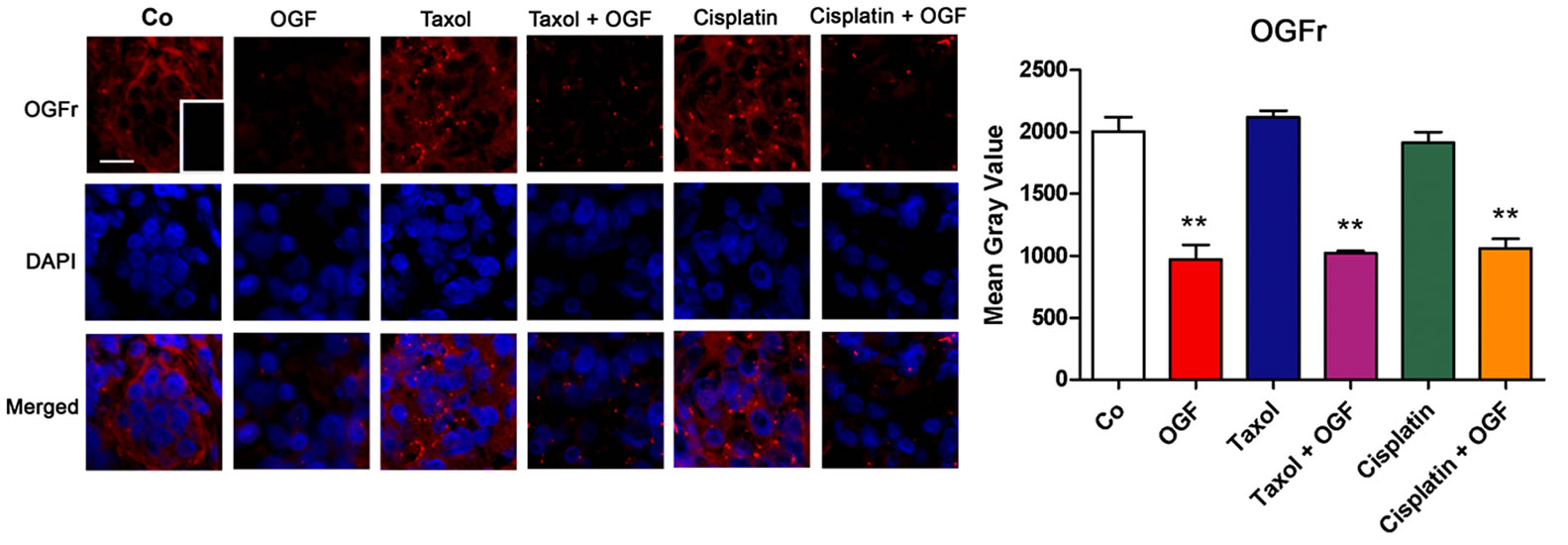 (b)
(b) (c)
(c)
Figure 6. The distribution and expression of OGF and OGFr in xenografts of SKOV-3 cells. Mice were treated with OGF, taxol, cisplatin, or OGF in combination with taxol or cisplatin when tumors were visible. (a) and (c) Photomicrographs taken at the same exposure time of tumors on the day of sacrifice (day 37) stained with antibodies (1:200) to OGF (a) or OGFr (c). Rhodamine conjugated IgG (1:1000) served as the secondary antibody and nuclei were visualized with DAPI. Preparations incubated with secondary antibodies only (insets). Bar = 10 μm. (b) and (d) Semiquantitative measurement of OGF (b) and OGFr (d) staining intensity (mean gray value) from at least 10 fields from 2 sections/tumor and 3 mice/group. (e) and (f) Western blot of the 62 kDa band of OGFr (e) and densitometric analysis (f) normalized to β-actin from 2 independent experiments. (g) Saturation isotherms calculating the binding capacity (Bmax) of OGFr in xenografts from at least 3 independent assays performed in duplicate. Data represent means + SE. Significantly different from saline at **p < 0.01 and ***p < 0.001.
OGF and chemotherapy with taxol or cisplatin, have a cooperative effect in retarding the growth of this lethal disease.
Although both OGF and taxol in the concentrations and regimens used in this study were not overtly toxic to mice with xenografts of ovarian cancer, animals subjected to cisplatin had a notable reduction in body weight. This systemic toxicity from cisplatin was diminished by simultaneous administration of OGF, indicating that this opioid peptide has the capacity to protect against toxicological insults. The amelioration of cisplatin toxicity by OGF, however, was not accompanied by a diminution of the antitumor action of cisplatin. In fact, the combination of OGF and cisplatin had an effect on tumor growth (i.e. weight, volume) that exceeded cisplatin or OGF alone. The mechanism of protection afforded by OGF against cisplatin toxicity is unknown. However, the alleviation of toxicity of one agent by the administration of another drug is not without precedence [37], and in fact has been observed when OGF was administered in combination with a toxic regimen of taxol in mice with xenografts of squamous cell carcinoma of the head and neck [38]. The finding of protection afforded by OGF from the side effects of cisplatin may allow higher doses of cisplatin to be administered to improve the therapeutic efficacy of this agent. This may be advantageous, as the success of chemotherapy is often limited by an intrinsic resistance of cancer cells [39], and the possibility of increasing the concentration of drugs without an accompanying increase in cytotoxicity would be beneficial.
In the present study, the mechanism for enhanced growth inhibition of SKOV-3 cells in tissue culture by a combination of OGF with taxol or cisplatin was not associated with induction of apoptosis or necrosis, at least at the low dosages of taxol and cisplatin used herein, but instead was related to an additive inhibitory effect on DNA synthesis. In vivo, the enhanced inhibition of tumorigenesis by OGF in combination with taxol, or both OGF and cisplatin, appears to be related to a number of mechanisms. First, OGF and/or taxol/cisplatin reduced DNA synthesis and angiogenesis in tumors. Second, taxol and cisplatin induced apoptosis. These effects of OGF, taxol, and cisplatin are consistent with previous observations. Taxol is a chemotherapeutic agent that stabilizes microtubules, thereby preventing microtubule depolymerization and chromosome segregation. These actions lead to an arresting of the cell cycle in the G2/M phase that ultimately results in cell death [40]. Cisplatin also arrests DNA replication in a cell-phase specific manner at G2/M, but induces apoptosis by binding to DNA and nuclear proteins to form DNA intra and interstrand crosslink [41,42]. OGF, on the other hand, stalls cells at the G1/S phase of the cell cycle through upregulation of p16/p21 CKI pathways, delaying cell proliferative processes (e.g., DNA synthesis, angiogenesis) without influencing cell survival [11-15,43,44]. Thus, the individual effects of biotherapy and chemotherapy are enhanced by combining agents that target similar and differing fundamental biological processes.
Another important finding of the present study is that OGF treatment administered either alone or in combination with taxol or cisplatin reduces the expression and binding of OGFr. Despite this reduction in OGFr, the OGF-OGFr axis remains functional, as demonstrated by the continued inhibition of tumor volume and weight seen with OGF treatment in animals with xenografts. This finding of a reduction in OGFr with OGF treatment is consonant with a previous report [27], wherein OGFr binding was reduced in pancreatic tumors removed from xenografted mice that were treated chronically with OGF. Furthermore, treatment with other opioids has been documented to reduce the expression of classic opioid receptors [45,46]. Therefore, in the present study it can be postulated that OGFr expression adjusts to the excess of OGF by administration of exogenous peptide, resulting in a downregulation of OGFr.
In previous studies investigating the effects of OGF on carcinogenesis, the paradigm used was to initiate administration of the peptide concomitant with tumor cell inoculation. These reports concluded that OGF could suppress tumor appearance and delay tumor progression of a wide variety of cancers when utilized in this manner [25-27]. In contrast to earlier investigations, treatment with OGF alone or in combination with taxol or cisplatin, in the present study commenced when tumors were measurable. These results reveal that OGF not only can influence early tumorigenic events, but can exert a potent action on an established cancer. With these observations in mind, it may be conjectured that OGF can be used as an antitumor agent in ovarian cancer prior to tumor expression as a prophylactic therapy. Moreover, our present findings would suggest that patients with established disease or following cytoreductive surgery could benefit from OGF biotherapy alone or in combination with standard of care drugs.
This report showing that OGF alone and in combination with two chemotherapeutic agents has a marked antitumor effect with regard to human ovarian cancer has several clinical implications. OGF has been documented to be non-toxic in Phase I trials, both by the infusion and s.c. routes of administration [47]. OGF also has been reported to have efficacy in extending survival of patients with advanced pancreatic cancer in a Phase II clinical trial [48]. These previous reports documenting that OGF is non-toxic and efficacious, make the transition from the laboratory to the clinic in terms of using OGF alone or combined with standard of care drugs more feasible for the treatment of ovarian cancer. Our results suggest that OGF could be used under three different circumstances: 1) as a prophylactic agent, particularly in patients with a family history of ovarian cancer, 2) as a first line treatment, alone or in combination with standard of care drugs, following cytoreductive surgery, and 3) following relapse when all other treatments are palliative.
5. Acknowledgements
We are grateful for the support by the Paul K. and Anna E. Shockey Family, Bonnie and Ken Shockey, and the Zagon/Kostel families.
REFERENCES
- P. Dinh, P. Harnett, M. J. Piccart-Gebhart and A. Awada, “New Therapies for Ovarian Cancer: Cytotoxics and Molecularly Targeted Agents,” Critical Reviews in Oncology/Hematology, Vol. 67, No. 2, August 2008, pp. 103-112. doi:10.1016/j.critrevonc.2008.01.012
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, “Global Cancer Statistics,” CA: A Cancer Journal for Clinicians, Vol. 61, No. 2, February 2011, pp. 69-90. doi:10.3322/caac.20107
- I. Boger-Megiddo and N. S. Weiss, “Histologic Subtypes and Laterality of Primary Ovarian Tumors,” Gynecologic Oncology, Vol. 97, No. 1, April 2005, pp. 80-83. doi:10.1016/j.ygyno.2004.11.054
- N. Chobanian and C. S. Dietrick, “Ovarian Cancer,” Surgical Clinics of North America, Vol. 88, No. 2, April 2008, pp. 285-299. doi:10.1016/j.suc.2007.12.002
- E. J. Nam and Y. T. Kim, “Alteration of Cell-Cycle Regulation in Epithelial Ovarian Cancer,” International Journal of Gynecological Cancer, Vol. 18, No. 6, November-December 2008, pp. 1169-1182.
- I. S. Zagon, M. F. Verderame and P. J. McLaughlin, “The Biology of the Opioid Growth Factor Receptor (OGFr),” Brain Research Reviews, Vol. 38, No. 3, February 2002, pp. 351-376. doi:10.1016/S0165-0173(01)00160-6
- P. J. McLaughlin, R. J. Levin and I. S. Zagon, “Regulation of Human Head and Neck Squamous Cell Carcinoma Growth in Tissue Culture by Opioid Growth Factor,” International Journal of Oncology, Vol. 14, No. 5, May 1999, pp. 991-998.
- I. S. Zagon, J. P. Smith and P. J. McLaughlin, “Human Pancreatic Cancer Cell Proliferation in Tissue Culture is Tonically Inhibited by Opioid Growth Factor,” International Journal of Oncology, Vol. 14, No. 3, March 1999, pp. 577-584.
- I. S. Zagon, S. D. Hytrek and P. J. McLaughlin, “Opioid Growth Factor Tonically Inhibits Human Colon Cancer Cell Proliferation in Tissue Culture,” American Journal of Physiology, Vol. 271, No. 3, Part 2, September 1996, pp. R511-R518.
- G. J. Bisignani, P. J. McLaughlin, S. D. Ordille, M. J. Jarowenko and I. S. Zagon, “Human Renal Cell Proliferation in Tissue Culture is Tonically Inhibited by Opioid Growth Factor,” Journal of Urology, Vol. 162, No. 6, December 1999, pp. 2186-2191. doi:10.1016/S0022-5347(05)68157-8
- R. N. Donahue, P. J. McLaughlin and I. S. Zagon, “Cell Proliferation of Human Ovarian Cancer is Regulated by the Opioid Growth Factor—Opioid Growth Factor Receptor Axis,” American Journal of Physiology-Regulatory Integrative and Comparative Physiology, Vol. 296, No. 6, June 2009, pp. R1716-R1725. doi:10.1152/ajpregu.00075.2009
- F. Cheng, I. S. Zagon, M. F. Verderame and P. J. McLaughlin, “The Opioid Growth Factor (OGF)-OGF Receptor Axis Utilizes the P16 Pathway to Inhibit Progression of Human Squamous Cell Carcinoma of the Head and Neck,” Cancer Research, Vol. 67, No. 21, November 2007, pp. 10511-10518.
- F. Cheng, P. J. McLaughlin, M. F. Verderame and I. S. Zagon, “The OGF-OGFr Axis Utilizes the P21 Pathway to Restrict Progression of Human Pancreatic Cancer,” Molecular Cancer, Vol. 7, No. 5, January 2008, pp. 5-17.
- F. Cheng, P. J. McLaughlin, M. F. Verderame and I. S. Zagon, “The OGF-OGFr Axis Utilizes the P16ink4a and P21waf1/CIP1 Pathways to Restrict Normal Cell Proliferation,” Molecular Biology of the Cell, Vol. 20, No. 1, January 2009, pp. 319-327. doi:10.1091/mbc.E08-07-0681
- I. S. Zagon and P. J. McLaughlin, “Opioids and the Apoptotic Pathway in Human Cancer Cells,” Neuropeptides, Vol. 37, No. 2, April 2003, pp. 79-88. doi:10.1016/S0143-4179(03)00007-6
- I. S. Zagon, M. F. Verderame, S. S. Allen and P. J. McLaughlin, “Cloning, Sequencing, Chromosomal Location, and Function of a Cdna Encoding the Opioid Growth Factor Receptor (Ogfr) in Humans,” Brain Research, Vol. 856, No. 1-2, February 2000, pp. 75-83. doi:10.1016/S0006-8993(99)02330-6
- F. Cheng, P. J. McLaughlin, M. F. Verderame and I. S. Zagon, “Dependence on Nuclear Localization Signals of the Opioid Growth Factor Receptor in the Regulation of Cell Proliferation,” Experimental Biology and Medicine, Vol. 234, No. 5, May 2009, pp. 532-541. doi:10.3181/0901-RM-16
- I. S. Zagon, T. B. Ruth, A. E. Leure-duPree, J. W. Sassani and P. J. McLaughlin, “Immunoelectron Microscopic Localization of the Opioid Growth Factor Receptor (Ogfr) and OGF in the Cornea,” Brain Research, Vol. 967, No. 1-2, March 2003, pp. 37-47. doi:10.1016/S0006-8993(02)04172-0
- I. S. Zagon, T. B. Ruth and P. J. McLaughlin, “Nucleocytoplasmic Distribution of Opioid Growth Factor (OGF) and Its Receptor (Ogfr) in Tongue Epithelium,” The Anatomical Record Part A: Discoveries in Molecular, Cellular, and Evolutionary Biology, Vol. 282, No. 1-2, March 2005, pp. 24-37.
- F. Cheng, P. J. McLaughlin and I. S. Zagon, “Regulation of Cell Proliferation by the Opioid Growth Factor is Dependent on Karyopherin Beta and Ran for Nucleocytoplasmic Trafficking,” Experimental Biology and Medicine, Vol. 235, No. 9, September 2010, pp. 1093-1101. doi:10.1258/ebm.2010.010139
- I. S. Zagon, R. N. Donahue and P. J. McLaughlin, “Opioid Growth Factor—Opioid Growth Factor Receptor Axis is a Physiological Determinant on Cell Proliferation in Diverse Human Cancers,” American Journal of Physiology-Regulatory Integrative and Comparative Physiology, Vol. 297, No. 4, October 2009, pp. R1154-R1161. doi:10.1152/ajpregu.00414.2009
- I. S. Zagon, P. J. McLaughlin, S. R. Goodman and R. E. Rhodes, “Opioid Receptors and Endogenous Opioids in Diverse Human and Animal Cancers,” Journal of the National Cancer Institute, Vol. 79, No. 5, November 1987, pp. 1059-1065.
- J. Fogh and G. Trempe, “New Human Tumor Cell Lines,” In: J. Fogh, Ed., Human Tumor Cells In Vitro, Plenum Publishing Corp., New York, 1975, pp. 115-159.
- W. S. Shim, M. Teh, P. O. Mack and R. Ge, “Inhibition of Angiopoietin-1 Expression in Tumor Cells by Antisence RNA Approach Inhibited Xenograft Tumor Growth in Immunodeficient Mice,” International Journal of Cancer, Vol. 94, No. 1, October 2001, pp. 6-15. doi:10.1002/ijc.1428
- I. S. Zagon, S. D. Hytrek, C. M. Lang, J. P. Smith, T. J. McGarrity, Y. Wu and P. J. McLaughlin, “Opioid Growth Factor ([Met5]Enkephalin) Prevents the Incidence and Retards the Growth of Human Colon Cancer,” American Journal of Physiology, Vol. 271, No. 3, Part 2, September 1996, pp. R780-R786.
- P. J. McLaughlin, R. J. Levin and I. S. Zagon, “Opioid Growth Factor (OGF) Inhibits the Progression of Human Squamous Cell Carcinoma of the Head and Neck Transplanted into Nude Mice,” Cancer Letters, Vol. 199, No. 2, September 2003, pp. 209-217. doi:10.1016/S0304-3835(03)00341-0
- I. S. Zagon, S. D. Hytrek, J. P. Smith and P. J. McLaughlin, “Opioid Growth Factor (OGF) Inhibits Human Pancreatic Cancer Transplanted into Nude Mice,” Cancer Letters, Vol. 112, No. 2, January 1997, pp. 167-175. doi:10.1016/S0304-3835(96)04566-1
- K. Yamamoto, Y. Kikuchi, K. Kudoh, J. Hirata, T. Kita and I. Nagata, “Treatment with Paclitaxel Alone, Rather than Combination with Paclitaxel and Cisplatin May be Selective for Cisplatin-Resistant Ovarian Carcinoma,” Japanese Journal of Clinical Oncology, Vol. 30, No. 10, October 2000, pp. 446-449. doi:10.1093/jjco/hyd116
- T. Maffucci, E. Piccolo, A. Cumashi, M. Iezzi, A. M. Riley, A. Saiardi, H. Y. Godage, C. Rossi, M. Broggini, B. V. Potter, P. Innocenti and M. Falasca, “Inhibition of the Phosphatidylinositol 3-Kinase/AKT Pathway by Inositol Pentakisphosphate Results in Antiangiogenic and Antitumor Effects,” Cancer Research, Vol. 65, No. 18, September 2005, pp. 8339-8349.
- I. S. Zagon and P. J. McLaughlin, “Production and Characterization of Polyclonal and Monoclonal Antibodies to the Zeta (Ζ) Opioid Receptor,” Brain Research, Vol. 630, No. 1-2, December 1993, pp. 295-302. doi:10.1016/0006-8993(93)90669-E
- I. S. Zagon, M. F. Verderame, J. L. Hankins and P. J. McLaughlin, “Overexpression of the Opioid Growth Factor Receptor Potentiates Growth Inhibition in Human Pancreatic Cancer Cells,” International Journal of Oncology, Vol. 30, No. 4, April 2007, pp. 775-783.
- P. J. McLaughlin, M. F. Verderame, J. L. Hankins and I. S. Zagon, “Overexpression of the Opioid Growth Factor Receptor Downregulates Cell Proliferation of Human Squamous Carcinoma Cells of the Head and Neck,” International Journal of Molecular Medicine, Vol. 19, No. 3, March 2007, pp. 421-428.
- A. Bandyopadhyay, F. López-Casillas, S. N. Malik, J. L. Montiel, V. Mendoza, J. Yang and L. Z. Sun, “Antitumor Activity of a Recombinant Soluble Betaglycan in Human Breast Cancer Xenograft,” Cancer Research, Vol. 62, No. 16, August 2002, pp. 4690-4695.
- R. Ravi, B. Mookerjee, Z. M. Bhujwalla, C. H. Sutter, D. Artemov, Q. Zeng, L. E. Dillehay, A. Madan, G. L. Semenza and A. Bedi, “Regulation of Tumor Angiogenesis by P53-Induced Degradation of Hypoxia-Inducible Factor 1alpha,” Genes & Development, Vol. 14, No. 1. January 2000, pp. 34-44.
- T. Egawa-Takata, H. Endo, M. Fujita, Y. Ueda, T. Miyatake, H. Okuyama, K. Yoshino, S. Kamiura, T. Enomoto, T. Kimura and M. Inoue, “Early Reduction of Glucose Uptake after Cisplatin Treatment is a Marker of Cisplatin Sensitivity in Ovarian Cancer,” Cancer Science, Vol. 101, No. 10. October 2010, pp. 2171-2178. doi:10.1111/j.1349-7006.2010.01670.x
- K. L. Wang, Y. C. Yang , J. C. Lai , T. H. Tsai, C. P. Lin, Y. T. Wu, Y. Y. Chen, S. C. Wang and Y. J. Chen, “Comparison in Purity and Antitumor Effect of Brand and Generic Paclitaxel Against Human Ovarian Cancer Cells by an In Vitro Experimental Model,” Drug Development and Industrial Pharmacy, Vol. 36, No. 10. October 2010, pp. 1253-1258.
- C. M. Kurbacher and P. K. Mallmann, “Chemoprotection in Anticancer Therapy: The Emerging Role of Amifostine,” Anticancer Research, Vol. 18, No. 3C, MayJune 1998, pp. 2203-2210.
- J. R. Jaglowski, I. S. Zagon, B. C. Stack, M. F. Verderame, A. E. Leure-duPree, J. D. Manning and P. J. McLaughlin, “Opioid Growth Factor Enhances Tumor Growth Inhibition and Increases the Survival of Paclitaxel-Treated Mice with Squamous Cell Carcinoma of the Head and Neck,” Cancer Chemother Pharmacol, Vol. 56, No. 1, July 2005, pp. 97-104. doi:10.1007/s00280-004-0929-4
- M. A. Whiteside, C. J. Piyathilake, T. M. Bushell and G. L. Johanning, “Intrinsic Cisplatin Resistance in Lung and Ovarian Cancer Cells Propagating in Medium Acutely Depleted of Folate,” Nutrition and Cancer, Vol. 54, No. 2, 2006, pp. 274-284. doi:10.1207/s15327914nc5402_14
- C. Rodriguez-Antona, “Pharmacogenomics of Paclitaxel,” Pharmacogenomics, Vol. 11, No. 5, May 2010, pp. 621-623. doi:10.2217/pgs.10.32
- S. F. Bellon, J. H. Coleman and S. J. Lippard, “DNA Unwinding Produced by Site-Specific Intrastrand CrossLinks of the Antitumor Drug Cis-Diamminedichloroplatinum(II),” Biochemistry, Vol. 30, No. 32, August 1991, pp. 8026- 8035.
- G. Chu, “Cellular Responses to Cisplatin; the Roles of DNA-Binding Proteins and DNA Repair,” The Journal of Biological Chemistry, Vol. 269, No. 2, January 1994, pp. 787-790.
- J. Blebea, J. E. Mazo, T. K. Kihara, J. H. Vu, P. J. McLaughlin, R. G. Atnip and I. S. Zagon, “Opioid Growth Factor Modulates Angiogenesis,” Journal of Vascular Surgery, Vol. 32, No. 2. August 2000, pp. 364-373. doi:10.1067/mva.2000.107763b
- J. Blebea, J. H. Vu, S. Assadnia, P. J. McLaughlin, R. G. Atnip and I. S. Zagon, “Differential Effects of Vascular Growth Factors on Arterial and Venous Angiogenesis,” Journal of Vascular Surgery, Vol. 35, No. 3, March 2002, pp. 532-538. doi:10.1067/mva.2002.120042
- K. Gach, M. Piestrzeniewicz, J. Fichna, B. Stefanska, J. Szemraj and A. Janecka, “Opioid-Induced Regulation of Mu-Opioid Receptor Gene Expression in the MCF-7 Breast Cancer Cell Line,” Biochemistry and Cell Biology, Vol. 86, No. 3, June 2008, pp. 217-226. doi:10.1139/O08-001
- D. S. Kim, H. Chin and W. A. Klee, “Agonist Regulation of the Expression of the Delta Opioid Receptor in NG108-15 Cells,” FEBS Letters, Vol. 376, No. 1-2, November 1995, pp. 11-14.
- J. P. Smith, R. L. Conter, S. I. Bingaman, H. A. Harvey, D. T. Mauger, M. Ahmad, L. M. Demers, W. B. Stanley, P. J. McLaughlin and I. S. Zagon, “Treatment of Advanced Pancreatic Cancer with Opioid Growth Factor: Phase I,” Anti-Cancer Drugs, Vol. 15, No. 3, March 2004, pp. 203-209. doi:10.1097/00001813-200403000-00003
- J. P. Smith, S. I. Bingaman, D. T. Mauger, H. A. Harvey, L. M. Demers and I. S. Zagon, “Opioid Growth Factor (OGF) Improves Clinical Benefit and Survival in Patients with Advanced Pancreatic Cancer,” Open Access Journal of Clinical Trials, Vol. 2010, No. 2, March 2010, pp. 1-12.