Health
Vol.5 No.5A(2013), Article ID:32001,10 pages DOI:10.4236/health.2013.55A005
ATP binding cassette C1 (ABCC1/MRP1)-mediated drug efflux contributes to disease progression in T-lineage acute lymphoblastic leukemia*
![]()
1Departments of Pediatrics Division of Pediatric Hematology/Oncology, Albuquerque, USA; #Corresponding Author: swinter@salud.unm.edu
2Department of Pathology, Pathology the UNM Clinical and Translation Science Center, Albuquerque, USA
3Department of Pathology, Biostatistics Core the UNM Clinical and Translation Science Center, Albuquerque, USA
4 The Department of Family and Community Medicine, The University of New Mexico Health Sciences Center, Albuquerque, USA
5Office of Research, The University of New Mexico Health Sciences Center, Albuquerque, USA
Copyright © 2013 Stuart S. Winter et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 18 March 2013; revised 19 April 2013; accepted 10 May 2013
Keywords: ATP Binding Cassette Proteins C1 and B1; Multi-Drug Resistance; T-Lineage Acute Lymphoblastic Leukemia
ABSTRACT
Purpose: In acute lymphoblastic leukemia (ALL), multidrug resistance is often mediated by ATPase Binding Cassette (ABC) proteins, which principally involve ABCC1 (multidrug resistance protein 1, MRP1) and ABCB1 (multidrug resistance 1, MDR1). However, direct comparisons between the differential effects of ABCC1 and ABCB1 have been difficult, since identical cell lines with differential expression of these transporters have not been developed. Experimental Design: In this study, we developed and compared the biological profiles of Jurkat cell lines that selectively over-expressed ABCC1 and ABCB1. Vincristine (VCR) plays an important role in the treatment of T-lineage ALL (T-ALL), and is often the first drug given to newly-diagnosed patients. Because of its importance in treatment, we provide descalating, sub-lethal doses of VCR to Jurkat cells, and extended our observations to expression profiling of newly diagnosed patients with T-ALL. Results: We found that VCR-resistant cells over-expressed ABCC1 nearly 30-fold. The calcein AM assay confirmed that VCR-resistant cells actively extruded VCR, and that ABCC1-mediated drug resistance conferred a different spectrum of multidrug resistance than other T-ALL induction agents. siRNA experiments that blocked ABCC1 export confirmed that VCR resistance could be reversed in vitro. Analyses of T-lymphoblasts obtained from 100 newly diagnosed T-ALL patients treated on Children’s Oncology Group Phase III studies 9404 and AALL0434 that induction failure could be could be partially explained by the over-expression of ABCC1 and ABCB1. Conclusions: Taken together, these results suggest that over-expression of ABC transporters plays a contributing role in mediating treatment failure in T-ALL, and underscore the need to employ alternate treatment approaches in patients for whom induction failed or for those with relapsed disease.
1. INTRODUCTION
With modern, dose-intensive chemotherapeutic approaches, nearly 80% of children, adolescents and young adults with acute lymphoblastic leukemia are now being cured of their diseases [1,2] . Despite this steady progress towards improved outcome, leukemic relapse remains a common problem. Inherently involved in relapse are a super-family of ATP Binding Cassette (ABC) drug transport proteins that actively extrude many chemically unrelated compounds out of dividing cancer cells [3-5] . The ABC transport proteins have been widely implicated in disease relapse in a variety of hematopoietic malignancies, including T-lineage acute lymphoblastic leukemia (T-ALL) [6-8] .
Malignant T-lymphoblast survival in the presence of chemotherapeutic agents is dependent on cellular and molecular alterations that are not present in chemotherapy-sensitive cells. The resistance of tumor cells to multiple, structurally or functionally dissimilar drugs is classified as multi-drug resistance (MDR) [9]. Experimentally-induced drug resistance in leukemic cell lines has shown that acquired MDR is commonly mediated by ATPbinding cassette (ABC) proteins ABCC1 (MRP1) and ABCB1 (MDR1) [7,8] . In T-ALL, over-expression of ABCB1 has not been uniformly associated with a poor outcome [10,11] , but we found that intrinsic up-regulation of ABCB1 did correlate with induction failure within a subset of patients who were treated on Children’s Oncology Group Phase III studies 8704 and 9404 [8]. The role for ABCC1 in mediating therapy resistance for T-ALL has been even less clear. While the family of multidrug resistance-associated proteins has been implicated in causing chemotherapy resistance in T-ALL [6] , others found that ABCC1 did not independently predict poor outcome [12] . The roles played by ABC transport proteins in T-ALL have yet to be fully understood, and may have differential effects on treatment within biologically-defined subsets of patients.
While treatment approaches have varied significantly over the preceding decades, induction therapy for T-ALL commonly begins with vincristine (VCR), which is typically given as 1.5 mg/m2 (maximum 2 mg) IVP once weekly [13-16] . Because of this consistent approach in VCR dosing, we developed a VCR-resistance cell line to investigate its resistance profile. We found that T-ALL cells acquire resistance to VCR through an up-regulation of ABCC1, and that the multi-drug resistance generated from ABCC1 expression is distinctly different from ABCB1 over-expression. We found that intrinsic overexpression of ABCC1 was associated with disease progression within the induction failure group, but not necessarily for individual patients, suggesting that additional resistance mechanism(s) remain to be defined. Taken together, these findings indicated that ABC proteins are important mediators of disease progression, and call for treatment approaches that bypass their resistance mechanisms.
2. PATIENTS MATERIALS AND METHODS
2.1. Cell Lines Chemotherapy Drugs Chemicals and Reagents
Jurkat T-ALL cells was purchased from American Tissue Culture Corporation (Manassas, VA) and grown in RPMI 1640 medium (Gibco BRL, Grand Island, NY) with 100 units/ml penicillin-streptomycin (Invitrogen/ Gibco, Carlsbad, CA), 5 µg/l ciprofloxacin (Bayer Pharmaceuticals, Berkeley, CA) and 10% heat-inactivated fetal bovine serum (HyClone, Logan, UT). Using a method adapted from others ADDIN EN.CITE ADDIN EN.CITE.DATA [7,8] , Jurkat cells were exposed to incrementally increasing doses of VCR (Faulding Pharmaceutical Company, Paramus, NJ) to induce drug resistance while maintaining at least 80% viability. Daunorubicin (DNR) prednisone (PRED), and l-asparaginase (L-ASP) were purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Quantification of Drug Resistance
As previously described [7,8] , drug resistance was quantified first by simply measuring duration of survival in 300 nM VCR. Progenitor (parental) and VCR-resistant cells were maintained separately at a concentration of 2 × 105 cells/ml with chemotherapy added at Day 0 and percent viability estimated with a hemacytometer (Hausser Scientific, Horsham, PA) and 0.4% trypan blue dye (Sigma). Secondly, the effective concentration resulting in 50% cell death (EC50) was measured by culturing 1 × 105 cells/ml with logarithmically increasing concentrations of VCR and assessing cell viability for parental and resistant cell lines after 7 days.
2.3. Microarray Analyses of Jurkat Cells
Total RNA was extracted from parental and resistant cell lines according to RNeasy Mini Kit manufacturer’s protocol (Qiagen, Baltimore, MD). Hybridization of RNA to the Human Genome U133 Plus 2.0 oligonucleotide microarray (Affymetrix, Santa Clara, CA) and data were normalized using the methods of the Robust Microarray Analyses, as previously described [17]. The differences in gene expression between parental and resistant cell lines were computed using GeneSpring 7.3.1 (Agilent Technologies, Inc., SantaClara, CA).
2.4. Flow Cytometry Analysis for Surface Expression and Functional Analysis of Transport Activity.
The ABCC1 transporter pump was detected at the cell surface by flow cytometry using PE-labeled mouse anti-human ABCC1 monoclonal antibody (clone QRCL-3, BD Biosciences, San Jose, CA). The antibody was incubated at 4˚C for 30 minutes in the dark with 1 × 106 parental and resistant cell lines and then were fixed and permeabilized according to the Cytofix/Cytoperm Kit (BD Biosciences) alternative protocol. In addition, to detect that the ABCC1 pumps were functional, we employed a calceinacetoxymethyl ester (calcein AM) duplex assay [18] . In living cells, calcein AM is cell permeant and non-fluorescent until it is hydrolyzed by intracellular esterases into a strongly green fluorescent anion, calcein. Calcein is retained within the cell cytoplasm, unless it is extruded by active protein transporters, such as ABCB1 or ABCC1. The Vybrant Multidrug Resistance Assay Kit (Invitrogen-Molecular Probes, Eugene, OR) was used to functionally test ABCC1 activity. Experiments were run according to manufacturer’s suggested protocol and at least 10,000 events were counted (FACscan cytometer) and analyzed with IDLeQuery software [19].
2.5. SiRNA-Mediated Suppression of Gene Expression
To investigate the effects of siRNA on ABCC1-mediated VCR resistance, 2 × 105 Jurkat cells were centrifuged and re-suspended in 75 µl siPORT electroporation buffer (Ambion, Austin, TX) and transferred into 1-mm gap electroporation cuvettes (Bio-Rad Laboratories, Hercules, CA). According to the manufacturer’s suggestions, 2.25 µg of siRNA was added and cells were then electroporated with a single square-wave pulse for 280 µs at 220 V delivered by a Gene PulserXCell (Bio-Rad). To preserve cell wall integrity, cells were incubated in the cuvette for 10 minutes at 37˚C before transfer. The siRNA sequence used to suppress ABCC1 transporter RNA was 5’-GGUGUUAUACAAGACCUUUtt-3’ (sense) and 5’- AAAGGUCUUGUAUAACACCtt-3” (antisense) at the optimal amount of 1 ug/sample (ABCC1-siRNA, #AM51334; Ambion). VCR resistant cell lines were used to establish experimental points, with and without GAPDH siRNA (Ambion) was used as a control. After 48 hours of maintaining viability, 500 nM VCR was added to each sample with percent viability and absolute cell concentration recorded on subsequent days. Total RNA was isolated with the RNeasy Mini Kit (Qiagen) and qRT-PCR performed using the SuperScript III Platinum Two-Step qRT-PCR Kit with SYBR Green (Invitrogen, Carlsbad, CA). The relative rate of gene expression was compared using the delta-delta method [20].
2.6. Clinical Case Preparation and Microarray Analyses
Children’s Oncology Group (COG) Cell Bank newlydiagnostic samples from ALL Classification studies 9900/ 9904 and AALL03B1/AALL0434 were enriched to >90% T-ALL blasts with ficoll-hypaque density centrifugation prior to cryopreservation. RNA processing and hybridization to the Human Genome U133 Plus 2.0 oligonucleotide microarray (Affymetrix) were performed as previously described [17], and 100 cases were examined for their relative expression of ABC proteins based on their outcome into three groups: 1) induction failure (n = 11); 2) post-remission relapse (n = 20); and 3) complete continuous remission (CCR, n = 69). Patient subset comparesons were performed using two-tailed Student’s t-test function in Prism 4.0 (GraphPad, La Jolla, CA). Expression profiles for the 9404 profiles are publically available under GEO accession number GSE14618 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14618).
3. RESULTS
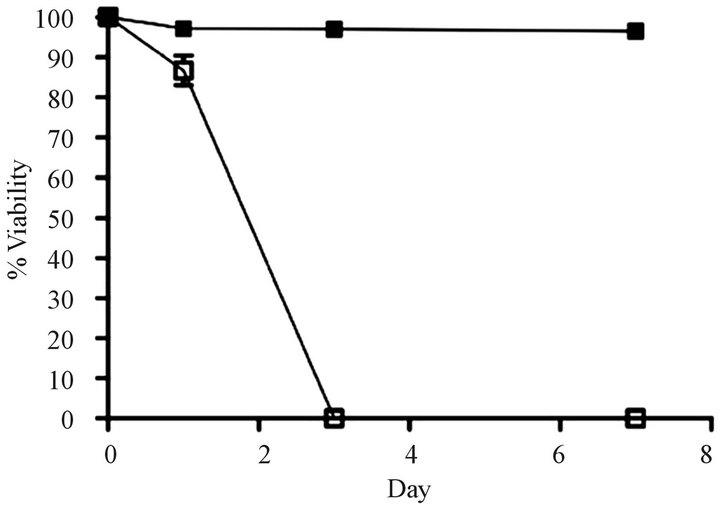
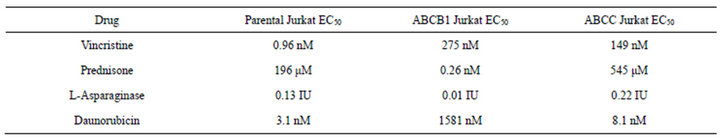
Development of a VCR-resistant T-ALL cell line. To investigate our hypothesis that VCR-resistance is an important underlying feature in disease progression/relapse in T-ALL, it was necessary to first develop a VCR resistance in a T-ALL cell line. This goal was accomplished by incrementally increasing VCR doses on a weekly basis over a time-span of approximately nine months; these conditions mimic those associated with leukemic relapse. Once the cell line became tolerant to VCR concentrations that are found within first and second order elimination therapeutic ranges in patient sera [21] ,we next tested Jurkat cell viability in analogous VCR concentrations (300 nM) and found 100% of the cells remained viable for one week in a tissue culture setting (Figure 1(a)). Following these experiments, we measured the ex vivo inhibitory concentration causing a 50% diminution of cell viability (EC50) in the wild-type, parental cells and the VCR-resistant clone. Compared to the parental cell line, the VCR-resistant cells showed a 155-fold increase in EC50 from 0.96 nM to 149 nM (Supplemental Table, and Figure 1(b)). These data show the Jurkat T-ALL cells are dynamically able to acquire a high level of VCR resistance when maintained in escalating doses of sublethal VCR concentrations, and at levels greater than the mean therapeutic concentration typically achieved during therapy.
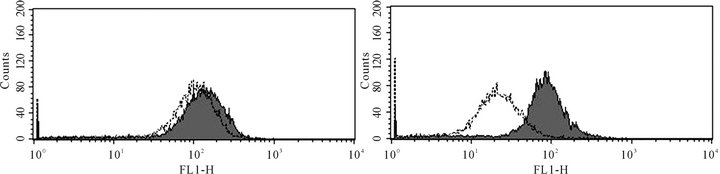
ABCC1 up-regulation in VCR-resistant cells. To assess whether over-expression of a membrane transporter protein might be responsible for VCR resistance, we next tested whether ABCC1 surface protein expression might be inducible under conditions of cytotoxic stress (Figures 2(a1) and (a2)). Wild type Jurkat cells did not show baseline expression of the ABCC1 protein relative to a control mAb (Panel a1), but cells maintained in longterm co-culture with VCR demonstrated an approximate 100-fold increase in expression of ABCC1 (Panel a2). We then tested whether additional ABC genes might be differentially regulated under the same experimental conditions. To accomplish this goal, we compared the RNA expression level of all the ABC-transporters in both the parental and VCR-resistant cells by gene expression profiling. ABCC1 was the only transporter that was significantly up-regulated out of the 48 ABC-transporters that were tested (Figure 2(b)). Specifically, ABCC1 was increased nearly 30-fold in VCR-resistant cells as compared to the parental controls, but no other ABC transporters
 (a)
(a)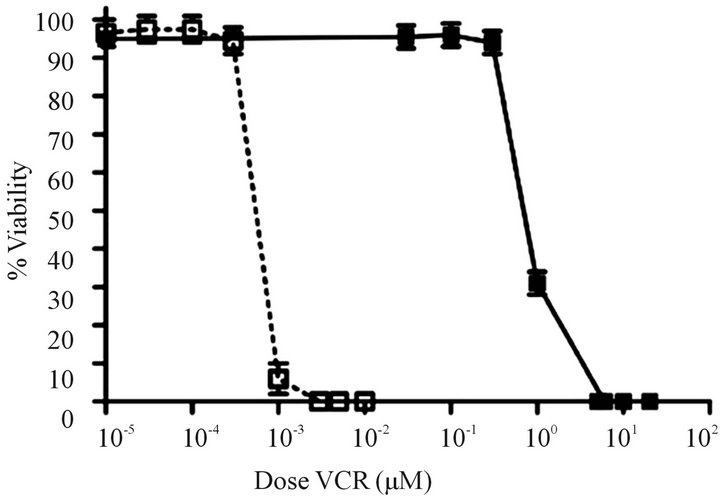 (b)
(b)
Figure 1. Comparison of survival rates for drug resistant and parental cell lines. (a) Survival over a 7 day period for parental cells that are sensitive to vincristine (VCR) (o) and cells that are resistant to VCR (n) in the presence of 300 nM VCR (n = 5 replicates). (b) Relative fold increases in tolerance of resistant cells to VCR (n) and parental cells to VCR (o) (n = three independent experiments). Error bars for survival are larger than the actual error so that they may be visualized on the scales shown here.
genes were differentially over-expressed. Interestingly, ABCB1 (MDR1) was not upregulated in response to VCRdependent disease selection, as we previously described in response to DNR exposure [8].
Up-regulated ABCC1 is an effective drug efflux protein. In order to demonstrate that ABCC1 expression is responsible for VCR resistance, we examined the efflux capability of parental and resistant cells using a chemical VCR surrogate, calcein AM (Figure 3). Calcein AM did not accumulate in VCR-resistant cells, but did so in parental, drug sensitive cells. In addition, the efflux in ABCC1-expressing, drug resistant cells could be blocked by verapamil, a known inhibitor of ABCC1 (Figure 3). [22] . These results show that VCR-induced chemo-resistance in Jurkat is mediated by the specific up-regulation of ABCC1, which then results in increased surface expression of function ABCC1 efflux protein.
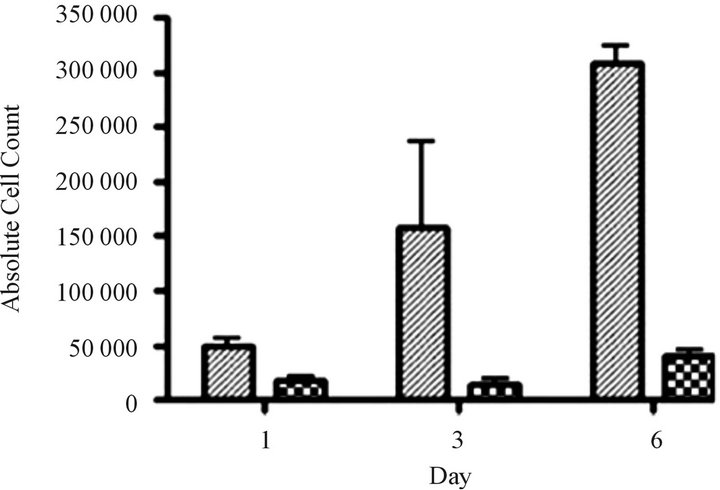
Silent interfering RNA inhibition of ABCC1 expression restores drug sensitivity. We performed siRNA experiments to show that the elimination of ABCC1 expression would return drug sensitivity. We exposed cells to siRNA directed against ABCC1 and quantified their growth over 6 days. Our measurements of ABCC1 mRNA levels in resistant cells showed that ABCC1 expression remained high, allowing cells to maintain proliferative growth. When ABCC1expression was markedly reduced, sensitivity to the cytotoxiic effects of VCR was restored (Figure 4(a)). To confirm the knock-out effects of siRNA, we measured the expression level of the ABCC1 mRNA over an 8-day period. Following a siRNA hiatus of 6 days, ABCC1 mRNA expression increased to pre-siRNA levels (Figure 4(b)). These results suggest that a single multidrug resistance transport protein (ABCC1) can be selectively up-regulated by VCR, which has been previously shown for ABCB1 in other tumor models [23].
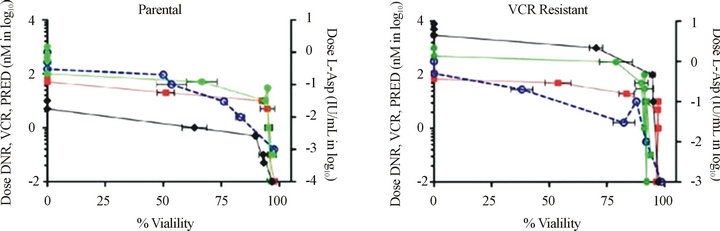
VCR resistant cells show multidrug resistance. Previously, it has been shown that a single agent can induce resistance to several dissimilar drugs. Therefore, we tested parental and drug-resistant cell lines for resistance to several common chemotherapeutic agents. We confirmed that VCR-resistant cells also displayed increased resistance to daunorubicin and prednisone, but not to L-asparaginase (Supplemental Table and Supplemental Figures 1(a) and (b)). In addition, the resistance to daunorubicin and prednisone increased mildly from 3.11 nM to 8.06 nM, and 196 µM to 545 μM, respectively, from VCR naïve to VCR-resistant Jurkat cells. These findings are consistent with the extensive observations, that ABC proteins can extrude a wide variety of molecules, hence the earlier designated name for ABCC1, “multi-resistance protein 1(MRP1)” [3,8,24] . In the resistant cells, L-asparaginase shows very minimal change, as it is not transported by ABC proteins [8]. Taken together, the results described for Figures 2-5 demonstrate two important findings: first, resistance to VCR was selectively mediated by ABCC1, and secondly, up-regulation of ABCC1 resulted in multi-drug resistance that included DNR and PRED, but not L-ASP. Importantly, these drugs are commonly employed in the induction phases for newly-diagnosed and relapsed T-ALL patients.
ABCC1 up-regulation is associated with treatment failure in patients treated for T-ALL. To investigate the clinical significance of ABCC1 up-regulation in a cohort of patients with newly diagnosed T-ALL, we next investigated de novo ABCC1 RNA levels in a cohort of 100 patients treated on COG T-ALL studies 9404 and AALL0434. The patient’s outcome status was characterized by those for whom induction failed, those who achieved a first remission, but later relapsed, and those who remained in complete continuous remission for at least four years from
 (a1) (a2)
(a1) (a2) 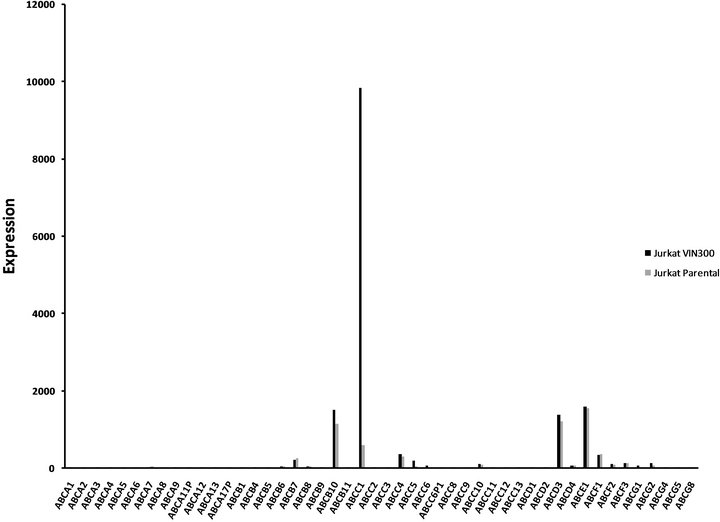 (b)
(b)
Figure 2. ABCC1 is up-regulated on the cell surface of vincristine resistant cells.(a1) Parental (therapy sensitive) cells do not express ABCC1 (grey peak: murine IgG2a PE isotypemAbs; white peak: PE-conjugated ABCC1 mAb). (a2) Cells resistant to VCR express increased levels of ABCB1, as demonstrated by a 2 log10 (100-fold) shift in peak fluorescence intensity (grey peak [41]: murine IgG2a PE isotypemAbs; white peak [right], PE-conjugated ABCC1 mAb). These data show that VCR induces the up-regulation of ABCC1 mRNA expression, resulting in increased cell surface expression of this efflux pump. (b) RMA-normalized gene profiling in parental (n) and VCR resistant (n) cells showed that ABCC1/MRP1 (202804_at) was approximately 16-fold up-regulated in the drug-resistant cell line. ABCB1/MDR1 (209993_at) was not up-regulated in response to VCR.
initial diagnosis. The one-way analysis of variance (ANOVA) test was performed and identified a significant difference of the ABCC1RNA levels among the three patient groups (P = 0.038). We also performed pairwise comparisons of the ABCC1 RNA levels using the Tukey-Kramer adjustment for multiple comparisons. As shown in Figure 5, a subset of 11 patients for whom induction failed had an RMA-normalized ABCC1 level of 9.695 ± 0.1646. This level was significantly higher than the 20 patients who relapsed, having a mean ABCC1 level of 8.883 ± 0.1573 (unpaired, two-tailed t test, P = 0.0025), or in comparison to 69 patients who remained in long-term remission, expressing a group mean level of 9.141 ± 0.0687 (unpaired, two-tailed t test, P = 0.0033).
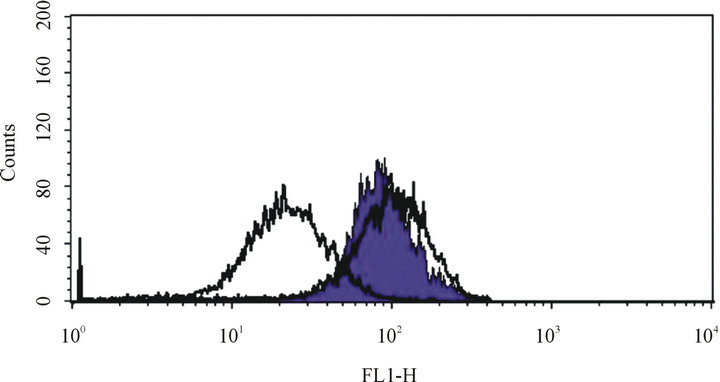
Figure 3. ABCC1 is a functional drug efflux transporter. Leftcentered white peak: VCR-resistantJurkat cells maintained in calcein AM fluorescent dye without verapamil. Right-centered white peak: VCR-sensitive Jurkat cell maintained in calcein AM fluorescent dye without verapamil. Purple peak: VCR-resistant Jurkat cells maintained in calceim AM fluorescent dye with verapamil. These peaks demonstrate that verapamil can block the ABCC1-mediated efflux of the calcein AM fluorescent dye, demonstrating that ABCC1 retains functional transporter physiology in Jurkat cells.
 (a)
(a)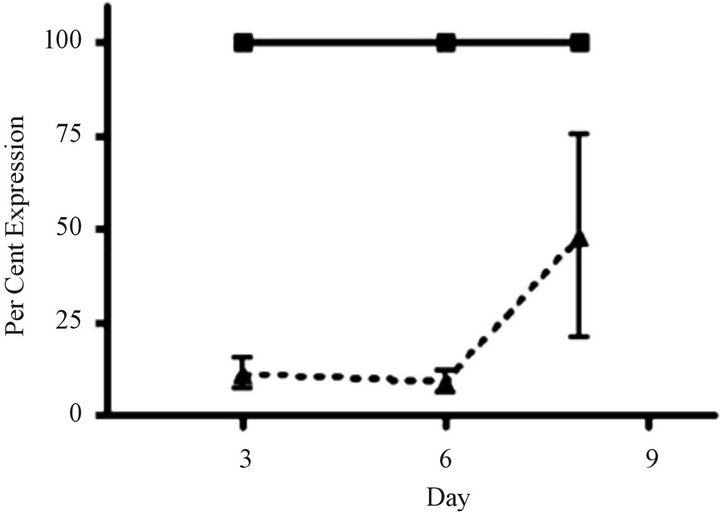 (b)
(b)
Figure 4. siRNA restores sensitivity to vincristine resistant cells over-expressing ABCC1. (a) Cell growth of parental and “knock-out” cells. VCR-resistant cells were transfected with control (GAPDH) and experimental (ABCC1 siRNA) conditions. Cells electroporated cells with GAPDHsiRNA (solid grey) and 2.25 µg of ABCC1 siRNA (checkered grey) showed distinctly different growth rates. (b) Expression of ABCC1 in either mock (solid line) or siRNA (dotted line) transfectant cells. ABCC1 expression was normalized to Day 1 levels (n = 3 for all samples).
While over-expression of ABCC1 could not alone explain disease progression amongst individual patients, these data show that de novo drug resistance can be explained in part by a group mean over-expression of ABCC1 within these outcome subsets.
4. DISCUSSION
ABC transport proteins mediate drug resistance by exporting chemotherapeutic agents out of proliferating cancer cells, including malignant T-lymphoblasts [3,5, 7-9] . We have previously reported that ABCB1 (MDR1) is selectively up regulated in T-ALL cells that have been exposed to sub-lethal concentrations of anthracyclines [8]. To further investigate the roles of structurally and functionally dissimilar ABC transporters in mediating drug resistance, we maintained cells in sublethal concentrations of VCR, a chemotherapeutic agent that is universally included during induction therapy for T-ALL and T non-Hodgkins Lymphoma [14,16,25] . Using molecular, cellular and functional assays, we identified that ABCC1 (MRP1) was specifically upregulated under VCRinduced cytotoxic pressure, but conferred resistance to other induction phase drugs, including PRED, and to a lesser extent, DNR. Our siRNA analyses showed that ABCC1 up-regulation in T-ALL cell lines resulted in profound resistance to VCR, suggesting that under conditions of that mimic sub-lethal drug exposure, ABCC1 may play a
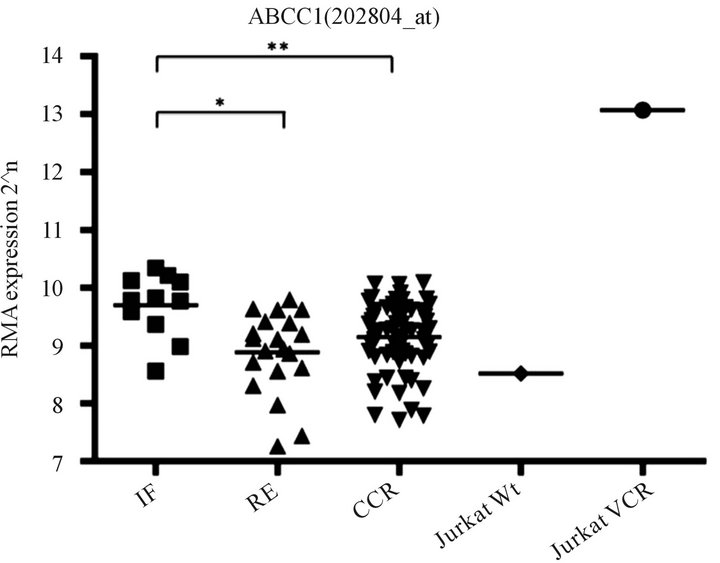
Figure 5. ABCC1 expression levels are elevated in patients for whom induction fails in Children’s Oncology Group T-ALL Studies 9404 and AALL0434. Patient cohorts were treated on COG studies 9404 and AALL0434. Patients for whom induction failed (IF; n = 11; box), relapsed (n = 20; up-triangle) and who remained in complete continuous remission (CCR; n = 69, down-triangle) displayed variance in ABCC1 expression at the time of first diagnosis. ABCC1 over-expression was significantly higher in the IF group as compared to the relapse and CCR groups. Comparative ABCC1 expression for VCR naïve (diamond) and VCR-resistant (circle) Jurkat are shown using the same RMA normalization scale.
key role in mediating disease progression. In this study, our subsequent analyses of the patient samples indicated that ABCC1 was over-expressed in patients who suffered disease progression with induction therapy. Because second-line, non-ablative therapies for induction failure are often unsuccessful [12,26,27] , our findings underscore the importance of early identification of such patients so that their treatment might be modified to utilize agents that bypass ABCB1 and ABCC1-mediated drug efflux.
For patients who do not achieve a first remission, de novo resistance is thought to be mediated by a number of biological events that control cell proliferation, including a disruption of cell division at the G0/G1 checkpoint, [17] molecular aberrations that prevent therapy-induced apoptosis [28] , or by extrusion of chemotherapeutic drugs by ABC transporters [8]. Over-expressed genes within our previously-described 116 member classifier and those over-expressed within the Jurkat VCR-resistant cell line included TMSB15A (205347_s_at), TJP2 (202085_at) and CDK1 (23153_at). We speculate that these genes maintain cell division in rapidly dividing leukemic blasts through their mediation of cytoskeletal integrity (TMSB15A), tight junction protein assembly (TJP2) and cell cycle progression (CDK1). Our experiments with sub-lethal VCR dosing resulted in highly VCR-resistant T-ALL cells, suggesting that in some patients for whom VCR dosing is not optimized, or for patients having primary VCR resistance, relapse or disease progression may be further complicated with the development of a multi-drug resistance phenotype early in treatment. Others have suggested that VCR under-dosing may be an important contributor to relapse, calling for further efforts to optimize VCR dosing in the treatment of ALL [29,30] . In contrast to the findings of Efferth et al. [7] , we found that under conditions of selective VCR exposure only ABCC1, and not ABCB1, was significantly up-regulated in our T-ALL cell line model. We are the first to demonstrate that ABCC1 contributes to de novo resistance against VCR in T-ALL, leading us to investigate its role in combination with other ABC transporters.
Multidrug resistance in T-ALL is conferred by a number of ABC transporters, which can mediate resistance to anthracyclines (ABCB1, ABCC1), methotrexate (ABCC1, ABCC2, and ABCG2) [31,32] and VCR (ABCC1). Compared to other ABC transporters, ABCC1 has several unique properties. While there is overlap between the xenobiotic substrates that are transported by ABCB1 and ABCC1, ABCC1 transports relatively broader ranges of compounds, including organic anions, heavy metals and vinca-alkaloids [31,33] . In a recent study of 72 T-ALL patients treated on DCOG ALL-7, -8 and -9, van Grotel and co-workers found CD34 expression was an important predictor of a poor 5 year disease free survival, but did not find an association between outcome and greater than median quantitative RT-PCR expression of ABCC1/
MRP1and ABCCB1/MDR1 [12] . In their study, patient outcomes were not described for early and late relapse, and since induction failure occurs in small subset of 2 to 5% of patients, over-expression of ABCC1 and ABCB1 in patients with early treatment failure may have been missed. Plasschaert and colleagues found that ABCB1- dependent disease resistance was prevalent in adults with T-ALL, but less so in children [10] . Similarly, Wuchter and coworkers [11] found no evidence for ABCB1-mediated chemotherapy resistance in nine children having T-ALL for whom induction failed when treated on ALLBFM 90/95. All patients in our studies were age 18 or less, and we used expression profiling to identify overexpression of ABCB1 and ABCC1, not the rhodamine- 123efflux assay [34] . Differences in patient age and assay sensitivity may account for discrepancies between our study and the findings of others. Among the patients with over-expression of ABCC1 transporter (Figure 5), one later relapsed on therapy, and three remained in CCR for more than four years, suggesting that additional molecular or phenotypic features may modulate ABC-mediated therapy resistance.
Patients with a phenotype characterized by CD1a (-), CD8(-), CD5(weak) with stem-cell or myeloid markers have been designated to have an “early T-precursor” (ETP) T-ALL [35] . Such patients have been found to have very poor outcomes, and either fail to achieve a first remission or relapse early in the treatment course. In most cases, they have had high MRD levels at the end of induction. The molecular features of ETP patients have yet to be completely defined, but may be characterized by specific expression profiling features, an absence of biallelic TCR gammadeletions (ABD), or have mutations of the MEF2C and PTEN genes [15,28,36] .We did not have access to the surface phenotypic features of those patients. However, a sub-set of our patient cohort were analyzed for molecular mutations by Gutierrez and colleagues [15] ; interestingly, all patients for whom induction failed and had molecular evidence for ABCC1 overexpression were also found to have an ABD and an ETP gene expression signature. One patient with ABCC1 overexpression and yet remained in CCR had an ABD, but did not have an ETP expression profile. In some cases, patients with an ETP expression signature did not overexpress ABCB1 or ABCC1, and did not fail induction therapy. These findings suggest that ETP patients have a variety of molecular aberrations, as recently extensively documented by Zhang and co-workers [37] . In several tumor models, cells arrested at early stages of development (e.g. patients with ETP) have been reported to have ABC transport activities, possibly to protect developing cells from cytotoxic damage, including chemotherapeutic agents [38,39] . We speculate that the genotypic mutations that result in ETP or induction failure T-ALL may also maintain affected progenitor T-lymphoblasts in a stem-cell like state, in which ABC transport molecules are highly expressed, resulting in de novo chemotherapy resistance. The collaborating roles between ABC transport proteins and other molecular features associated with induction failure or early relapse will require further study as potential biomarkers for high-risk disease.
Until recently, re-induction regimens containing VCR, DNR and other ABCC1 substrates produced only transient remissions in a fraction of patients, calling for the introduction of a novel set of agents that were not ABC substrates. Nelarabine, an RNA nucleoside analog that is not known to be a substrate of ABCC1, induced remissions that were achieved and maintained in up to 50% of first-time relapse patients, suggesting that relapse regimens must utilize cytotoxic drugs that might escape the transporting effects of ABC proteins [40] . Our findings suggest that patients who are at risk to not achieve a first remission should be identified early in the course of induction, as they may benefit from alternate treatment strategies that bypass ABC-mediated drug efflux.
5. ACKNOWLEDGEMENTS
This research was supported by the COG Cell Bank grant, U24 CA114766, the COG grant U24 CA114766 and the COG Statistics and Data Center grant, U10 CA98413. Children’s Oncology Group Cell Bank grant U24 CA114766, COG grant U10 CA98543, and the COG Statistics and Data Center grant U10 CA98413. Some experiments used the facilities or services provided by the Keck-UNM Genomics Resource, a facility supported by the State of New Mexico and the UNM Cancer Center support grant 5P30CA118100. Special thanks to Drs. Alejandro Gutierrez and A. Thomas Look (Dana Farber Cancer Institute, Boston, MA) for contributing information regarding the ABD and ETP status for selected cases in our shared patient databases. SSW is funded by NIH R01 5CA114589-07, and RSL by UL1 TR000041.
REFERENCES
- Trigg, M.E., Sather, H.N., Reaman, G.H., et al. (2008) Tenyear survival of children with acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leuk Lymphoma, 49, 1142-1154. doi:10.1080/10428190802074593
- Vrooman, L.M. and Silverman, L.B. (2009) Childhood acute lymphoblastic leukemia: Update on prognostic factors. Current Opinion in Pediatrics, 21, 1-8. doi:10.1097/MOP.0b013e32831f1f24
- Chang, G. (2003) Multidrug resistance ABC transporters. FEBS Letters, 555, 102-105. doi:10.1016/S0014-5793(03)01085-8
- Altenberg, G.A. (2004) Structure of multidrug-resistance proteins of the ATP-binding cassette (ABC) superfamily. Current Medicine Chemistry and Anti-Cancer Agents, 4, 53-62. doi:10.2174/1568011043482160
- Szakacs, G., Annereau, J.P., Lababidi, S., et al. (2004) Pre- dicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell, 6, 129-137. doi:10.1016/j.ccr.2004.06.026
- Steinbach, D., Wittig, S., Cario, G., et al. (2003) The multidrug resistance-associated protein 3 (MRP3) is associated with a poor outcome in childhood ALL and may account for the worse prognosis in male patients and T-cell immunophenotype. Blood, 102, 4493-4498. doi:10.1182/blood-2002-11-3461
- Efferth, T., Gillet, J.P., Sauerbrey, A., et al. (2006) Expres- sion profiling of ATP-binding cassette transporters in childhood T-cell acute lymphoblastic leukemia. Molecular Cancer Therapy, 5, 1986-1994. doi:10.1158/1535-7163.MCT-06-0086
- Estes, D.A., Lovato, D.M., Khawaja, H.M., et al. (2007) Genetic alterations determine chemotherapy resistance in childhood T-ALL: Modelling in stage-specific cell lines and correlation with diagnostic patient samples. British Journal of Haematology, 139, 20-30. doi:10.1111/j.1365-2141.2007.06763.x
- Leonard, G.D., Fojo, T. and Bates, S.E. (2003) The role of ABC transporters in clinical practice. Oncologist, 8, 411- 424. doi:10.1634/theoncologist.8-5-411
- Plasschaert, S.L., Vellenga, E., de Bont, E.S., et al. (2003) High functional P-glycoprotein activity is more often present in T-cell acute lymphoblastic leukaemic cells in adults than in children. Leuk Lymphoma, 44, 85-95. doi:10.1080/1042819021000040288
- Wuchter, C., Karawajew, L., Ruppert, V., et al. (2000) Constitutive expression levels of CD95 and Bcl-2 as well as CD95 function and spontaneous apoptosis in vitro do not predict the response to induction chemotherapy and relapse rate in childhood acute lymphoblastic leukaemia. British Journal of Haematology, 110, 154-160. doi:10.1046/j.1365-2141.2000.02147.x
- van Grotel, M., van den Heuvel-Eibrink, M.M., van Wering, E.R., et al. (2008) CD34 expression is associated with poor survival in pediatric T-cell acute lymphoblastic leukemia. Pediatric Blood Cancer, 51, 737-740. doi:10.1002/pbc.21707
- Amylon, M.D., Shuster, J., Pullen, J., et al. (1999) Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: A Pediatric Oncology Group study. Leukemia, 13, 335- 342. doi:10.1038/sj.leu.2401310
- Goldberg, J.M., Silverman, L.B., Levy, D.E., et al. (2003) Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. Journal of Clinical Oncology, 21, 3616-3622. doi:10.1200/JCO.2003.10.116
- Gutierrez, A., Dahlberg, S.E., Neuberg, D.S., et al. (2010) Absence of biallelic TCR gamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. Journal of Clinical Oncology, 28, 3816-3823. doi:10.1200/JCO.2010.28.3390
- Winter, S.S., Holdsworth, M.T., Devidas, M., et al. (2006) Antimetabolite-based therapy in childhood T-cell acute lymphoblastic leukemia: A report of POG study 9296. Pediatric Blood Cancer, 46, 179-186. doi:10.1002/pbc.20429
- Winter, S.S., Jiang, Z., Khawaja, H.M., et al. (2007) Identification of genomic classifiers that distinguish induction failure in T-lineage acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood, 110, 1429-1438. doi:10.1182/blood-2006-12-059790
- Ivnitski-Steele, I., Holmes, A.R., Lamping, E., et al. (2009) Identification of Nile red as a fluorescent substrate of the Candida albicans ATP-binding cassette transporters Cdr1p and Cdr2p and the major facilitator superfamily transporter Mdr1p. Analytical Biochemistry, 394, 87-91. doi:10.1016/j.ab.2009.07.001
- Edwards, B.S., Bologa, C., Young, S.M., et al. (2005) Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Molecular Pharmacology, 68, 1301- 1310. doi:10.1124/mol.105.014068
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402- 408. doi:10.1006/meth.2001.1262
- Holleman, A., Cheok, M.H., den Boer, M.L., et al. (2004) Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. New England Journal of Medicine, 351, 533-542. doi:10.1056/NEJMoa033513
- Perrotton, T., Trompier, D., Chang, X.B., et al. (2007) (R)- and (S)-verapamil differentially modulate the multidrug-resistant protein MRP1. Journal of Biological Chemistry, 282, 31542- 31548. doi:10.1074/jbc.M703964200
- Den Boer, M.L., Kapaun, P., Pieters, R., et al. (1999) Myeloid antigen co-expression in childhood acute lymphoblastic leukaemia: Relationship with in vitro drug resistance. British Journal of Haematology, 105, 876-882. doi:10.1046/j.1365-2141.1999.01440.x
- Efferth, T., Sauerbrey, A., Steinbach, D., et al. (2003) Ana- lysis of single nucleotide polymorphism C3435T of the multidrug resistance gene MDR1 in acute lymphoblastic leukemia. International Journal of Oncology, 23, 509-517.
- Breit, S., Stanulla, M., Flohr, T., et al. (2006) Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood, 108, 1151-1157. doi:10.1182/blood-2005-12-4956
- Schroeder, H., Garwicz, S., Kristinsson, J., et al. (1995) Outcome after first relapse in children with acute lymphoblastic leukemia: A population-based study of 315 patients from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Medicine Pediatric Oncology, 25, 372-378. doi:10.1002/mpo.2950250503
- Raetz EA, Borowitz MJ, Devidas M, et al. Reinduction platform for children with first marrow relapse of acute lymphoblastic Leukemia: A children’s oncology group study. Journal of Clinical Oncology, 26, 3971-3978. doi:10.1200/JCO.2008.16.1414
- Gutierrez, A., Sanda, T., Grebliunaite, R., et al. (2009) High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood, 114, 647- 650. doi:10.1182/blood-2009-02-206722
- Kobayashi, H., Takemura, Y., Holland, J.F., et al. (1998) Vincristine saturation of cellular binding sites and its cytotoxic activity in human lymphoblastic leukemia cells: Mechanism of inoculum effect. Biochemical Pharmacology, 55, 1229-1234. doi:10.1016/S0006-2952(97)00615-1
- McCune, J.S. and Lindley, C. (1997) Appropriateness of maximum-dose guidelines for vincristine. American Journal of Health-System Pharmacy, 54, 1755-1758.
- Szakacs, G., Varadi, A., Ozvegy-Laczka, C., et al. (2008) The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discovery Today, 13, 379-393. doi:10.1016/j.drudis.2007.12.010
- Fotoohi, A.K., Assaraf, Y.G., Moshfegh, A., et al. (2009) Gene expression profiling of leukemia T-cells resistant to methotrexate and 7-hydroxymethotrexate reveals alterations that preserve intracellular levels of folate and nucleotide biosynthesis. Biochemical Pharmacology, 77, 1410-1417. doi:10.1016/j.bcp.2008.12.026
- Munoz, M., Henderson, M., Haber, M., et al. (2007) Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life, 59, 752-757. doi:10.1080/15216540701736285
- Wuchter, C., Karawajew, L., Ruppert, V., et al. (1999) Cli- nical significance of CD95, Bcl-2 and Bax expression and CD95 function in adult de novo acute myeloid leukemia in context of P-glycoprotein function, maturation stage, and cytogenetics. Leukemia, 13, 1943-1953. doi:10.1038/sj.leu.2401605
- Coustan-Smith, E., Mullighan, C.G., Onciu, M., et al. (2009) Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncology, 10, 147-156. doi:10.1016/S1470-2045(08)70314-0
- Homminga, I., Pieters, R., Langerak, A.W., et al. (2011) Integrated Transcript and Genome Analyses Reveal NKX2- 1 and MEF2C as Potential Oncogenes in T Cell Acute Lymphoblastic Leukemia. Cancer Cell, 19, 484-497. doi:10.1016/j.ccr.2011.02.008
- Zhang, J., Ding, L., Holmfeldt, L., et al. (2012) The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature, 481, 157-163. doi:10.1038/nature10725
- Donnenberg, V.S. and Donnenberg, A.D. (2005) Multiple drug resistance in cancer revisited: The cancer stem cell hypothesis. Journal of Clinical Oncology, 45, 872-877. doi:10.1177/0091270005276905
- Donnenberg, V.S., Landreneau, R.J. and Donnenberg, A.D. (2007) Tumorigenic stem and progenitor cells: Implications for the therapeutic index of anti-cancer agents. Journal of Control Release, 122, 385-391. doi:10.1016/j.jconrel.2007.05.005
- Berg, S.L., Blaney, S.M., Devidas, M., et al. (2005) Phase II study of nelarabine (compound 506U78) in children and young adults with refractory T-cell malignancies: A report from the Children’s Oncology Group. Journal of Clinical Oncology, 23, 3376-3382. doi:10.1200/JCO.2005.03.426
- Bermudez, M., Fuster, J.L., Llinares, E., et al. (2005) Itraconazole-related increased vincristine neurotoxicity: Case report and review of literature. Journal of Pediatric Hematology/Oncology, 27, 389-392. doi:10.1097/01.mph.0000172751.06286.5b
Supplement
(a) (b)
Figure S1. VCR resistant cells with up-regulated ABCC1 express the multi-drug resistance phenotype. Resistance to prednisone (PRED), vincristine (VCR), and daunorubicin (DNR) are shown on the left axis, and resistance for L-asparagine (L-ASP) is shown on the right axis. (a) Parental cells demonstrate sensitivity to DNR (n), VCR (u), and PRED (l), but not for L-ASP (¡). (b) ABCC1 over-expressing cells maintained in VCR show greater resistance to DNR (n) and a significantly greater resistance to VCR (u) than the parental cells. Cells over-expressing ABCB1 maintained in DNR show relatively greater resistance to DNR (n) and VCR (u) [8].
NOTES
*Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.