Agricultural Sciences
Vol.06 No.01(2015), Article ID:53106,28 pages
10.4236/as.2015.61005
Aluminum and Glyphosate Can Synergistically Induce Pineal Gland Pathology: Connection to Gut Dysbiosis and Neurological Disease
Stephanie Seneff1*, Nancy Swanson2, Chen Li1
1Computer Science and Artificial Intelligence Laboratory, MIT, Cambridge, MA, USA
2Independent Researcher, Abacus Enterprises, Lummi Island, WA, USA
Email: *seneff@csail.mit.edu
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 October 2014; revised 10 November 2014; accepted 10 December 2014
ABSTRACT
Many neurological diseases, including autism, depression, dementia, anxiety disorder and Par- kinson’s disease, are associated with abnormal sleep patterns, which are directly linked to pineal gland dysfunction. The pineal gland is highly susceptible to environmental toxicants. Two pervasive substances in modern industrialized nations are aluminum and glyphosate, the active ingredient in the herbicide, Roundup®. In this paper, we show how these two toxicants work synergistically to induce neurological damage. Glyphosate disrupts gut bacteria, leading to an overgrowth of Clostridium difficile. Its toxic product, p-cresol, is linked to autism in both human and mouse models. p-Cresol enhances uptake of aluminum via transferrin. Anemia, a result of both aluminum disruption of heme and impaired heme synthesis by glyphosate, leads to hypoxia, which induces increased pineal gland transferrin synthesis. Premature birth is associated with hypoxic stress and with substantial increased risk to the subsequent development of autism, linking hypoxia to autism. Glyphosate chelates aluminum, allowing ingested aluminum to bypass the gut barrier. This leads to anemia-induced hypoxia, promoting neurotoxicity and damaging the pineal gland. Both glyphosate and aluminum disrupt cytochrome P450 enzymes, which are involved in melatonin metabolism. Furthermore, melatonin is derived from tryptophan, whose synthesis in plants and microbes is blocked by glyphosate. We also demonstrate a plausible role for vitamin D3 dysbiosis in impaired gut function and impaired serotonin synthesis. This paper proposes that impaired sulfate supply to the brain mediates the damage induced by the synergistic action of aluminum and glyphosate on the pineal gland and related midbrain nuclei.
Keywords:
Pineal Gland, Aluminum, Glyphosate, Genetically Engineered Crops, Autism, Sulfate

1. Introduction
Sleep disorders are associated with a large number of neurological diseases and conditions, including autism [1] , attention deficit hyperactivity disorder (ADHD) [2] - [4] , depression [5] , Parkinson’s disease [6] - [8] , schizophrenia [9] - [11] , anxiety disorder [12] , amyotrophic lateral sclerosis (ALS) [13] , and Alzheimer’s disease [14] . A large survey of office-based physicians found that 69% of patients presented with secondary insomnia, often related to depression or other mental diseases [15] . A study based in Hong Kong found that psychiatric conditions were the strongest predictors of insomnia in both men and women [16] .
There have been alarming increases in the rate of autism among children in the United States over the past two decades. The most recent estimate in 2014 by the Centers for Disease Control (CDC) is one in 68 for 12- year-old children [17] . Two recent papers have dispelled the myth that this increase is mostly due to increased diagnosis. Two economists reached the conclusion that the increase is real on the basis of an analysis driven by market theory [18] [19] . Nevison analyzed data spanning several decades to reach the conclusion that 75% - 80% of the increase is real, and she hypothesized that this increase is due to environmental chemicals [19] . In her analysis of the temporal trends of a large number of different chemicals considered to be plausibly causal in autism, she identified only three with significant increases in exposure over time that aligned well with the increasing trend in autism: glyphosate, aluminum vaccine adjuvant and polybrominated diphenyl ethers (used as flame retardants) [19] .
Sleep is regulated by the neurotransmitter, melatonin, released by the pineal gland at night [20] [21] . The pineal gland is situated in the middle of the brain behind the optic chiasma. It is outside of the blood brain barrier (BBB) and is highly perfused―receiving a blood flow rate that is second only to that of the kidney [22] . These considerations likely explain why it is especially susceptible to exposure to environmental toxicants such as aluminum, mercury, cadmium and fluoride [23] [24] . A postmortem study of aluminum levels in various brain regions revealed that the pineal gland accumulates aluminum at a rate that is at least twice that of other brain regions [25] .
Aluminum is a well-established neurotoxicant [26] - [28] . Progressive encephalopathy develops in association with high aluminum levels [29] . Accidental exposure to aluminum in dialysis fluid is associated with microcytic anemia followed by encephalopathy and dementia [30] - [32] . Premature infants exposed to aluminum in intravenous therapy [33] [34] experience neurological damage and mental impairment [35] . A study on the neurological effects of occupational exposure to aluminum revealed sleep disturbance as a common complaint, with insomnia being reported in 22.4% of cases and sleepiness in 14.9% [36] . A review of the underlying mechanisms of aluminum toxicity is presented in [26] where an argument for a direct role of pineal gland disruption is developed. We do not mean to imply that pineal gland dysfunction is the only neuropathology following aluminum exposure, but we believe it plays a significant role, as sleep disorder is associated with most neurological diseases.
Much less well studied is the herbicide, glyphosate, currently the most heavily used herbicide worldwide. Glyphosate application to weeds has grown enormously in the last decade in the US, in step with the increased adoption of genetically engineered (GE) crops such as corn, soy and canola that are engineered to be “Roundup Ready”. Epidemiological data readily available on the web from US government sources indicate a remarkably strong correlation between glyphosate application to corn and soy and multiple neurological disorders, as will be shown later in this paper.
Early in vivo toxicology studies allegedly proved that glyphosate is nearly nontoxic to mammals. However, there are two major concerns about these studies: 1) the studies are typically too short (lasting only 3 months), and 2) glyphosate was used in pure form rather than in combination with the surfactants/adjuvants that are always present in the formulas of herbicide products. A study that exposed rats to GM Roundup-ready corn and soy feed over the entire course of their lifetime showed significant damage, including massive mammary tumors in the females and liver and kidney damage in the males [37] . Two studies examining the toxicology profile of the surfactants in Roundup, such as the polyethoxylated alkylamines (POEAs), showed both that these additives are very toxic in and of themselves, and that they work synergistically with glyphosate to increase glyphosate’s toxicity, by an amount estimated to be at least 125-fold [38] [39] . These authors wrote ( [39] Abstract, p. 1): “Despite its reputation, Roundup was by far the most toxic among the herbicides and insecticides tested”.
In the remainder of this paper, we first present evidence of strong correlations between glyphosate usage and several diseases and conditions with rising trends over time. In Section 3, we discuss how glyphosate and aluminum can work synergistically to disrupt pineal gland function, in part through induction of anemia and hypoxia. In Sections 4 and 5, we show that both aluminum and glyphosate cause anemia and interfere with the CYP class of enzymes, leading to disrupted melatonin homeostasis. The next two sections focus on impaired sulfate supplies to the gut and to the brain respectively, and we discuss the implications in terms of gut health and brain health. Section 8 concerns the evidence of pineal gland dysfunction in association with Alzheimer’s disease, and the implications this has for Alzheimer’s pathology. Finally we summarize our findings in the discussion section and the conclusion.
2. Disease Trends
Most if not all of the major neurological diseases have been rising in incidence in recent decades, and it is imperative that we identify environmental factors that may be causative in this trend. In this section, we present several graphs showing remarkable correlations between many modern diseases and glyphosate application to corn and soy crops.
2.1. Methods
Time-trend data for various neurologically-related diseases and glyphosate applications to corn and soy crops were obtained as outlined in this section.
2.1.1. Glyphosate Data
Data for glyphosate applications to corn and soy crops were obtained from the US Department of Agriculture [40] . The procedure for gathering and analyzing these data are explained in detail in [41] . In this paper, the additional step was taken of dividing the total glyphosate applied to corn and soy crops by the total acreage planted in the survey states for each year to obtain an average application rate (in lbs/acre). The glyphosate average application rate data were then plotted against each of the data sets for disease as described below. The correlation coefficients between the data trend for the disease and glyphosate applications were calculated for each data set as previously outlined in detail [41] .
2.1.2. Hospital Discharge Data
One can get a sense of the trends in various diseases over time by looking at the hospital discharge diagnoses collected from hundreds of hospitals by the United States Centers for Disease Control and Prevention (CDC). These data are available for free download from the web at: ftp://ftp.cdc.gov/pub/Health_Statistics/NCHS/Datasets/NHDS/
Raw data files were available from 1998 through 2010. We downloaded the files and documentation from the CDC website. Each data file contains thousands of discharge records collected from hospitals using a statistically random sampling procedure [42] . The records contain information about the age, sex, race, geographic location and diagnoses for each discharge. The diagnoses are recorded by the International Classification of Diseases, Ninth Revision (ICD-9) codes. Up to seven diagnostic codes can be recorded for each discharge, with the first listed being the primary reason for hospital admission. We included in the set for any particular ICD-9 code any event which mentioned that code as one of the diagnostic codes for the event; i.e., we did not treat the first- mentioned code in any special way.
A computer program was written to query the data file for specific ICD codes for each year. A rate of increase, as an estimate of prevalence, over time for each particular diagnosis was obtained as follows:
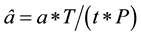
where â is the normalized number of hospital discharges of a disease in a year; a is the total number of the hospital discharge records of the disease in the year computed from the raw files; T represents the total number of hospital discharges in that year in the US; t is the total number of hospital discharge records in the sampled hospitals in that same year, which is computed from the raw files; and P is the total population in the US for that year. Population estimates were obtained from the CDC mortality database [43] .
2.1.3. Autism Data
We requested and received the individual Child Count data from the US Department of Education (USDE). These data are collected for children with disabilities under the Individuals with Disabilities Education Act (IDEA). The Child Count data are recorded by disability and age range. We extracted the data for the number of 6-year-old children with autism for each year from 1995-2010.
2.1.4. Mortality Data
Age-adjusted death data were obtained directly from the CDC mortality files [44] .
2.2. Results
Our first interest was in tracking the adoption of genetically engineered (GE) herbicide resistant (Roundup Ready) crops. As can be seen from Figure 1, only cotton had greater than 10% coverage in 1996, whereas by 2012 all crops had become around 90% herbicide resistant. The increase in glyphosate use on soy and corn crops closely tracks the adoption of herbicide-resistant, GE crops, as shown in Figure 2 and Figure 3. The total amount of glyphosate applied to soy, corn and cotton crops in the states surveyed by the USDA is shown in Figure 4. The survey states comprise approximately 90% of the total of these crops planted in the US
As can be seen from Figure 5, during the interval from 1998 to 2010, the estimated number of diagnoses at hospital discharge with ICD9 codes related to sleep disorder (ICD 327, 780.50 - 59, 307.41 - 49) have been increasing steadily, precisely in step with the increase in glyphosate usage on corn and soy crops (R = 0.97; p < 0.000013). Figures 6-10 show similar statistics drawn from the hospital discharge data for the following neurological diseases, which are all associated with insomnia: autism (R = 0.98), ADHD (R = 0.95), anxiety (R = 0.95), schizophrenia (R = 0.88) and dementia (R = 0.93). In addition, anemia, which we identify as a key initiat- ing factor in sleep disorder and in neurological disease, is also rising in step with glyphosate usage on corn and soy crops, as shown in Figure 11. Data from death certificates are consistent with hospital discharge data. Suici- de by overdose of prescription drugs (R = 0.95) is shown in Figure 12. Age-adjusted deaths from Alzheimer’s

Figure 1. Percentage of corn, soy and cotton crops that are genetically engineered to be herbicide tolerant from 1996 to 2012.

Figure 2. Percentage of GE soy crops planted and the percentage of acreage treated with glyphosate. Interpolated values are indicated with an asterisk next to the year. Data were not available for glyphosate applications to soy from 2007-2011. Data were only used through 2010; therefore the data point at 2011 was not interpolated. Data point for 2012 is shown for reference.

Figure 3. Percent GE corn crops and percentage of acreage treated with glyphosate; interpolated values indicated with asterisk.
disease (Figure 13) and from dementia (Figure 14) are both also highly correlated with glyphosate usage. The correlation with deaths from dementia is 0.991, with p-value ≤ 2.3E−9.
Finally, autism rates can be estimated independently from the hospital discharge data by looking at the number of children enrolled in the autism category under the Individuals with Disabilities Act (IDEA). The consistency
![]()
Figure 4. Glyphosate applied to corn, cotton and soy crops in the surveyed states.
![]()
Figure 5. Correlation between sleep disorder prevalence and glyphosate applications to corn and soy crops.
between the two data sets is remarkable. Figure 15 shows the number of first grade children diagnosed with autism in public schools in the US plotted along with glyphosate usage on corn and soy crops cumulated over the previous four years, thus estimating the child’s exposure rates from the age of two to the age of six. These two plots align almost perfectly: R = 0.997, p-value ≤ 2.37E−7.
3. Evidence of Harm
If we assume, based on the evidence in Figures 5-15, that there might be a link between glyphosate exposure
![]()
Figure 6. Correlation between autism prevalence, from hospital discharge data, and glyphosate applications to corn and soy crops.
![]()
Figure 7. Correlation between ADHD prevalence and glyphosate applications to corn and soy crops.
and neurological diseases, how can we explain it physiologically? One possibility is that glyphosate might enhance the bioavailability of other toxicants such as aluminum. A thorough literature review reveals a plausible me- chanism for such a hypothesis, as we will later show. It is also likely that glyphosate would also directly disrupt the synthesis of melatonin. The precursor to melatonin is serotonin, and the sole precursor to serotonin is trypto- phan, one of the three aromatic amino acids whose synthesis by plants and microbes is disrupted by glyphosate
![]()
Figure 8. Correlation between anxiety prevalence and glyphosate applications to corn and soy crops.
![]()
Figure 9. Correlation between schizophrenia prevalence and glyphosate applications to corn and soy crops.
through the shikimate pathway [45] . Finally, glyphosate also disrupts sulfur metabolism, especially the homeostasis of sulfate in the body [46] . We propose here that melatonin plays an important role in distributing sulfate to the brain, and that sulfate plays an essential role in disposing of cellular debris during sleep.
Because human cells do not possess the shikimate pathway, it has been argued that glyphosate is nearly nontoxic to humans. But this argument overlooks the fact that we depend upon our plant-based diet and our gut bacteria to provide the essential nutrients produced by the shikimate pathway: the aromatic amino acids, tryptophan, tyrosine, and phenylalanine. A study comparing 14 patients with early stage Alzheimer’s disease with 17
![]()
Figure 10. Correlation between dementia prevalence and glyphosate applications to corn and soy crops.
![]()
Figure 11. Correlation between anemia prevalence and glyphosate applications to corn and soy crops.
age-matched controls showed significantly reduced levels of tryptophan and methionine in patients compared to controls [47] . Plasma amino acid analyses, performed on 138 autistic children and 138 controls, showed that the autistic cases had significantly lower levels of phenylalanine, tryptophan, and methionine [48] . It was specifically mentioned in the abstract that tryptophan would be less available for serotonin synthesis, due to the low serum ratio of tryptophan to large neutral amino acids.
Although Monsanto has maintained that glyphosate does not bioaccumulate, studies on the blackworm species,
![]()
Figure 12. Correlation between suicide by overdose and glyphosate applications to corn and soy crops.
![]()
Figure 13. Correlation between deaths due to Alzheimer’s disease and glyphosate applications to corn and soy crops. Data discontinuity between 1998 and 1999 has been removed by subtracting a constant from 1999-2010 data points. Discontinuity arises in some data because of the ICD code change from the ninth revision to the tenth (ICD-9, ICD-10).
Lumbriculus variegatus have shown bioaccumulation of glyphosate [49] . Studies on humans and cows found glyphosate residue in urine [50] . The humans on an organic diet had significantly lower urinary levels, and chronically ill humans had a higher urinary level than those not chronically ill.
Furthermore, the cows were dissected and glyphosate residues in the tissues of the kidney, liver, lung, spleen, muscles and intestines were comparable to that found in the urine. This means that the glyphosate is not being pass- ed through the urine without affecting the organism, and that meat and dairy are an additional source of dietary

Figure 14. Correlation between deaths due to senile dementia and glyphosate applications to corn and soy crops.

Figure 15. Graph of US rates of autism prevalence in first grade served under IDEA compared with glyphosate usage on corn and soy over the previous four years.
glyphosate for humans. In another study by Krüger et al., glyphosate residues were detected in the lungs, liver, kidney, brain, gut wall and heart, in malformed one-day-old piglets [51] .
Sulfation of melatonin depends on cytochrome P450 (CYP) enzymes, and it was proposed in [46] that CYP enzyme disruption may be a key component of pathology associated with glyphosate in humans. Sulfate deficiency in the brain is associated with neuropathology, and such deficiency arises in part due to impaired CYP
function. Furthermore, as we will show later, glyphosate impairs sulfate supplies through other mechanisms besides CYP enzyme inhibition. Glyphosate toxicity also leads to anemia, which enhances aluminum retention in the brain and induces hypoxia.
The notion of a gut-brain axis has become a “hot topic” in the recent biomedical literature [52] . The key idea is that enteric microbiota interact with the brain through signaling mechanisms that can influence homeostasis. A stunning set of experiments was recently performed on mice engineered to express many behavioral features of autism [53] . The autistic phenotype was introduced through maternal immune activation during gestation. A detailed profile was determined for the gut microbiome and for the plasma metabolome. The gut microbial analysis indicated a different distribution in the specific species of Clostridium and Bacteroides that were present in the autistic mice compared to controls, along with a distinct serum biomarker manifested as a 46-fold increase in serum levels of 4-ethylphenylsulfate (4EPS), which was, remarkably, completely normalized by probiotic treatment with B. fragilis. Germ-free mice display nearly undetectable levels of serum 4EPS, indicating that it is derived from products of the microbiota [53] . It has been shown that 4EPS is produced by microbiota in rats in response to heavy metals [54] . It is closely related to p-cresol sulfate (4-methyl phenylsulfate: 4MPS), which has been identified as a urinary marker for autism [55] [56] . Injection of 4EPS into control mice led to manifestations of an autistic phenotype, showing a direct causal effect of this one metabolite in autism.
4. Glyphosate and Aluminum Synergy: The Anemia Factor
In this section, we provide evidence that glyphosate works synergistically with aluminum to cause anemia, and that anemia leads to hypoxia, which enhances the effects of aluminum on neurons. The hypoxic conditions often associated with preterm birth promote aluminum uptake by the pineal gland, and infants born prematurely are highly susceptible to later developing autism [57] [58] . Hypoxia is an important factor in the pathological processes that lead to endothelial cell migration and angiogenesis. Melatonin protects from the adverse effects of hypoxia, but this appears to exhaust the supply in the pineal gland.
In addition to its antioxidant properties, melatonin inhibits hypoxia-induced cell migration by suppressing hypoxia-inducible factor-1α (HIF-1α) [59] . A study on the effects of hypoxia on the pineal gland showed that the expression of HIF-1α, vascular endothelial growth factor (VEGF), endothelial nitric oxide synthase (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS) were all increased, while melatonin content was significantly reduced [60] . The authors suggested that increased vascular permeability may allow free access of serum-de- rived substances in the pineal gland to disrupt secretory function. This could enhance the toxicity of aluminum and glyphosate present in the serum, as well as reducing the bioavailability of melatonin.
Mineral deficiencies in iron [61] and cobalt [62] both contribute to anemia, a direct cause of hypoxia due to insufficient oxygen-carrying capacity. Iron deficiency is prevalent among children with autism [63] . Cobalamin deficiency is a common cause of macrocytic anemia [62] , and cobalamin depends on cobalt. A Danish study on cows showed severe cobalt deficiency in conjunction with exposure to glyphosate as evidenced by glyphosate presence in the urine [64] . A case study showed optic neuropathy following severe cobalamin deficiency in three children with autism [65] .
Aluminum causes a microcytic hypoproliferative anemia which contributes to the anemia in patients with end stage renal disease, and the mechanism includes cytochrome-oxidase inhibition, impaired iron transport, and ineffective erythropoieisis. It appears to involve inhibition of heme synthesis, either via inhibition of enzyme activity or by interference with iron incorporation [66] .
Anemia and hypoxia were observed in 6 out of 16 human patients exposed to acute glyphosate intoxication [67] . There are many mechanisms by which glyphosate might induce anemia. Ferric reductase is required for iron absorption in the human gut. Glyphosate reduced the activity of ferric reductase in plants by 50% within six hours of treatment [68] , so it might have a similar effect in mammals. Evidence from plants supports the idea that glyphosate may directly interfere with heme synthesis. Glyphosate inhibits 5-Enolpyruvyl Shikimate 3- Phosphate synthase (EPSP synthase) and it interferes with the reaction of the enzyme with pyridoxal 5’-phos- phate, probably by binding at the same site [69] . This suggests that glyphosate might have the same effect for δ- aminolevulinic acid synthase (ALAS), the enzyme that catalyzes the first step of pyrrole synthesis, which is dependent upon pyridoxal 5’-phosphate as a catalyst. Indeed, inhibition of ALAS by glyphosate has been noted in many studies in plants [70] - [73] .
This leads directly to impaired chlorophyll synthesis, since chlorophyll is assembled from pyrrole units that encase the magnesium atom at the center. Some have suggested that this may be more important than shikimate pathway inhibition in glyphosate’s pathological effects on plants. Impaired pyrrole synthesis would have a significant effect in animals, since both cobalamin (vitamin B12) and heme depend upon pyrrole rings for their construction. Impaired pyrrole synthesis would then lead to both hemoglobin-deficiency anemia and the anemia associated with vitamin B12 deficiency.
In addition to the independent effects of glyphosate and aluminum on hypoxia and their neurotoxicity, glyphosate will likely enhance the toxic effects of aluminum through multiple factors. There is strong evidence that glyphosate chelates aluminum [74] , as it does many other metal cations: it can form “aluminum cages” through several different configurations involving multiple coordinated glyphosate molecules. It is well established that citrate similarly chelates aluminum, and that this allows ingested aluminum to penetrate the gut barrier [75] [76] .
Citrate also chelates free calcium, and this is known to disrupt cellular tight junction integrity. Small un- charged molecules such as Al-citrate can then freely pass through a paracellular pathway, and this greatly in- creases the rate of absorption of aluminum in the proximal bowel [77] . Glyphosate also chelates calcium [78] , and, like citrate, it is a small molecule. Therefore, it can be expected that glyphosate would similarly allow oral- ly delivered aluminum to gain access to the vasculature, although this has not yet been demonstrated experimen- tally.
Pigs fed a diet of GM Roundup-Ready corn and soy suffered from an inflamed stomach [79] , clearly suggesting gut dysbiosis. A leaky gut, associated with gastrointestinal disorders, will facilitate the passage of both Al- citrate and Al-glyphosate past the impaired tight junctions of the gut boundary. Glyphosate is a microbicide, and it preferentially kills beneficial gut bacteria [46] [80] , allowing pathogens like Clostridium difficile to overgrow. As previously mentioned, Clostridium difficile produces the toxic phenol, p-cresol, which has been shown to enhance aluminum uptake in hepatocytes [81] , mediated by transferrin receptors.
The pineal gland has significant transferrin receptor protein expression, which is enhanced under hypoxic conditions [82] . Neuronal injury due to hypoxia resembles the molecular abnormalities seen in Alzheimer’s disease [83] . There is a concern of aluminum exposure from parenteral nutrition in preterm infants, as demonstrated from long-term follow-up study of their bone health [84] . Transferrin expression in the pineal gland is repressed by melatonin [82] , so melatonin deficiency would work synergistically with hypoxia to promote aluminum uptake.
5. Hepatic Toxicity and CYP Enzymes
In vivo experiments on aluminum hepatotoxicity in rats revealed that aluminum reduces the levels of CYP enzymes in the liver by as much as 50%, and that this is associated with a reduced ability to metabolize drugs [85] [86] . Jeffery et al. [85] suggested that the toxicity of aluminum might be mediated in part through the release of free iron from heme groups due to competitive ligand displacement. This might also explain its ability to interfere with CYP enzyme function, as CYP enzymes contain a heme group. This idea is further supported by the observation of aluminum-induced anemia, which also appears to involve inhibition of heme synthesis, possibly through interference with iron incorporation or utilization in heme [66] .
Glyphosate has also been shown to disrupt CYP enzymes in plants [87] , rat liver [88] , and human hepatic microsomes [89] . Its mechanism of action is likely through nitrogen binding to heme, and therefore it acts synergistically with aluminum’s interference with iron. Glyphosate would also be anticipated to impair CYP enzyme synthesis through its disruption of heme synthesis, as discussed previously. Obviously, CYP enzyme impairment in the liver results in enhanced toxicity of multiple environmental toxins, including both drugs and pesticides. Thus, hepatic encephalopathy [90] may arise from increased vulnerability of the brain, particularly the region outside the BBB, to toxins that were not adequately cleared by the liver.
CYP enzymes are heavily involved in the metabolism of melatonin [91] . CYP1A2 and CYP2C19 are largely responsible for plasma clearance of melatonin by the liver. 6-hydroxylation of melatonin, a necessary intermediary step towards sulfation, is likely achieved by CYP1B1 in the brain and the liver. Intriguingly, supplementary melatonin in rats accumulates in the brain almost exclusively as melatonin sulfate, whereas the plasma melatonin component was found to be only 75% sulfated [92] . Thus, melatonin sulfation would be expected to be impaired in the context of dysfunctional CYP enzymes.
Sections 4 and 5 will develop more fully the argument that sulfate serves a critical role in both the gut and the brain, and that aluminum and glyphosate work synergistically to disrupt sulfate synthesis and sulfate transport.
6. The Significance of Sulfate in the Gut
Gastrointestinal symptoms are strongly correlated with autism [93] , and this could lead to treatment with aluminum-containing antacids, thus providing a ready source of aluminum, chelated by either citrate or glyphosate, to gain entry into the general circulation. Aluminum-containing antacids can lead to constipation [94] , which is a common symptom among autistic children [95] . Serotonin initiates peristalsis by activating serotonin receptors on sensory neurons in the intestine [96] , and hence a deficiency in serotonin could further promote constipation issues. Such a deficiency could easily arise in the presence of glyphosate, since serotonin is derived from tryptophan, a product of the shikimate pathway.
It has long been known that children with autism have abnormal sulfation chemistry, with extremely low levels of free serum sulfate [97] . A study of metabolic biomarkers of autism revealed lower baseline plasma concentrations of methionine, homocysteine, cystathionine, cysteine, and glutathione [98] . Studies on Escherichia coli (E. coli) have shown that glyphosate suppresses two enzymes involved in sulfate activation and metabolism. Adenosine 5’-phosphosulfate (APS) kinase is reduced 2.55-fold and 3’-phosphoadenosine 5’-phosphosulfate (PAPS) reductase activity is reduced 3.75-fold [98] . APS kinase produces PAPS, which is then reduced by PAPS reductase to yield PAP and sulfite. PAPS synthesis is an essential step in the process of assimilating sulfate into biologically important molecules, such as the sulfur-containing amino acids or the glucosinolates in plants. Defects in PAPS metabolism in members of the Brassicaceae family of plants produces a pseudo-sulfate deficiency despite adequate sulfate in the medium, along with reduced glutathione levels [99] . Thus we can predict that glyphosate exposure in plants will lead to depleted supplies of these important nutrients in food sources. Furthermore, it is likely that this same inhibition will also negatively impact E. coli and other gut bacteria, and it might well explain the overgrowth of desulfovibrio found in association with autism [100] . Desulfovibrio is able to utilize sulfate as an energy source by reducing it to hydrogen sulfide gas (H2S) through an alternate pathway. Excess H2S is highly toxic, and this could lead to gastrointestinal discomfort, as well as depleting sulfur supplies.
Celiac disease has become an epidemic in the last decade [101] , and it is characterized by villous atrophy in the small intestine, interfering with absorption of multiple micronutrients. Samsel and Seneff [102] developed a hypothesis that glyphosate residues in food may promote celiac disease, and much of their argument was based on disrupted CYP enzyme function. The destroyed epithelial cells lead to an accumulation of protein debris which is then partially degraded by anaerobic gut bacteria in the large intestine, producing toxic phenolic compounds like p-cresol as derivative molecules of tyrosine [103] [104] . In this section, we discuss an important role for CYP enzymes in maintaining the health of the crypts and the villi in the small intestine, and we will argue that CYP dysfunction in the gut and liver leads to impaired supply of serotonin, melatonin and sulfate to the brain.
Dysfunctional vitamin D homeostasis likely plays an important role. It has recently been acknowledged that there is an epidemic of vitamin D3 deficiency in the US today, as determined by measuring serum levels of 25(OH)D3 [105] . Figure 16 shows a rapid rise in hospital discharge diagnoses of vitamin D deficiency beginning in 2006. Maternal vitamin D3 deficiency has been implicated in autism [106] . Vitamin D activation is a two-step process, beginning with a CYP-mediated oxidation to 25(OH)D3 (calcidiol), and followed by a CYP- mediated oxidation to 1,25(OH)2D3 (calcitriol). Two distinct CYP enzymes in the liver, one microsomal and one mitochondrial, perform the first step in vitamin D activation, and two CYP enzymes in the kidneys, also one microsomal and one mitochondrial, perform the second step [107] . Thus, impaired CYP function (especially in the liver but also in the kidney), due to exposure to aluminum and/or glyphosate, could be a contributor to celiac disease and related gut disorders, as well as the epidemic in vitamin D3 deficiency.
Vitamin D regulates sulfate homeostasis through its influence on sulfate reuptake in the kidney [108] . The kidney normally controls serum levels of free sulfate through reabsorption in the proximal tubules of over 80% of the sulfate in urine. Vitamin D-deficient rats exhibit reduced serum sulfate levels along with markedly increased renal excretion of sulfate [109] . Experiments on mice with impaired vitamin D receptor (VDR) activity have confirmed that VDRs in the kidney regulate the amount of sulfate that is reabsorbed [108] . These mice showed a 42% increase in urinary sulfate levels, a decrease of 50% in serum sulfate, reduced glutathione content in the liver, and, most importantly, 45% lower sulfated proteoglycan content in the skeleton. Thus, a low level of

Figure 16. Temporal trends in vitamin D deficiency according to CDC hospital discharge data.
calcitriol, as would be expected with impaired CYP activity, can be predicted to lead to a deficiency in sulfated proteoglycans, which is, in turn, associated with colitis and Crohn’s disease [110] .
VDR expression is reduced in diseased colonic regions of both Crohn’s disease and ulcerative colitis patients, and mice engineered to have increased expression of VDR in the colon are resistant to colitis [111] . Epithelial VDR signaling inhibited epithelial cell apoptosis by blocking NF-κB, thus protecting from mucosal permeability. Calcitriol induces autophagy in human monocytes/macrophages as well as the capture of bacteria within autophagosomes, promoting antimicrobial activity [112] .
Colston et al. [113] proposed that the main function of calcitriol in the small intestine is to promote differentiation of enterocytes in the crypts at the base of the villi. Thus, a deficiency in calcitriol will lead to impoverished enterocyte renewal. Serotonin is primarily produced in the gastrointestinal system, specifically, the enterochromaffin (EC) cells of the intestinal mucosa [114] , which are interspersed with the enterocytes in the villi. Serotonin activates neural reflexes in the gut through 5-HT3 and 5-HT4 receptor action to induce intestinal motility through peristalsis and secretion [115] . Inflammatory bowel disease, irritable bowel syndrome, and constipation are all associated with altered serotonin signaling. It has been proposed that the diarrhea and vomiting associated with the very common infective agent, Rotavirus, is due to the fact that the virus stimulates enterocytes to produce an excess of serotonin [116] [117] . Of note, serotonin is found in the cerebral spinal fluid of the brain conjugated with sulfate [118] , and thus it may be a transport mechanism for sulfate from the gut to the brain.
A recent in vivo study on mice, involving a drug, (R)-DOI, with a very specific agonistic effect on serotonin 5-HT2A receptors, revealed a remarkable anti-inflammatory effect localized to the small intestine [119] . The authors wrote: “Remarkably, we observed a near complete blockade of TNF-α-induced proinflammatory marker gene expression in this tissue at the lowest dose of (R)-DOI (0.01 mg/kg), mirroring the super potencies observed in vitro” ( [119] p. 3). The dose was administered through injection in the abdominal region, likely giving the drug direct access to this tissue. However, this demonstrates that the small intestine depends upon serotonin bioavailability to protect from TNF-α-induced inflammation that leads to inflammatory bowel syndrome.
Much can be learned from a set of rare genetic defects associated with impaired protein glycosylation. A case in point is a boy who suffered from a “protein losing enteropathy” (PLE) due to a congenital glycosylation disorder [120] . Intestinal biopsy revealed a complete absence of heparan sulfate on the basolateral surface of enterocytes, which would ordinarily be attached to membrane-bound syndecan-1 proteoglycans [121] [122] . It was noted that the most severe gastrointestinal manifestation of the disorder was observed in the rapidly dividing enterocytes associated with gastroenteritis-induced stress. Three other examples of this condition are described in [123] . Three infant boys required total parenteral nutrition due to massive enteric protein loss and secretory diarrhoea. A gross abnormality was detected in the small intestinal glycosaminoglycans (GAGs) in all three infants, once again with complete absence of enterocyte heparan sulfate.
These observations attest to the importance of heparan sulfate in the maintenance of proper bowel function. Substantial loss of sulfated GAGs from the subepithelial basal lamina has been found in association with both colitis and Crohn’s disease [123] . Increased enterocyte apoptosis is a hallmark of celiac disease [124] [125] , and it could be due to vulnerabilitiesfollowing insufficient sulfate supply to the matrix proteins. Insufficient calcitriol supply likely also plays a role here, as nuclear calcitriol (1,25(OH)2D3) receptor activity in the small intestine is localized to crypts in the duodenum and jejunum, where precursor cells begin their maturation into enterocytes [113] . A depleted renewal of the supply of enterocytes can lead to excess wearing of the brush border associated with celiac disease [126] . These authors wrote: “Taken together these findings suggest that the major function of calcitriol on small intestine is to promote differentiation of immature enterocytes in the crypts and at the base of villi” ( [126] p. 1224).
It has been demonstrated that the liver produces sulfated versions of both vitamin D2 and vitamin D3, and that vitamin D3 sulfate appears in rabbit urine after oral administration of vitamin D3 [127] . Furthermore, it appears that only the liver is capable of synthesizing vitamin D3 sulfate, and that one pathway for secretion is through the bile acids [128] . Given the strong dependency upon heparan sulfate for proper formation of intestinal villi, and given the strong dependency of villus maturation on VDRs, it can be hypothesized that vitamin D delivers sulfate to the villi, which is then used to produce heparan sulfate proteoglycans (HSPGs), which are so critical for the healthy function of the enterocytes in the crypts.
7. The Significance of Sulfate in the Brain
A much-overlooked factor in neurological disease is the critical role of sulfate in maintaining the health of the brain. In a post-mortem study, among all lipids examined, only the sulfonated lipid, sulfatide, was found to be severely depleted in association with Alzheimer’s disease [129] , and this defect existed even in the earliest stages. Mice engineered to be defective in the synthesis of heparan sulfate in the brain exhibit all the features of “mouse autism” [130] . Postmortem studies have confirmed that heparan sulfate is depleted in the cerebrospinal fluid in the lateral ventricles of autistic children [131] . HSPGs are attached to syndecans and glypicans in the plasma membrane of all cells, and they play critical roles in metabolism, ion and nutrient transport, recycling of damaged molecules, and signaling cascades [132] . Experiments in rats have shown that low serotonin during early brain development can lead to fewer dendritic spines and reduced synaptic density, as well as excess cortical brain growth [133] - [135] , and these developmental abnormalities are features of autism in humans [136] [137] .
The tryptophan hydroxylases, TPH1 and TPH2, are responsible for serotonin synthesis from tryptophan. Vitamin D activates the transcription of TPH2 in the brain, and suppresses the transcription of TPH1, which exists at sites outside the brain, including the placenta during pregnancy. It has been proposed that this regulation of THP1 and TYP2 by vitamin D3 explains three major characteristics of autism that follow from the observation that autism is associated with low vitamin D status in both the mother [106] and the child [138] . These are: 1) low serotonin concentrations in the autistic brain, 2) the high male prevalence of autism, and 3) the presence of maternal antibodies against fetal brain tissue [139] .
Obviously, low vitamin D status in the child would lead to reduced serotonin expression in the brain. Studies on mice have reproduced an autism phenotype through selective neonatal forebrain serotonin depletion [136] . Males would be more adversely affected by the loss of brain serotonin due to low vitamin D because estrogen also stimulates serotonin expression and can therefore rescue the female brain from deficiency. Low vitamin D in the placenta due to the mother’s deficiency could cause TPH1 activity to act as a tryptophan trap, shunting tryptophan away from the kynurenine pathway, which would result in a decreased number of regulatory T cells to protect the fetal brain from immune attack by maternal antibodies. Vitamin D, tryptophan, and estrogen are all sulfoconjugated when transported in the blood.
An experiment that involved administering 40 ppm melatonin for 11 weeks to mice demonstrated that 99.9% of the melatonin taken up into the brain had been 6-hydroxylated and then conjugated with sulfate [92] . Pinealocytes in the pineal gland augment the sulfate content in their HSPGs during the daylight hours, along with light-induced upregulation of the enzyme, 3-O-sulfotransferase [140] . The pineal gland is situated at the base of the third ventricle, and it delivers sulfated melatonin to the tissues of the brain during the night cycle. Furthermore, all of the monoamine neurotransmitters present in the cerebrospinal fluid are predominately conjugated with sulfate [141] .
In a postmortem study of the brains of subjects with autism, the principal anomalous finding was diminished grey matter in the region of the hypothalamus directly adjacent to the optic chiasma [142] , i.e., the median eminence. Studies on melatonin receptor locations in rat brain revealed that the median eminence has the highest receptor concentrations [143] . Multiple regulatory hormones are released from the median eminence to the posterior part of the pituitary gland, including corticotropin releasing factor, gonadotropin releasing hormone, thyrotropin releasing hormone, growth hormone releasing hormone and dopamine. The median eminence is situated adjacent to the optic chiasma, and therefore in close proximity to the pineal gland, and thus has ready access to melatonin. Thus, reduced grey matter in this critical region, possibly as a result of reduced melatonin supply, would predictably lead to disrupted pituitary regulation.
Intriguingly, only a few regions of the brain have been shown to exhibit circadian rhythms in their metabolic activities, and these are all regions that have access to light stimuli through the optic chiasma. They include the suprachiasmic, supraoptic and raphe nuclei of the hypothalamus. All exhibit substantially higher glucose uptake during the daylight hours [144] . We hypothesize that this is related to the increased bioavailability of sulfate during daylight, which can be exploited to produce sulfated GAGs for temporary storage of glucose as glucosamine sulfate in the extracellular space [145] .
Dehydroepiandrosterone (DHEA) and DHEA sulfate are neuroactive steroids that are produced from cholesterol in the brain [146] . DHEA sulfate and pregnenolone sulfate display memory-enhancing properties in aged rodents [147] , and they play an important regulatory role in the brain [148] [149] . Decreased levels of brain DHEA sulfate are correlated with reduced perfusion in the hippocampus in association with Alzheimer’s disease [150] . Remarkably, DHEA sulfate, but not DHEA, increases the melatonin secretion by the pineal gland by 50% to 80%, but only if it is present during the daylight phase of the diurnal cycle [151] . One can surmise that it is providing sulfate for conjugation with melatonin.
Cholesterol sulfate interacts with the omega-3 fatty acid, docosahexaenoic acid (DHA) in response to light stimulus [152] to induce electron flow. DHA is important in the pineal gland [153] , and dietary enrichment in DHA in rats resulted in increased levels of DHA in the pineal gland, which was associated with increased melatonin sulfate secretion in urine at night [154] . DHA likely plays an important role in catalyzing sulfate synthesis, through its induction of electron flow.
A study on keratinocytes, specialized cells in the skin that produce an abundance of cholesterol sulfate, have shown that activation of liver X receptors (LXRs) in the keratinocytes leads to a substantial upregulation of the cholesterol sulfotransferase, SULT21b, and therefore induces synthesis of cholesterol sulfate [155] . Mice engineered to have defective LXR receptors (both the α and β isoforms) exhibited a remarkable phenotypic expression which amounted to completely collapsed lateral and third ventricles in the brain, and the ventricles were lined with an accumulation of lipid-laden cells [156] . These two considerations, taken together, imply that sulfation of cholesterol may be essential for cholesterol export, a logical deduction due to the fact that sulfation makes cholesterol amphiphilic. Further support for this conclusion comes from a study on human aortic cells, which showed that over expression of SULT2B1b led to the release of intracellular lipids from the aortic cells [157] . Intriguingly, 25-hydroxycholesterol-3-sulfate (25HC3S) (but not 25HC) suppresses the inflammatory response through a signaling cascade involving nuclear PPARγ and NF-κB [155] . Inflammation induces the acute phase response which leads to pathological lipid and lipoprotein metabolism [158] [159] .
It has recently been proposed that the restorative function of sleep may be a consequence of the enhanced removal of cellular debris such as oxidized/glycated/misfolded proteins, oxidized lipids, and amyloid-β plaque [160] . Endocytosis and subsequent digestion through the endosomal-lysosomal progression are critical for neuronal function, and are known to be impaired in association with Alzheimer’s disease and other neurological disorders [161] [162] . Debris such as amyloid β and α synuclein that accumulate in association with neurological diseases induce a pro-inflammatory response that is neurotoxic [163] . An arylsulfatase localized to lysoso- mes breaks down GAGs, including the HSPGs that are temporarily housed in the immediate surrounding area of most cells, and an impaired version of this enzyme leads to lysosomal storage disorders associated with severe neurological impairment [164] .
Iron is the most abundant transition metal on the earth’s crust and it easily cycles between ferric (Fe(III)) and ferrous (Fe(II)) oxidation states, making it very useful for redox reactions but also very dangerous to exposed biomolecules. Most lysosomes contain large amounts of iron, which makes them susceptible to rupture [165] . DNA, membrane lipids and cellular proteins are highly susceptible to damage from exposure to ferrousiron through the generation of reactive oxygen species (ROS) [166] . Lysosomes are intimately involved in iron recycling, and they utilize ferrous iron to help break down cellular debris. Heparin, the most highly sulfated molecule known to biology, protects lipids such as linolenic acid from peroxidation damage through exposure to ferrous iron, and it is hypothesized that it does so by providing an acidic environment where water is produced instead of the ROS, superoxide, when iron is oxidized [167] . Removal of sulfate from the heparin disables its protective effects. These authors further proposed that this acidification effect may be an important role for the HSPGs that are endocytosed into lysosomes along with cellular debris to be broken down [168] .
In summary, evidence from multiple fronts suggests that sulfate insufficiency is a critical factor in neurological disease. A deficiency in vitamin D, which induces sulfate wasting through the kidneys, leads to suppressed serotonin synthesis in the brain and excess serotonin synthesis in the placenta, both of which contribute to the pathology of autism in the fetus and the neonate. Cholesterol sulfate suppresses inflammation [155] , which is a key component of neuronal degeneration [169] . Cholesterol sulfate synthesis is mediated through LXRs [155] , and defective LXRs lead to severe pathology in the ventricles [156] . Melatonin is transported within the ventricles as melatonin sulfate, and melatonin induces the sleep cycle which enables the clearance of cellular debris, whose recycling depends critically upon heparan sulfate in the lysosome, via the protection it affords from oxidative damage from ferrous iron. Both oxidative stress and impaired clearance of cellular debris are implicated in neurological diseases.
8. Pineal Gland Calcification and “Alumification”
Seneff et al. [145] proposed that endothelial nitric oxide synthase (eNOS), in addition to its well established role in synthesizing nitric oxide, produces sulfate in specialized cells in the skin, catalyzed by sunlight, using mechanisms that are similar to those used by sulfur-oxidizing bacteria. In this paper, we propose that the pineal gland similarly utilizes nitric oxide synthase to synthesize sulfate in response to light, following the same arguments detailed in [145] . eNOS is a CYP enzyme, although it is the only CYP enzyme that produces nitric oxide, and it does not appear to use its CYP domain for this task. Many other CYP enzymes are involved in sulfur oxidation [170] , and so this dual function would legitimize the presence of a CYP domain in eNOS. As a CYP enzyme, it would be subject to disruption by both aluminum and glyphosate.
It is well established that, in a “decoupled” mode, eNOS produces superoxide instead of nitric oxide, and this superoxide can be utilized to oxidize reduced sulfur under appropriate physiological conditions [145] . Here, we further propose that both eNOS and nNOS in the pineal gland also produce sulfate in response to sunlight, which is used to resupply the HSPGs with sulfate that is depleted following melatonin sulfoconjugation and distribution.
The pineal gland contains an abundance of eNOS, nNOS and iNOS isoforms [60] , present in both pinealocytes and in nerve fibers innervating the gland, and it remains a mystery as to what role they play [171] . Following hypoxic stress, the pineal gland melatonin content was significantly reduced, along with increased vascular permeability and increased expression of eNOS, nNOS, and iNOS [60] , suggesting that melatonin sulfate was distributed to the neural tissues through this process. Remarkably, the amount of nNOS synthesized by the pineal gland is regulated by the light/dark cycle. When the gland is exposed to constant light or to a light:dark cycle of 20:4, its synthesis of nNOS is suppressed [172] . If it utilizes its nNOS to synthesize sulfate, it would be necessary to exploit the night cycle to dispense with the synthesized sulfate via the melatonin carrier. Without a night cycle, excess sulfate would accumulate and nNOS synthesis suppression is expected through a feedback regulatory mechanism.
Through the release of melatonin, the pineal gland regulates reciprocal relationships between the neuroendocrine system and the immune system [173] . The pineal gland produces melatonin under control from the circadian clock, maintained by the suprachiasmic nucleus (SCN). Melatonin is an important antioxidant and neuroprotector that also regulates the sleep-wake cycle [174] . Melatonin exerts efficient protection against oxidative stress and nitrosative damage, through its metal-binding effects, free-radical scavenging, and stimulation of antioxidative enzymes [175] . van Rensburg et al. [176] proposed a model for AD pathology whereby excess aluminum is taken up into the brain where it exasperates iron-induced lipid peroxidation in the lysosomes. The study also demonstrated that melatonin was protective against such iron-induced lipid peroxidation, possibly through free radical scavenging.
It has become very clear that Alzheimer’s is associated with calcification of the pineal gland, and with sharply decreased synthesis of melatonin. When melatonin levels were measured postmortem in the cerebrospinal fluid (CSF) of 85 Alzheimer’s patients and 82 age-matched controls [177] , it was found that CSF melatonin levels in the AD patients were only 20% of the levels found in the control subjects. This was equally true for early-stage Alzheimer’s as for late-stage. Those with two copies of the ApoE-4 marker for Alzheimer’s risk had even lower levels of melatonin than the other Alzheimer’s patients. Computer tomography was used [178] to show that the amount of calcified pineal tissue in patients with Alzheimer’s disease was significantly more than that in either normal controls, patients with depression, or patients with other types of dementia.
Melatonin is a potent antioxidant, and its reaction with hydrogen peroxide produces another scavenger that is at least as effective as melatonin itself [179] . It also stimulates antioxidative enzymes and stimulates ATP production in the inter-mitochondrial membrane. Since oxidation damage and mitochondrial dysfunction are hallmarks of Alzheimer’s disease [180] , it is plausible that melatonin insufficiency is a key factor in Alzheimer’s pathology. Melatonin, as well as its precursors, tryptophan and serotonin, all have been shown to bind aluminum, and it was argued that loss of melatonin with age may be a key factor in increased susceptibility to aluminum toxicity in the brain leading to Alzheimer’s disease [181] . Furthermore, researchers have used several different methods to confirm that melatonin protects cultured cells from death following exposure to toxic fragments of amyloid-β [182] , another hallmark of Alzheimer’s disease. Finally, in vivo chemical-induced tau hyperphosphorylation in the rat lateral ventricle could be partially blocked by prior exposure to melatonin [183] .
Calcification of the pineal gland can begin very early in life, and there is a trend towards increased calcification with increasing age [174] . A study in 1974 showed that Blacks in America had much slower rates of calcification than Whites, and that Blacks in West Africa had substantially less calcification than their counterparts in the US [184] . These differences might be attributed in part to differentials in sunlight exposure. Calcification likely has a significant negative effect on the pineal gland’s ability to produce sulfate following light stimulation. This is because calcium binds to calmodulin, triggering a signaling cascade that causes eNOS to detach from the plasma membrane and switch to nitric oxide synthesis, following phosphorylation [185] [186] .
As we have previously stated, the pineal gland also tends to bioaccumulate aluminum more easily than other parts of the brain [25] . Aluminum gains entry into cells as a calcium mimetic, and it binds calmodulin with a ten-fold higher binding strength than calcium [187] . So it can be expected that aluminum will have a severe adverse effect on sulfate synthesis and melatonin synthesis in the pineal gland.
9. Discussion
In this paper, we have developed the argument that a synergistic interaction between aluminum and glyphosate can lead to severe impairment of pineal gland function, and that this could explain the association of sleep disorder with a wide range of neurological diseases, and likely even help explain the related pathologies of those diseases. We began by showing the remarkably strong correlations between glyphosate application to corn and soy in the US and sleep disorder, autism, ADHD, suicide, anxiety disorder, and dementia. We have shown how glyphosate could assist entry of aluminum into the general circulation through chelation, and how glyphosate would indirectly enhance aluminum absorption by the pineal gland via its induction of p-cresol synthesis by pathogenic gut bacteria. p-Cresol enhances aluminum uptake via transferrin, which is present in large amounts in the pineal gland, and in enhanced amounts under hypoxic stress conditions, induced by both glyphosate and aluminum. We have also discussed the important role of CYP enzyme dysfunction in disrupting vitamin D3 homeostasis and disrupting sulfation and metabolism of melatonin. We have shown that vitamin D3 normally plays an important role in maintaining the health of the intestinal villi in the small intestine, and we have argued that part ofits role may be to supply sulfate to the enterocytes. Serotonin is the sole precursor to melatonin, and serotonin itself is produced by ECcells in the intestinal villi from tryptophan, one of the three aromatic amino acids whose synthesis is disrupted by glyphosate, through its interference with the shikimate pathway. Since tryptophan is an essential amino acid, we depend upon supply from food sources and/or from our gut bacteria. Thus, it can be expected that glyphosate residues in the food crops introducing exposure to gut microbes would lead to deficiencies in tryptophan and therefore in serotonin and melatonin.
An important contribution of this paper is to raise awareness of the essential roles of sulfate in the brain, and to argue that sulfate deficiency in the cerebrospinal fluid in the ventricles plays a crucial role in neurological disease. Research has shown that CSF deficiency in heparan sulfate is associated with autism [130] , and that deficiency in sulfatide is a feature of Alzheimer’s disease [129] . Lysosomal recycling of cellular debris, which has been argued to be a key role for the sleep cycle [160] , is impaired in Alzheimer’s disease [161] [162] . Melatonin sulfate can supply sulfate to the HSPGs in the brain at night to induce the low pH needed to safely break down proteins using iron-based redox chemistry, while protecting the cell from the ROS that are produced through iron oxidation under non-acidic conditions [167] . And the melatonin provides essential antioxidant capabilities to further protect from collateral damage [174] . Melatonin has been shown to be protective against amyloid β toxicity and to prevent the formation of tau tangles, both hallmarks of Alzheimer’s disease [182] .
Glyphosate disrupts sulfur metabolism through its suppression of both CYP enzymes and enzymes involving the synthesis and metabolism of PAPS, the activated form of sulfate. Besides reducing the bioavailability of essential sulfur-containing amino acids, this also leads to impaired sulfate supplies, systemically. As we have shown, this has implications for both the gut and the brain, and it can help explain the deficiencies in sulfate in the gut inassociation with inflammatory intestinal diseases and deficiencies in the brain associated with neurological diseases.
We have proposed here that the pineal gland uses its eNOS and nNOS enzymes to produce sulfate from reduced sulfur sources. The pineal gland becomes calcified with age, and calcification of the pineal gland is highly correlated with Alzheimer’s and with reduced melatonin synthesis [178] . The pineal gland also bioaccumulates aluminum [25] , and aluminum behaves as an analog to calcium in inducing eNOS to detach from the membrane and switch from sulfate synthesis to nitric oxide synthesis [187] . This would have catastrophic effects on the ability of the pineal gland to produce sulfate, and would therefore interfere with the synthesis of melatonin sulfate at night. Thus, both aluminum and calcium would be expected to negatively impact the pineal gland’s ability to maintain supplies of melatonin and sulfate to the CSF. We hypothesize that this is a key component of most neurological diseases, and that it links them to the associated sleep disorders.Future research is needed to specifically investigate pineal gland pathologies in association with aluminum, melatonin, and sulfate.
10. Conclusion
In this paper, we have developed the argument that glyphosate, the active ingredient in the herbicide, Roundup, and aluminum, a pervasive toxic metal in our environment, operate synergistically to induce dysfunction in the pineal gland leading to the sleep disorder that is characteristic of multiple neurological diseases, including autism, ADHD, depression, Alzheimer’s disease, ALS, anxiety disorder and Parkinson’s disease. We further argue that impaired supply of melatonin and sulfate to the brain as a consequence of pineal damage can explain how the disrupted sleep can lead to more general neurological damage, and we propose that this is a significant component of the disease process. The steady increase in glyphosate usage on corn and soy crops aligns remarkably well with the increase in sleep disorder and in autism, as well as other neurological diseases. We have shown how disruption of CYP enzymes and promotion of anemia and hypoxia, due to both aluminum and glyphosate, and disruption of gut bacteria by glyphosate, can cause a pathology leading to deficiencies in both melatonin and sulfate in the cerebrospinal fluid that is characteristic of autism and Alzheimer’s disease. Insufficient sulfate leads to impaired lysosomal recycling of cellular debris, and insufficient melatonin leads to sleep disorder, vascular disease and impaired protection from ROS damage in the brain.
Disclosure/Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
This research was funded in part by Quanta Computer, Inc., Taiwan, under the auspices of the Qmulus project. This paper benefitted from discussions with Anthony Samsel, James Beecham, Eric Waymann, and Gerald Koenig.
References
- Taira, M., Takase, M. and Sasaki, H. (1998) Sleep Disorder in Children with Autism. Psychiatry and Clinical Neurosciences, 52, 182-183. http://dx.doi.org/10.1111/j.1440-1819.1998.tb01018.x
- Bartholemew, K. and Owens, J. (2006) Sleep and ADHD: A Review. Medicine and Health Rhode Island, 89, 91-93.
- Corkum, P.H., Tannock, R. and Moldofsy, H. (1998) Sleep Disturbances in Children with Attention Deficit/Hyperactivity Disorder. Journal of the American Academy of Child and Adolescent Psychiatry, 37, 637-646. http://dx.doi.org/10.1097/00004583-199806000-00014
- Gruber, R. and Sadeh, A. (2004) Sleep and Neurobehavioral Functioning in Boys with Attention-Deficit/Hyperactivity Disorder and No Reported Breathing Problems. Sleep, 27, 267-273.
- Song, C., Lin, A., Bonaccorso, S., Heide, C., Verkerk, R., Kenis, G., et al. (1998) The Inflammatory Response System and the Availability of Plasma Tryptophan in Patients with Primary Sleep Disorders and Major Depression. Journal of Affective Disorders, 49, 211-219. http://dx.doi.org/10.1016/S0165-0327(98)00025-1
- Comella, C.L. (2008) Sleep Disorders in Parkinson’s Disease. Current Treatment Options in Neurology, 10, 215-221. http://dx.doi.org/10.1007/s11940-008-0023-z
- Gjerstad, M.D., Wentzel-Larsen, T., Aarsland, D. and Larsen, J.P. (2007) Insomnia in Parkinson’s Disease: Frequency and Progression over Time. Journal of Neurology, Neurosurgery, and Psychiatry, 78, 476-479.
- Tandberg, E., Larsen, J.P. and Karlsen, K. (1998) A Community-Based Study of Sleep Disorders in Patients with Parkinson’s Disease. Movement Disorders, 13, 895-899. http://dx.doi.org/10.1002/mds.870130606
- Cohrs, S. (2008) Sleep Disturbances in Patients with Schizophrenia: Impact and Effect of Antipsychotics. CNS Drugs, 22, 939-962. http://dx.doi.org/10.2165/00023210-200822110-00004
- Benca, R.M., Obermeyer, W.H., Thisted, R.A. and Gillin, J.C. (1992) Sleep and Psychiatric Disorders: A Meta-Analysis. Archives of General Psychiatry, 49, 651-668. http://dx.doi.org/10.1001/archpsyc.1992.01820080059010
- Tandon, R., Shipley, J.E., Taylor, S., Greden, J.F., Eiser, A., DeQuardo, J. and Goodson, J. (1992) Electroencephalographic Sleep Abnormalities in Schizophrenia: Relationship to Positive/Negative Symptoms and Prior Neuroleptic Treatment. Archives of General Psychiatry, 49, 185-194. http://dx.doi.org/10.1001/archpsyc.1992.01820030017003
- Ohayon, M.M. and Roth, T. (2003) Place of Chronic Insomnia in the Course of Depressive and Anxiety Disorders. Journal of Psychiatric Research, 37, 9-15. http://dx.doi.org/10.1016/S0022-3956(02)00052-3
- Coco, D.L., Cannizzaro, E., Spataro, R., Taiellod, A.C. and La Bella, V. (2012) Sleep-Wake Problems in Patients with Amyotrophic Lateral Sclerosis: Implications for Patient Management. Neurodegenerative Disease Management, 2, 315-324. http://dx.doi.org/10.2217/nmt.12.28
- Bliwise, D.L. (2004) Sleep Disorders in Alzheimer’s Disease and Other Dementias. Sleep Disorders, 6, 16-28.
- Radecki, S.E. and Brunton, S.A. (1993) Management of Insomnia in Office-Based Practice. National Prevalence and Therapeutic Patterns. Archives of Family Medicine, 2, 1129-1134. http://dx.doi.org/10.1001/archfami.2.11.1129
- Li, R.H., Wing, Y.K., Ho, S.C. and Fong, S.Y. (2002) Gender Differences in Insomnia―A Study in the Hong Kong Chinese Population. Journal of Psychosomatic Research, 53, 601-609. http://dx.doi.org/10.1016/S0022-3999(02)00437-3
- CDC (2014) Autism Spectrum Disorder. http://www.cdc.gov/ncbddd/autism/data.html
- Dave, D.M. and Fernandez, J.M. (2015) Rising Autism Prevalence: Real or Displacing Other Mental Disorders? Evidence from Demand for Auxiliary Healthcare Workers in California. Economic Inquiry, 53, 448-468. http://dx.doi.org/10.1111/ecin.12137
- Nevison, C.D. (2014) A Comparison of Temporal Trends in United States Autism Prevalence to Trends in Suspected Environmental Factors. Environmental Health, 13, 73. http://dx.doi.org/10.1186/1476-069X-13-73
- Macchi, M.M. and Bruce, J.N. (2004) Human Pineal Physiology and Functional Significance of Melatonin. Frontiers in Neuroendocrinology, 25, 177-195. http://dx.doi.org/10.1016/j.yfrne.2004.08.001
- Arendt, J., Middleton, B., Stone, B. and Skene, D. (1999) Complex Effects of Melatonin: Evidence for Photoperiodic Responses in Humans? Sleep, 22, 625-635.
- Goldman, H. and Wurtman, R.J. (1964) Flow of Blood to the Pineal Body of the Rat. Nature, 203, 87-88. http://dx.doi.org/10.1038/203087a0
- Falnoga, I., Tusek-Znidric, M., Horvat, M. and Stegnar, P. (2000) Mercury, Selenium and Cadmium in Human Autopsy Samples from Idrija Residents and Mercury Mine Workers. Environmental Research, 84, 211-218. http://dx.doi.org/10.1006/enrs.2000.4116
- Luke, J. (2001) Fluoride Deposition in the Aged Human Pineal Gland. Caries Research, 35, 125-128. http://dx.doi.org/10.1159/000047443
- Lang, S.B., Marino, A.A., Berkovic, G., Fowler, M. and Abreo, K.D. (1996) Piezoelectricity in the Human Pineal Gland. Bioelectrochemistry and Bioenergetics, 41, 191-195. http://dx.doi.org/10.1016/S0302-4598(96)05147-1
- Shaw, C.A., Kette, S.D., Davidson, R.M. and Seneff, S. (2013) Aluminum’s Role in CNS-Immune System Interactions Leading to Neurological Disorders. Immunome Research, 9, 069.
- Strong, M.J. (2001) Chapter 9―Aluminum as an Experimental Neurotoxicant: The Neuropathology and Neurochemistry. In: Exley, C., Ed., Aluminuium and Alzheimer’s Disease: The Science that Describes the Link, Elsevier Science, Amsterdam. http://dx.doi.org/10.1016/B978-044450811-9/50034-3
- Yokel, R.A. and McNamara, P.J. (2001) Aluminium Toxicokinetics: An Updated Minireview. Pharmacology & Toxicology, 88, 159-167.
- Alfrey, A.C., LeGendre, G.R. and Kaehny, W.D. (1976) The Dialysis Encephalopathy Syndrome Possible Aluminum Intoxication. The New England Journal of Medicine, 294, 184-188. http://dx.doi.org/10.1056/NEJM197601222940402
- Bia, M.J., Cooper, K., Schnall, S., Duffy, T., Hendler, E., Malluche, H. and Solomon, L. (1989) Aluminum Induced Anemia: Pathogenesis and Treatment in Patients on Chronic Hemodialysis. Kidney International, 36, 852-858. http://dx.doi.org/10.1038/ki.1989.271
- Dunea, G., Mahurkar, S.D., Mamdani, B. and Smith, E.C. (1978) Role of Aluminum in Dialysis Dementia. Annals of Internal Medicine, 88, 502-504. http://dx.doi.org/10.7326/0003-4819-88-4-502
- Sideman, S. and Manor, D. (1982) The Dialysis Dementia Syndrome and Aluminum Intoxication. Nephron, 31, 1-10. http://dx.doi.org/10.1159/000182595
- Heaf, J.G., Pdenphant, J. and Andersen, J.R. (1986) Bone Aluminum Deposition in Maintenance Dialysis Patients Treated with Aluminium-Free Dialysate: Role of Aluminium Hydroxide Consumption. Nephron, 42, 210-216. http://dx.doi.org/10.1159/000183669
- Sedman, A.B., Klein, G.L., Merritt, R.J., Miller, N.L., Weber, K.O., Gill, W.L., et al. (1985) Evidence of Aluminum Loading in Infants Receiving Intravenous Therapy. The New England Journal of Medicine, 312, 1337-1343. http://dx.doi.org/10.1056/NEJM198505233122101
- Bishop, N.J., Morley, R., Day, J.P. and Lucas, A. (1997) Aluminum Neurotoxicity in Preterm Infants Receiving Intravenous-Feeding Solutions. The New England Journal of Medicine, 336, 1557-1561. http://dx.doi.org/10.1056/NEJM199705293362203
- Si?czuk-Walczak, H., Szymczak, M., Raźniewska, G., Matczak, W. and Szymczak, W. (2003) Effects of Occupational Exposure to Aluminum on Nervous System: Clinical and Electroencephalographic Findings. International Journal of Occupational Medicine and Environmental Health, 16, 301-310.
- Séralini, G.E., Clair, E., Mesnage, R., Gress, S., Defarge, N., Malatesta, M., et al. (2014) Republished Study: Long- Term Toxicity of a Roundup Herbicide and a Roundup-Tolerant Genetically Modified Maize. Environmental Sciences Europe, 26, 14. http://dx.doi.org/10.1186/s12302-014-0014-5
- Mesnage, R., Bernay, B. and Séralini, G.E. (2013) Ethoxylated Adjuvants of Glyphosate-Based Herbicides Are Active Principles of Human Cell Toxicity. Toxicology, 313, 122-128. http://dx.doi.org/10.1016/j.tox.2012.09.006
- Mesnage, R., Defarge, N., de Vendômois, J.S. and Séralini, G.E. (2014) Major Pesticides Are More Toxic to Human Cells Than Their Declared Active Principles. BioMed Research International, 2014, Article ID: 179691.
- USDA (2013) NASS Agricultural Chemical Usage-Field Crops and Potatoes. Agricultural Chemical Usage-Field Crops USDA Economics, Statistics and Market Information System. Albert R. Mann Library, Cornell University. http://usda.mannlib.cornell.edu/MannUsda/viewDocumentInfo.do?documentID=1560
- Swanson, N., Leu, A., Abrahamson, J. and Wallet, B. (2014) Genetically Engineered Crops, Glyphosate and the Deterioration of Health in the United States of America. Journal of Organic Systems, 9, 6-37.
- Dennison, C. and Pokras, R. (2000) Design and Operation of the National Hospital Discharge Survey: 1988 Redesign. Vital and Health Statistics. http://www.cdc.gov/nchs/data/series/sr01/sr01039.pdf
- CDC (2013) Population Estimates. http://wonder.cdc.gov/wonder/help/cmf.html#Population%20Inform
- CDC (2013) Death Data. Centers for Disease Control and Prevention, National Center for Health Statistics. Compressed Mortality Files on CDC WONDER Online Database. 1999-2010 Data Are Compiled from Compressed Mortality File 1999-2010 Series 20, No. 2P, Released January 2013. 1989-1998 Data Are Compiled from Compressed Mortality File CMF 1989-1998, Series 20, No. 2E, 2003. http://wonder.cdc.gov/mortSQL.html
- de Mara, N., Becerril, J.M., Garca-Plazaola, J.I., Hernandez, A., De Felipe, M.R. and Fernandez-Pascual, M. (2006) New Insights on Glyphosate Mode of Action in Nodular Metabolism: Role of Shikimate Accumulation. Journal of Agricultural and Food Chemistry, 54, 2621-2628. http://dx.doi.org/10.1021/jf058166c
- Samsel, A. and Seneff, S. (2013) Glyphosate’s Suppression of Cytochrome P450 Enzymes and Amino Acid Biosynthesis by the Gut Microbiome: Pathways to Modern Diseases. Entropy, 15, 1416-1463. http://dx.doi.org/10.3390/e15041416
- Fekkes, D., van der Cammen, T.J., van Loon, C.P., Verschoor, C., van Harskamp, F., de Koning, I., et al. (1998) Abnormal Amino Acid Metabolism in Patients with Early Stage Alzheimer Dementia. Journal of Neural Transmission, 105, 287-294. http://dx.doi.org/10.1007/s007020050058
- Naushad, S.M., Jain, J.M., Prasad, C.K., Naik, U. and Akella, R.R. (2013) Autistic Children Exhibit Distinct Plasma Amino acid Profile. Indian Journal of Biochemistry and Biophysics, 50, 474-478.
- Contardo-Jara, V., Klingelmann, E. and Wiegand, C. (2009) Bioaccumulation of Glyphosate and Its Formulation Roundup Ultra in Lumbriculus variegatus and Its Effects on Biotransformation and Antioxidant Enzymes. Environmental Pollution, 157, 57-63. http://dx.doi.org/10.1016/j.envpol.2008.07.027
- Krüger, M., Schledorn, P., Schrödl, W., Hoppe, H.W., Lutz, W. and Shehata, A.A. (2014) Detection of Glyphosate Residues in Animals and Humans. Journal of Analytical Toxicology, 4, 2.
- Krüger, M., Schrödl, W., Pedersen, I. and Shehata, A.A. (2014) Detection of Glyphosate in Malformed Piglets. Journal of Environmental & Analytical Toxicology, 4, 5.
- Cryan, J.F. and O’Mahony, S.M. (2011) The Microbiome-Gut-Brain Axis: From Bowel to Behavior. Neurogastroenterology & Motility, 23, 187-192. http://dx.doi.org/10.1111/j.1365-2982.2010.01664.x
- Hsiao, E.Y., McBride, S.W., Hsien, S., Sharon, G., Hyde, E.R., McCue, T., et al. (2013) Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell, 155, 1451-1463. http://dx.doi.org/10.1016/j.cell.2013.11.024
- Lafaye, A., Junot, C., Ramounet-Le Gall, B., Fritsch, P., Ezan, E. and Tabet, J.C. (2004) Profiling of Sulfoconjugates in Urine by Using Precursor Ion and Neutral Loss Scans in Tandem Mass Spectrometry. Application to the Investigation of Heavy Metal Toxicity in Rats. Journal of Mass Spectrometry, 39, 655-664. http://dx.doi.org/10.1002/jms.635
- Altieri, L., Neri, C., Sacco, R., Curatolo, P., Benvenuto, A., Muratori, F., et al. (2011) Urinary p-Cresol Is Elevated in Small Children with Severe Autism Spectrum disorder. Biomarkers, 16, 252-260. http://dx.doi.org/10.3109/1354750X.2010.548010
- Persico, A.M. and Napolioni, V. (2013) Urinary p-Cresol in Autism Spectrum Disorder. Neurotoxicology and Teratology, 36, 82-90. http://dx.doi.org/10.1016/j.ntt.2012.09.002
- Johnson, S., Hollis, C., Kochhar, P., Hennessy, E., Wolke, D. and Marlow, N. (2010) Autism Spectrum Disorders in Extremely Preterm Children. The Journal of Pediatrics, 156, 525-531. http://dx.doi.org/10.1016/j.jpeds.2009.10.041
- Limperopoulos, C., Bassan, H., Sullivan, N.R., Soul, J.S., Robertson Jr., R.L., Moore, M., et al. (2008) Positive Screening for Autism in Ex-Preterm Infants: Prevalence and Risk Factors. Pediatrics, 121, 758-765. http://dx.doi.org/10.1542/peds.2007-2158
- Yang, L., Zheng, J., Xu, R., Zhang, Y., Gu, L., Dong, J., et al. (2014) Melatonin Suppresses Hypoxia-Induced Migration of HUVECs via Inhibition of ERK/Rac1 Activation. International Journal of Molecular Sciences, 15, 14102- 14121. http://dx.doi.org/10.3390/ijms150814102
- Kaur, C., Sivakumar, V., Lu, J. and Ling, E.A. (2007) Increased Vascular Permeability and Nitric Oxide Production in Response to Hypoxia in the Pineal Gland. Journal of Pineal Research, 42, 338-349. http://dx.doi.org/10.1111/j.1600-079X.2007.00424.x
- Clark, S.F. (2008) Iron Deficiency Anemia. Nutrition in Clinical Practice, 23, 128-141. http://dx.doi.org/10.1177/0884533608314536
- Oh, R. and Brown, D.L. (2003) Vitamin B12 Deficiency. American Family Physician, 67, 979-986.
- Latif, A., Heinz, P. and Cook, R. (2002) Iron Deficiency in Autism and Asperger Syndrome. Autism, 6, 103-114. http://dx.doi.org/10.1177/1362361302006001008
- Krüger, M., Schrödl, W., Neuhaus, J. and Shehata, A.A. (2013) Field Investigations of Glyphosate in Urine of Danish Dairy Cows. Journal of Environmental & Analytical Toxicology, 3.
- Pineles, S.L., Avery, R.A. and Liu, G.T. (2010) Vitamin B12 Optic Neuropathy in Autism. Pediatrics, 126, 967-970. http://dx.doi.org/10.1542/peds.2009-2975
- Kaiser, L. and Schwartz, K.A. (1985) Aluminum-Induced Anemia. American Journal of Kidney Diseases, 6, 348-352. http://dx.doi.org/10.1016/S0272-6386(85)80092-5
- Lee, J.Y., Hwang, K.Y., Nam, I.S., Yim, H.J., Kim, E.N., Jung, J.H., et al. (2001) Clinical Observation of Acute Glyphosate Intoxication. The Korean Journal of Medicine, 60, 383-387. (In Korean)
- Ozturk, L., Yazici, A., Eker, S., Gokmen, O., Rmheld, V. and Cakmak, I. (2008) Glyphosate Inhibition of Ferric Reductase Activity in Iron Deficient Sunflower Roots. New Phytologist, 177, 899-906. http://dx.doi.org/10.1111/j.1469-8137.2007.02340.x
- Huynh, Q.K., Kishore, G.M. and Bild, G.S. (1988) 5-Enolpyruvyl Shikimate 3-Phosphate Synthase from Escherichia coli. Journal of Biological Chemistry, 263, 735-739.
- Cebeci, O. and Budak, H. (2009) Global Expression Patterns of Three Festuca Species Exposed to Different Doses of Glyphosate Using the Affymetrix GeneChip Wheat Genome Array. Comparative and Functional Genomics, 2009, Article ID: 505701. http://dx.doi.org/10.1155/2009/505701
- Cole, D.J. (1985) Mode of Action of Glyphosate―A Literature Analysis. In: Grossbard, E. and Atkinson, D., Eds., The Herbicide Glyphosate, Butterworths, London, 48-74.
- Kearney, P.C. and Kaufman, D.D. (1988) Herbicides Chemistry: Degradation and Mode of Action. CRC Press, Boca Raton.
- Zaidi, A., Khan, M.S. and Rizvi, P.Q. (2005) Effect of Herbicides on Growth, Nodulation and Nitrogen Content of Greengram. Agronomy for Sustainable Development, 25, 497-504. http://dx.doi.org/10.1051/agro:2005050
- Purgel, M., Takács, Z., Jonsson, C.M., Nagy, L., Andersson, I., Bányai, I., et al. (2009) Glyphosate Complexation to Aluminium(III). An Equilibrium and Structural Study in Solution Using Potentiometry, Multinuclear NMR, ATR-FTIR, ESI-MS and DFT Calculations. Journal of Inorganic Biochemistry, 103, 1426-1438. http://dx.doi.org/10.1016/j.jinorgbio.2009.06.011
- Coburn, J.W., Mischel, M.G., Goodman, W.G. and Salusky, I.B. (1991) Calcium Citrate Markedly Enhances Aluminum Absorption from Aluminum Hydroxide. American Journal of Kidney Diseases, 17, 708-711. http://dx.doi.org/10.1016/S0272-6386(12)80356-8
- Slanina, P., Frech, W., Ekström, L.G., Lööf, L., Slorach, S. and Cedergren, A. (1986) Dietary Citric Acid Enhances Absorption of Aluminum in Antacids. Clinical Chemistry, 32, 539-541.
- Froment, D.P., Molitoris, B.A., Buddington, B., Miller, N. and Alfrey, A.C. (1989) Site and Mechanism of Enhanced Gastrointestinal Absorption of Aluminum by Citrate. Kidney International, 36, 978-984. http://dx.doi.org/10.1038/ki.1989.290
- Cakmak, I., Yazici, A., Tutus, Y. and Ozturk, L. (2009) Glyphosate Reduced Seed and Leaf Concentrations of Calcium, Manganese, Magnesium, and Iron in Non-Glyphosate Resistant Soybean. European Journal of Agronomy, 31, 114-119. http://dx.doi.org/10.1016/j.eja.2009.07.001
- Carman, J.A., Vlieger, H.R., Ver Steeg, L.J., Sneller, V.E., Robinson, G.W., Clinch-Jones, C.A., et al. (2013) A Long- Term Toxicology Study on Pigs Fed a Combined Genetically Modified (GM) Soy and GM Maize Diet. Journal of Organic Systems, 8, 38-54.
- Shehata, A.A, Schrödl, W., Aldin, A.A., Hafez, H.M. and Krüger, M. (2013) The Effect of Glyphosate on Potential Pathogens and Beneficial Members of Poultry Microbiota in Vitro. Current Microbiology, 66, 350-358. http://dx.doi.org/10.1007/s00284-012-0277-2
- Abreo, K., Sella, M., Gautreaux, S., De Smet, R., Vogeleere, P., Ringoir, S., et al. (1997) P-Cresol, a Uremic Compound, Enhances the Uptake of Aluminum in Hepatocytes. Journal of the American Society of Nephrology, 8, 935-942.
- Kaur, C., Sivakumar, V. and Ling, E.A. (2007) Expression of Tranferrin Receptors in the Pineal Gland of Postnatal and Adult Rats and Its Alteration in Hypoxia and Melatonin Treatment. Glia, 55, 263-273.
- de la Monte, S.M., Neely, T.R. and Wands Jr., C.J. (2000) Oxidative Stress and Hypoxia-Like Injury Cause Alzheimer- Type Molecular Abnormalities in Central Nervous System Neurons. Cellular and Molecular Life Sciences CMLS, 57, 1471-1481. http://dx.doi.org/10.1007/PL00000630
- Fewtrell, M.S., Bishop, N.J., Edmonds, C.J., Isaacs, E.B. and Lucas, A. (2009) Aluminum Exposure from Parenteral Nutrition in Preterm Infants: Bone Health at 15-Year Follow-Up. Pediatrics, 124, 1372-1379. http://dx.doi.org/10.1542/peds.2009-0783
- Jeffery, E.H., Jansen, H.T. and Dellinger, J.A. (1987) In Vivo Interactions of Aluminum with Hepatic Cytochrome P-450 and Metallothionein. Fundamental and Applied Toxicology, 8, 541-548. http://dx.doi.org/10.1016/0272-0590(87)90139-4
- Bidlack, W.R., Brown, R.C., Meskin, M.S., Lee, T.C. and Klein, G.L. (1987) Effect of Aluminum on the Hepatic Mixed Function Oxidase and Drug Metabolism. Nutrient-Drug Interactions, 5, 33-42.
- Lamb, D.C., Kelly, D.E., Hanley, S.Z., Mehmood, Z. and Kelly, S.L. (1998) Glyphosate Is an Inhibitor of Plant Cytochrome P450: Functional Expression of Thlaspi arvensae Cytochrome P45071B1/Reductase Fusion Protein in Escherichia coli. Biochemical and Biophysical Research Communications, 244, 110-114. http://dx.doi.org/10.1006/bbrc.1997.7988
- Hietanen, E., Linnainmaa, K. and Vainio, H. (1983) Effects of Phenoxyherbicides and Glyphosate on the Hepatic and Intestinal Biotransformation Activities in the Rat. Acta Pharmacologica et Toxicologica, 53, 103-112. http://dx.doi.org/10.1111/j.1600-0773.1983.tb01876.x
- Abass, K., Turpeinen, M. and Pelkonen, O. (2009) An Evaluation of the Cytochrome P450 Inhibition Potential of Selected Pesticides in Human Hepatic Microsomes. Journal of Environmental Science and Health, Part B: Pesticides, Food Contaminants, and Agricultural Wastes, 44, 553-563. http://dx.doi.org/10.1080/03601230902997766
- Poordad, F.F. (2007) The Burden of Hepatic Encephalopathy. Alimentary Pharmacology & Therapeutics, 25, 3-9. http://dx.doi.org/10.1111/j.1746-6342.2006.03215.x
- Ma, X., Idle, J.R., Krausz, K.W. and Gonzalez, F.J. (2005) Metabolism of Melatonin by Human Cytochromes P450. Drug Metabolism and Disposition, 33, 489-494. http://dx.doi.org/10.1124/dmd.104.002410
- Lahiri, D.K., Ge, Y.W., Sharman, E.H. and Bondy, S.C. (2004) Age-Related Changes in Serum Melatonin in Mice: Higher Levels of Combined Melatonin and 6-Hydroxymelatonin Sulfate in the Cerebral Cortex Than Serum, Heart, Liver and Kidney Tissues. Journal of Pineal Research, 36, 217-223. http://dx.doi.org/10.1111/j.1600-079X.2004.00120.x
- Adams, J.B., Johansen, L.J., Powell, L.D., Quig, D. and Rubin, R.A. (2011) Gastrointestinal Flora and Gastrointestinal Status in Children with Autism―Comparisons to Typical Children and Correlation with Autism Severity. BMC Gastroenterology, 11. http://dx.doi.org/10.1186/1471-230X-11-22
- Gorsky, J.E., Dietz, A.A., Spencer, H. and Os, D. (1979) Metabolic Balance of Aluminum Studied in Six Men. Clinical Chemistry, 25, 1739-1743.
- Afzal, N., Murch, S., Thirrupathy, K., Berger, L., Fagbemi, A. and Heuschkel, R. (2003) Constipation with Acquired Megarectum in Children with Autism. Pediatrics, 112, 939-942. http://dx.doi.org/10.1542/peds.112.4.939
- Foxx-Orenstein, A.E., Kuemmerle, J.F. and Grider, J.R. (1996) Distinct 5-HT Receptors Mediate the Peristaltic Reflex Induced by Mucosal Stimuli in Human and Guinea Pig Intestine. Gastroenterology, 111, 1281-1290. http://dx.doi.org/10.1053/gast.1996.v111.pm8898642
- Kern, J.K., Grannemann, B.D., Trivedi, M.H., Waring, R.H., Ramsden, D.B. and Garver, C.R. (2004) Abnormal Sulfation Chemistry in Autism. In: Ryaskin, O.T., Ed., Trends in Autism Research, Chapter XI, Nova Publishers, Hauppauge.
- James, S.J., Cutler, P., Melnyk, S., Jernigan, S., Janak, L., Gaylor, D.W., et al. (2004) Metabolic Biomarkers of Increased Oxidative Stress and Impaired Methylation Capacity in Children with Autism. The American Journal of Clinical Nutrition, 80, 1611-1617.
- Lee, B.R., Huseby, S., Koprivova, A., Chételat, A., Wirtz, M., Mugford, S.T., et al. (2012) Effects of fou8/fry1 Mutation on Sulfur Metabolism: Is Decreased Internal Sulfate the Trigger of Sulfate Starvation Response? PLOS ONE, 7, e39425. http://dx.doi.org/10.1371/journal.pone.0039425
- Finegold, S.M. (2011) Desulfovibrio Species Are Potentially Important in Regressive Autism. Medical Hypotheses, 77, 270-274. http://dx.doi.org/10.1016/j.mehy.2011.04.032
- Tack, G.J., Verbeek, W.H.M., Schreurs, M.W.J. and Mulder, C.J.J. (2010) The Spectrum of Celiac Disease: Epidemiology, Clinical Aspects and Treatment. Nature Reviews Gastroenterology and Hepatology, 7, 204-213. http://dx.doi.org/10.1038/nrgastro.2010.23
- Samsel, A. and Seneff, S. (2013) Glyphosate, Pathways to Modern Diseases II: Celiac Sprue and Gluten Intolerance. Interdisciplinary Toxicology, 6, 159-184. http://dx.doi.org/10.2478/intox-2013-0026
- Smith, A. and Macfarlane, G.T. (1997) Formation of Phenolic and Indolic Compounds by Anaerobic Bacteria in the Human Large Intestine. Microbial Ecology, 33, 180-188. http://dx.doi.org/10.1007/s002489900020
- Mathus, T.L., Townsend, D.E. and Miller, K.W. (1995) Anaerobic Biogenesis of Phenol and p-Cresol from L-Tyrosine. Fuel, 74, 1505-1508. http://dx.doi.org/10.1016/0016-2361(95)00109-I
- Holick, M.F. (2005) The Vitamin D Epidemic and Its Health Consequences. Journal of Nutrition, 135, 2739-2748.
- Grant, W.B. and Soles, C.M. (2009) Epidemiologic Evidence Supporting the Role of Maternal Vitamin D Deficiency as a Risk Factor for the Development of Infantile Autism. Dermato-Endocrinology, 1, 223-228. http://dx.doi.org/10.4161/derm.1.4.9500
- Wikvall, K. (2001) Cytochrome P450 Enzymes in the Bioactivation of Vitamin D to Its Hormonal Form. International Journal of Molecular Medicine, 7, 201-209.
- Bolt, M.J.G., Liu, W., Qiao, G., Kong, J., Zheng, W., Krausz, T., et al. (2004) Critical Role of Vitamin D in Sulfate Homeostasis: Regulation of the Sodium-Sulfate Cotransporter by 1,25-Dihydroxyvitamin D3. American Journal of Physiology―Endocrinology and Metabolism, 287, E744-E749. http://dx.doi.org/10.1152/ajpendo.00151.2004
- Fernandes, I., Hampson, G., Cahours, X., Morin, P., Coureau, C., Couette, S., et al. (1997) Abnormal Sulfate Metabolism in Vitamin D-Deficient Rats. Journal of Clinical Investigation, 100, 2196-2203. http://dx.doi.org/10.1172/JCI119756
- Murch, S.H., MacDonald, T.T., Walker-Smith, J.A., Lionetti, P., Levin, M. and Klein, N.J. (1993) Disruption of Sulphated Glycosaminoglycans in Intestinal Inflammation. The Lancet, 341, 711-714. http://dx.doi.org/10.1016/0140-6736(93)90485-Y
- Liu, W., Chen, Y., Golan, M.A., Annunziata, M.L., Du, J., Dougherty, U., et al. (2013) Intestinal Epithelial Vitamin D Receptor Signaling Inhibits Experimental Colitis. Journal of Clinical Investigation, 123, 3983-3996. http://dx.doi.org/10.1172/JCI65842
- Yuk, J.M., Shin, D.M., Lee, H.M., Yang, C.S., Jin, H.S., Kim, K.K., et al. (2009) Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host and Microbe, 6, 231-243. http://dx.doi.org/10.1016/j.chom.2009.08.004
- Colston, K.W., Mackay, A.G., Finlayson, C., Wu, J.C. and Maxwell, J.C. (1994) Localisation of Vitamin D Receptor in Normal Human Duodenum and in Patients with Coeliac Disease. Gut, 35, 1219-1225. http://dx.doi.org/10.1136/gut.35.9.1219
- Costedio, M.M., Hyman, N. and Mawe, G.M. (2007) Serotonin and Its Role in Colonic Function and in Gastrointestinal Disorders. Diseases of the Colon & Rectum, 50, 376-388. http://dx.doi.org/10.1007/s10350-006-0763-3
- Wade, P.R., Chen, J., Jaffe, B., Kassem, I.S., Blakely, R.D. and Gershon, M.D. (1996) Localization and Function of a 5-HT Transporter in Crypt Epithelia of the Gastrointestinal Tract. The Journal of Neuroscience, 16, 2352-2364.
- Hagbom, M., Istrate, C., Engblom, D., Karlsson, T., Rodriguez-Diaz, J., Buesa, J., et al. (2011) Rotavirus Stimulates Release of Serotonin (5-HT) from Human Enterochromaffin Cells and Activates Brain Structures Involved in Nausea and Vomiting. PLOS Pathogens, 7, e1002115. http://dx.doi.org/10.1371/journal.ppat.1002115
- Lundgren, O. and Svensson, L. (2001) Pathogenesis of Rotavirus diarrhea. Microbes and Infection, 3, 1145-1156. http://dx.doi.org/10.1016/S1286-4579(01)01475-7
- Suominen, T., Uutela, P., Ketola, R.A., Bergquist, J., Hillered, L., Finel, M., et al. (2013) Determination of Serotonin and Dopamine Metabolites in Human Brain Microdialysis and Cerebrospinal Fluid Samples by UPLC-MS/MS: Discovery of Intact Glucuronide and Sulfate Conjugates. PLOS ONE, 8, e68007. http://dx.doi.org/10.1371/journal.pone.0068007
- Nau Jr., F., Yu, B., Martin, D. and Nichols, C.D. (2013) Serotonin 5-HT2A Receptor Activation Blocks TNF-α Mediated Inflammation in Vivo. PLOS ONE, 8, e75426. http://dx.doi.org/10.1371/journal.pone.0075426
- Westphal, V., Murch, S., Kim, S., Srikrishna, G., Winchester, B., Day, R. and Freeze, H.H. (2000) Reduced Heparan Sulfate Accumulation in Enterocytes Contributes to Protein-Losing Enteropathy in a Congenital Disorder of Glycosylation. American Journal of Pathology, 157, 1917-1925. http://dx.doi.org/10.1016/S0002-9440(10)64830-4
- Day, R., Ilyas, M., Daszak, P., Talbot, I. and Forbes, A. (1999) Expression of Syndecan-1 in Inflammatory Bowel Disease and a Possible Mechanism of Heparin Therapy. Digestive Diseases and Sciences, 44, 2508-2515. http://dx.doi.org/10.1023/A:1026647308089
- Tanabe, H., Yokota, K. and Kohgo, Y. (1999) Localization of Syndecan-1 in Human Gastric Mucosa Associated with Ulceration. The Journal of Pathology, 187, 338-344.
- Murch, S.H., Phillips, D., Walker-Smith, J.A., Winyard, P.J.D., Meadows, N., Koletzko, S., et al. (1997) Congenital Enterocyte Heparan Sulphate Deficiency with Massive Albumin Loss, Secretory Diarrhoea, and Malnutrition. The Lancet, 347, 1299-1301. http://dx.doi.org/10.1016/S0140-6736(96)90941-1
- Ciccocioppo, R., Di Sabatino, A., Parroni, R., Muzi, P., D’Alò, S., Ventura, T., et al. (2001) Increased Enterocyte Apoptosis and Fas-Fas Ligand System in Celiac Disease. American Journal of Clinical Pathology, 115, 494-503. http://dx.doi.org/10.1309/UV54-BHP3-A66B-0QUD
- Adriaanse, M.P., Tack, G.J., Passos, V.L., Damoiseaux, J.G., Schreurs, M.W., van Wijck, K., et al. (2013) Serum I-FABP as Marker for Enterocyte Damage in Coeliac Disease and Its Relation to Villous Atrophy and Circulating Autoantibodies. Alimentary Pharmacology & Therapeutics, 37, 482-490. http://dx.doi.org/10.1111/apt.12194
- Bracken, S., Byrne, G., Kelly, J., Jackson, J. and Feighery, C. (2008) Altered Gene Expression in Highly Purified Enterocytes from Patients with Active Coeliac Disease. BMC Genomics, 9. http://dx.doi.org/10.1186/1471-2164-9-377
- Higaki, M., Takahashi, M., Suzuki, T. and Sahashi, Y. (1965) Biological Activities in Animals of Vitamin D III. Biogenesis of Vitamin D Sulfate in Animal Tissues. The Journal of Vitaminology, 11, 261-265. http://dx.doi.org/10.5925/jnsv1954.11.261
- Higaki, M., Takahashi, M., Suzuki, T. and Sahash, Y. (1965) Metabolic Activities of Vitamin D in Animals. IV. Distribution of Vitamin D Sulfokinase in Animal Tissues and Its Isolation. The Journal of Vitaminology, 11, 266-270. http://dx.doi.org/10.5925/jnsv1954.11.266
- Han, X.M., Holtzman, D., McKeel Jr., D.W., Kelley, J. and Morris, J.C. (2002) Substantial Sulfatide Deficiency and Ceramide Elevation in Very Early Alzheimer’s Disease: Potential Role in Disease Pathogenesis. Journal of Neurochemistry, 82, 809-818. http://dx.doi.org/10.1046/j.1471-4159.2002.00997.x
- Irie, F., Badie-Mahdavi, H. and Yamaguchi, Y. (2012) Autism-Like Socio-Communicative Deficits and Stereotypies in Mice Lacking Heparan Sulfate. Proceedings of the National Academy of Sciences of the United States of America, 109, 5052-5056. http://dx.doi.org/10.1073/pnas.1117881109
- Pearson, B.L., Corley, M.J., Vasconcellos, A., Blanchard, D.C. and Blanchard, R.J. (2013) Heparan Sulfate Deficiency in Autistic Postmortem Brain Tissue from the Subventricular Zone of the Lateral Ventricles. Behavioural Brain Research, 243, 138-145. http://dx.doi.org/10.1016/j.bbr.2012.12.062
- Bishop, J.R., Schuksz, M. and Esko, J.D. (2007) Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature, 446, 1030-1037. http://dx.doi.org/10.1038/nature05817
- Bennett-Clarke, C.A., Leslie, M.J., Lane, R.D. and Rhoades, R.W. (1994) Effect of Serotonin Depletion on Vibrissa- Related Patterns of Thalamic Afferents in the Rats Somatosensory Cortex. The Journal of Neuroscience, 14, 7594- 7607.
- Mazer, C., Muneyyirci, J., Taheny, K., Raio, N., Borella, A. and Whitaker-Azmitia, P. (1997) Serotonin Depletion during Synaptogenesis Leads to Decreased Synaptic Density and Learning Deficits in the Adult Rat: A Possible Model of Neurodevelopmental Disorders with Cognitive Deficits. Brain Research, 760, 68-73. http://dx.doi.org/10.1016/S0006-8993(97)00297-7
- Casanova, M.F., Buxhoeveden, D.P., Switala, A.E. and Roy, E. (2002) Minicolumnar Pathology in Autism. Neurology, 58, 428-432. http://dx.doi.org/10.1212/WNL.58.3.428
- Boylan, C.B., Blue, M.E. and Hohmann, C.F. (2007) Modeling Early Cortical Serotonergic Deficits in Autism. Behavioural Brain Research, 176, 94-108. http://dx.doi.org/10.1016/j.bbr.2006.08.026
- Hohmann, C.F., Walker, E.M., Boylan, C.B. and Blue, M.E. (2007) Neonatal Serotonin Depletion Alters Behavioral Responses to Spatial Change and Novelty. Brain Research, 1139, 163-177. http://dx.doi.org/10.1016/j.brainres.2006.12.095
- Kočovská, E., Fernell, E., Billstedt, E., Minnis, H. and Gillberg, C. (2012) Vitamin D and Autism: Clinical Review. Research in Developmental Disabilities, 33, 1541-1550. http://dx.doi.org/10.1016/j.ridd.2012.02.015
- Patrick, R.P. and Ames, B.N. (2014) Vitamin D Hormone Regulates Serotonin Synthesis. Part 1: Relevance for Autism. FASEB Journal, 28, 2398-2413. (ePub Ahead of Print) http://dx.doi.org/10.1096/fj.13-246546
- Kuberan, B., Lech, M., Borjigin, J. and Rosenberg, R.D. (2004) Light-Induced 3-O-Sulfotransferase Expression Alters Pineal Heparan Sulfate Fine Structure. A Surprising Link to Circadian Rhythm. Journal of Biological Chemistry, 279, 5053-5054. http://dx.doi.org/10.1074/jbc.C300492200
- Tyce, G.M., Messick, J.M., Yaksh, T.L., Byer, D.E., Danielson, D.R. and Rorie, D.K. (1986) Amine Sulfate Formation in the Central Nervous System. Federation proceedings, 45, 2247-2253.
- Kurth, F., Narr, K.L., Woods, R.P., O’Neill, J., Alger, J.R., Caplan, R., et al. (2011) Diminished Gray Matter within the Hypothalamus in Autism Disorder: A Potential Link to Hormonal Effects? Biological Psychiatry, 70, 278-282. http://dx.doi.org/10.1016/j.biopsych.2011.03.026
- Weaver, D.R., Rivkees, S.A. and Reppert, S.M. (1989) Localization and Characterization of Melatonin Receptors in Rodent Brain by in Vitro Autoradiography. The Journal of Neuroscience, 9, 2581-2590.
- Rosenwasser, A.M., Trubowitsch, G. and Adler, N.T. (1985) Circadian Rhythm in Metabolic Activity of Suprachiasmatic, Supraoptic and Raphe Nuclei. Neuroscience Letters, 58, 183-187. http://dx.doi.org/10.1016/0304-3940(85)90161-2
- Seneff, S., Lauritzen, A., Davidson, R. and Lentz-Marino, L. (2012) Is Endothelial Nitric Oxide Synthase a Moon- lighting Protein Whose Day Job Is Cholesterol Sulfate Synthesis? Implications for Cholesterol Transport, Diabetes and Cardiovascular Disease. Entropy, 14, 2492-2530. http://dx.doi.org/10.3390/e14122492
- Baulieu, E.E. and Robel, P. (1998) Dehydroepiandrosterone (DHEA) and Dehydroepiandrosterone Sulfate (DHEAS) as Neuroactive Neurosteroids. Proceedings of the National Academy of Sciences of the United States of America, 95, 4089-4091. http://dx.doi.org/10.1073/pnas.95.8.4089
- Vallée, M., Mayo, W. and Le Moal, M. (2001) Role of Pregnenolone, Dehydroepiandrosterone and Their Sulfate Esters on Learning and Memory in Cognitive Aging. Brain Research Reviews, 37, 301-312. http://dx.doi.org/10.1016/S0165-0173(01)00135-7
- Beaujean, D., Mensah-Nyagan, A.G., Van Luu-The, J.L.D., Pelletier, G. and Vaudry, H. (1999) Immunocytochemical Localization and Biological Activity of Hydroxysteroid Sulfotransferase in the Frog Brain. Journal of Neurochemistry, 72, 848-857. http://dx.doi.org/10.1046/j.1471-4159.1999.720848.x
- Robel, P. and Baulieu, E.E. (1994) Neurosteroids: Biosynthesis and Function. Trends in Endocrinology and Metabolism, 5, 1-8. http://dx.doi.org/10.1016/1043-2760(94)90114-7
- Murialdo, G., Nobili, F., Rollero, A., Gianelli, M.V., Copello, F., Rodriguez, G., et al. (2000) Hippocampal Perfusion and Pituitary-Adrenal Axis in Alzheimer’s Disease. Neuropsychobiology, 42, 51-57. http://dx.doi.org/10.1159/000026672
- San Martin, M. and Touitou, Y. (2000) DHEA-Sulfate Causes a Phase-Dependent Increase in Melatonin Secretion: A Study of Perifused Rat Pineal Glands. Steroids, 65, 491-496. http://dx.doi.org/10.1016/S0039-128X(00)00111-2
- Schafield, M., Jenski, L.J., Dumaual, A.C. and Stillwell, W. (1998) Cholesterol versus Cholesterol Sulfate: Effects on Properties of Phospholipid Bilayers Containing Docosahexaenoic Acid. Chemistry and Physics of Lipids, 95, 23-36. http://dx.doi.org/10.1016/S0009-3084(98)00065-6
- Sawazaki, S., Salem Jr., N. and Kim, H.Y. (1994) Lipoxygenation of Docosahexaenoic Acid by the Rat Pineal Body. Journal of Neurochemistry, 62, 2437-2447. http://dx.doi.org/10.1046/j.1471-4159.1994.62062437.x
- Zaouali-Ajina, M., Gharib, A., Durand, G., Gazzah, N., Claustrat, B., Gharib, C., et al. (1999) Dietary Docosahexaenoic Acid-Enriched Phospholipids Normalize Urinary Melatonin Excretion in Adult (n-3) Polyunsaturated Fatty Acid- Deficient Rats. Journal of Nutrition, 129, 2074-2080.
- Jiang, Y.J., Kim, P., Elias, P.M. and Feingold, K.R. (2005) LXR and PPAR Activators Stimulate Cholesterol Sulfotransferase Type 2 Isoform 1b in Human Keratinocytes. Journal of Lipid Research, 46, 2657-2666. http://dx.doi.org/10.1194/jlr.M500235-JLR200
- Wang, L., Schuster, G.U., Hultenby, K., Zhang, Q., Andersson, S. and Gustafsson, J.A. (2002) Liver X Receptors in the Central Nervous System: From Lipid Homeostasis to Neuronal Degeneration. Proceedings of the National Academy of Sciences of the United States of America, 99, 13878-13883. http://dx.doi.org/10.1073/pnas.172510899
- Bai, Q., Xu, L., Kakiyama, G., Runge-Morris, M.A., Hylemon, P.B., Yin, L., et al. (2011) Sulfation of 25-Hydroxy- cholesterol by SULT2B1b Decreases Cellular Lipids via the LXR/SREBP-1c Signaling Pathway in Human Aortic Endothelial Cells. Atherosclerosis, 214, 350-356. http://dx.doi.org/10.1016/j.atherosclerosis.2010.11.021
- Erlinger, T.P., Miller III., E.R., Charleston, J. and Appel, L.J. (2003) Inflammation Modifies the Effects of a Reduced- Fat Low-Cholesterol Diet on Lipids: Results from the DASH-Sodium Trial. Circulation, 108, 150-154. http://dx.doi.org/10.1161/01.CIR.0000080288.30567.86
- Heliovaara, M.K., Teppo, A.M., Karonen, S.L. and Ebeling, P. (2006) Inflammation Affects Lipid Metabolism during Recovery from Hyperinsulinaemia. European Journal of Clinical Investigation, 36, 860-865. http://dx.doi.org/10.1111/j.1365-2362.2006.01730.x
- Xie, L., Kang, H., Xu, Q., Chen, M.J., Liao, Y., Thiyagarajan, M., et al. (2013) Sleep Drives Metabolite Clearance from the Adult Brain. Science, 342, 373-377. http://dx.doi.org/10.1126/science.1241224
- Nixon, R.A. (2005) Endosome Function and Dysfunction in Alzheimer’s Disease and Other Neurodegenerative Diseases. Neurobiology of Aging, 26, 373-382. http://dx.doi.org/10.1016/j.neurobiolaging.2004.09.018
- Nixon, R.A., Wegiel, J., Kumar, A., Yu, W.H., Peterhoff, C., Cataldo, A., et al. (2005) Extensive Involvement of Autophagy in Alzheimer’s Disease: An Immunoelectron Microscopy Study. Journal of Neuropathology & Experimental, 64, 113-122.
- Sokolowski, J.D. and Mandell, J.W. (2011) Phagocytic Clearance in Neurodegeneration. American Journal of Pathology, 178, 1416-1428. http://dx.doi.org/10.1016/j.ajpath.2010.12.051
- Wiegmann, E.M., Westendorf, E., Kalus, I., Pringle, T.H., Lübke, T. and Dierks, T. (2013) Arylsulfatase K, a Novel Lysosomal Sulfatase. Journal of Biological Chemistry, 288, 30019-30028. http://dx.doi.org/10.1074/jbc.M113.499541
- Kurz, T., Eaton, J.W. and Brunk, U.T. (2011) The Role of Lysosomes in Iron Metabolism and Recycling. The International Journal of Biochemistry & Cell Biology, 43, 1686-1697. http://dx.doi.org/10.1016/j.biocel.2011.08.016
- Kehrer, J.P. (2000) The Haber-Weiss Reaction and Mechanisms of Toxicity. Toxicology, 149, 43-50. http://dx.doi.org/10.1016/S0300-483X(00)00231-6
- Ross, M.A., Long, W.F. and Williamson, F.B. (1992) Inhibition by Heparin of Fe(II)-Catalysed Free-Radical Peroxidation of Linolenic Acid. Biochemical Journal, 286, 717-720.
- Egeberg, M., Kjeken, R., Kolset, S.O., Berg, T. and Prydz, K. (2001) Internalization and Stepwise Degradation of Heparan sulfate Proteoglycans in Rat Hepatocytes. Biochimica et Biophysica Acta (BBA)―Molecular Cell Research, 1541, 135-149.
- Scali, C., Prosperi, C., Vannucchi, M.G., Pepeu, G. and Casamenti, F. (2000) Brain Inflammatory Reaction in an Animal Model of Neuronal Degeneration and Its Modulation by an Anti-Inflammatory Drug: Implication in Alzheimer’s Disease. European Journal of Neuroscience, 12, 1900-1912. http://dx.doi.org/10.1046/j.1460-9568.2000.00075.x
- Teyssier, C., Guenot, L., Suschetet, W. and Siess, M.H. (1999) Metabolism of Diallyl Disulfide by Human Liver Microsomal Cytochromes P-450 and Flavin-Containing Monooxygenases. Drug Metabolism and Disposition, 27, 835- 841.
- López-Figueroa, M.O. and Møller, M. (1996) Nitric Oxide Synthase in the Pineal Gland. Histology and Histopathology, 11, 1089-1100.
- Spessert, R., Layes, E., Schollmayer, A., Reuss, S. and Vollrath, L. (1995) In the Rat Pineal Gland, but Not in the Suprachiasmatic Nucleus, the Amount of Constitutive Neuronal Nitric Oxide Synthase Is Regulated by Environmental Lighting Conditions. Biochemical and Biophysical Research Communications, 212, 70-76. http://dx.doi.org/10.1006/bbrc.1995.1937
- Skwarlo-Sonta, K. (2002) Melatonin in Immunity: Comparative Aspects. Neuroendocrinology Letters, 23, 61-66.
- Wu, Y.H. and Swaab, D.F. (2005) The Human Pineal Gland and Melatonin in Aging and Alzheimer’s Disease. Journal of Pineal Research, 38, 145-152. http://dx.doi.org/10.1111/j.1600-079X.2004.00196.x
- Romero, A., Ramos, E., de Los Ros, C., Egea, J., Del Pino, J. and Reiter, R.J. (2014) A Review of Metal-Catalyzed Molecular Damage: Protection by Melatonin. Journal of Pineal Research, 56, 343-370. http://dx.doi.org/10.1111/jpi.12132
- van Rensburg, S.J., Daniels, W.M., Potocnik, F.C., van Zyl, J.M., Taljaard, J.J. and Emsley, R.A. (1997) A New Model for the Pathophysiology of Alzheimer’s Disease. Aluminium Toxicity Is Exacerbated by Hydrogen Peroxide and Attenuated by an Amyloid Protein Fragment and Melatonin. South African Medical Journal, 7, 1111-1115.
- Liu, R.Y., Zhou, J.N., van Heerikhuize, J., Hofman, M.A. and Swaab, D.F. (1999) Decreased Melatonin Levels in Postmortem Cerebrospinal Fluid in Relation to Aging, Alzheimer’s Disease, and Apolipoprotein E-ε4/4 Genotype. Journal of Clinical Endocrinology & Metabolism, 84, 323-327.
- Mahlberg, R., Walther, S., Kalus, P., Bohner, G., Haedel, S., Reischies, F.M., et al. (2008) Pineal Calcification in Alzheimer’s Disease: An in Vivo Study Using Computed tomography. Neurobiology of Aging, 29, 203-209. http://dx.doi.org/10.1016/j.neurobiolaging.2006.10.003
- Reiter, R.J., Tan, D.X., Manchester, L.C. and El-Sawi, M.R. (2002) Melatonin Reduces Oxidant Damage and Promotes Mitochondrial Respiration: Implications for Aging. Annals of the New York Academy of Sciences, 959, 238-250. http://dx.doi.org/10.1111/j.1749-6632.2002.tb02096.x
- Maruszak, A. and Żekanowski, C. (2011) Mitochondrial Dysfunction and Alzheimer’s Disease. Progress in Neuro- Psychopharmacology and Biological Psychiatry, 35, 320-330. http://dx.doi.org/10.1016/j.pnpbp.2010.07.004
- Limson, J., Nyokong, T. and Daya, S. (1998) The Interaction of Melatonin and Its Precursors with Aluminium, Cadmium, Copper, Iron, Lead, and Zinc: An Adsorptive Voltammetric Study. Journal of Pineal Research, 24, 15-21. http://dx.doi.org/10.1111/j.1600-079X.1998.tb00361.x
- Pappolla, M.A., Sos, M., Omar, R.A., Bick, R.J., Hickson-Bick, D.L.M., Reiter, R.J., et al. (1997) Melatonin Prevents Death of Neuroblastoma Cells Exposed to the Alzheimer Amyloid Peptide. The Journal of Neuroscience, 17, 1683- 1690.
- Liu, S.J. and Wang, J.Z. (2002) Alzheimer-Like Tau Phosphorylation Induced by Wortmannin in Vivo and Its Attenuation by Melatonin. Acta Pharmacologica Sinica, 23, 183-187.
- Adeloye, A. and Felson, B. (1974) Incidence of Normal Pineal Gland Calcification in Skull Roentgenograms of Black and White Americans. American Journal of Roentgenology, 122, 503-507. http://dx.doi.org/10.2214/ajr.122.3.503
- Seneff, S., Davidson, R.M. and Liu, J. (2012) Is Cholesterol Sulfate Deficiency a Common Factor in Preeclampsia, Autism, and Pernicious Anemia? Entropy, 14, 2265-2290. http://dx.doi.org/10.3390/e14112265
- Michel, J.B., Feron, O., Sacks, D. and Michel, T. (1997) Reciprocal Regulation of Endothelial Nitric-Oxide Synthase by Ca2+-Calmodulin and Caveolin. Journal of Biological Chemistry, 272, 15583-15586. http://dx.doi.org/10.1074/jbc.272.25.15583
- Siegel, N. and Haug, A. (1983) Aluminum Interaction with Calmodulin. Evidence for Altered Structure and Function from Optical and Enzymatic Studies. Biochimica et Biophysica Acta (BBA)―Protein Structure and Molecular Enzymology, 744, 36-45. http://dx.doi.org/10.1016/0167-4838(83)90337-0
NOTES

*Corresponding author.