Neuroscience & Medicine
Vol.4 No.2(2013), Article ID:33239,8 pages DOI:10.4236/nm.2013.42013
Absence of Nitric Oxide Synthase 3 Increases Amyloid β-Protein Pathology in Tg-5xFAD Mice
![]()
Departments of Neurosurgery & Medicine, Stony Brook, Stony Brook University, New York, USA.
Email: *William.VanNostrand@sbumed.org
Copyright © 2013 Zishuo Ian Hu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original work is properly cited.
Received March 21st, 2013; revised April 25th, 2013; accepted May 10th, 2013
Keywords: Nitric Oxide Synthase 3; Amyloid β-Protein, Alzheimer’s Disease; Transgenic Mice; Deposition
ABSTRACT
Aim: The abnormal accumulation, assembly and deposition of the amyloid β-protein (Aβ) are prominent pathological features of patients with Alzheimer’s disease (AD) and related disorders. A number of factors in the brain can influence Aβ accumulation and associated pathologies. The aim of the present study was to determine the consequences of deleting nitric oxide synthase (NOS) 3, the endothelial form of NOS, in Tg-5xFAD mice, a model of parenchymal AD-like amyloid pathology. Methods: Tg-5xFAD mice were bred with NOS3−/− mice. Cohorts of Tg-5xFAD mice and bigenic Tg-5xFAD/NOS3−/− mice were aged to six months followed by collection of the blood and brain tissues from the mice for biochemical and pathological analyses. Results: ELISA analyses show that the absence of NOS3 results in elevated levels of cerebral and plasma Aβ peptides in Tg-5xFAD mice. Immunohistochemical analyses show that the absence of NOS3 increased the amount of parenchymal Aβ deposition and fibrillar amyloid accumulation in Tg-5xFAD mice. The elevated levels of Aβ were not due to changes in the expression levels of transgene encoded human amyloid precursor protein (APP), endogenous β-secretase, or increased proteolytic processing of APP. Conclusions: The results from this study suggest that the loss of NOS3 activity enhances Aβ pathology in Tg-5xFAD mice. These findings are similar to previous studies of NOS2 deletion suggesting that reduced NOS activity and NO levels enhance amyloid-associated pathologies in human APP transgenic mice.
1. Introduction
Alzheimer’s disease (AD), a neurodegenerative disorder and leading cause of dementia, is presently the 6th leading cause of death afflicting over 5 million individuals in the United States [1]. Pathologically, the disease is characterized by a progressive accumulation of cerebral amyloid β-protein (Aβ) assemblies and hyperphosporylated tau species leading to neuronal dysfunction and cell death culminating in loss of memory and cognitive function [2,3]. In AD, cerebral Aβ deposition occurs primarily in the form of parenchymal amyloid plaques. The deposition of Aβ peptides also occurs in cerebral blood vessels, a condition known as cerebral amyloid angiopathy (CAA) [4,5].
The amyloid cascade hypothesis proposes that the generation and aggregation of Aβ is the initiating event that leads to AD [2,3]. The assembly of Aβ into soluble oligomeric forms and amyloid plaques is thought to promote further pathological events including the formation of intraneuronal tau tangles and disruption of synaptic connections. Aβ is a 40-42 amino acid peptide produced by proteolytic cleavage of a type I transmembrane protein known as amyloid precursor protein (APP) [6,7]. The amyloidogenic processing of APP initially involves a cleavage at the amino terminus of Aβ peptide sequence by β-secretase, an aspartyl proteinase named BACE [8]. Subsequent cleavage of the remaining amyloidogenic membrane spanning APP carboxyl terminal fragment by β-secretase liberates the predominant Aβ40 or Aβ42 residue peptides. The proteolytic active site of the multiprotein β-secretase complex is contained in Presenilin (PS) proteins [9,10].
Nitric oxide (NO) is a multifunctional molecule that possesses both concentration-dependent and cell-specific cytoprotective and cytoxic properties [11,12]. For example, NO has been shown to promote oxidative damage by reacting with superoxide anion to form peroxynitrite [13,14]. Alternatively, NO has been shown to be neuroprotective by overexpressing heme oxygenase, promoting Creb and Akt survival and protecting excitotoxicity through S-nitrosylation [15,16].
Nitric oxide synthases (NOS) are the enzymes responsible for producing NO in the body. There are three isoforms of NOS present in the brain: NOS1, NOS2 and NOS3 [17]. NOS1 is largely constitutively expressed by neurons in the brain. NOS2 is found predominantly in macrophages and microglia and is inducible. NOS3, found in endothelial cells, is closely associated with cerebral vascular function. Previous studies showed that removing NOS2 in specific APP transgenic mouse models enhanced AD-like pathologies including hyperphosphylated tau and neuronal loss [18,19]. In another study it was shown that NOS3−/− mice had increased levels of BACE1 and elevated levels of endogenous mouse APP compared to wild-type mice [20]. Based on these findings, the main goal of the present study was to determine if the absence of NOS3 modifies development of ADlike features in Tg-5xFAD mice, an APP transgenic mouse of parenchymal amyloid pathology.
Here we show that deletion of NOS3 in Tg-5xFAD mice resulted in elevated Aβ levels in brain. This was reflected by increased parenchymal Aβ deposition and fibrillar amyloid. The elevated Aβ in brain was not due to changes in the expression levels of transgene encoded human APP, endogenous BACE1, or increased processing of APP. Furthermore, there were increased levels of plasma Aß that mirrored the increased levels of brain Aβ, suggesting that efflux of cerebral Aβ into blood is not impaired in the absence of NOS3. Together, the present findings with NOS3 deletion are similar to the previous studies of NOS2 deletion, suggesting that reduced NOS activity and NO levels enhance amyloid-associated pathologies in human APP transgenic mice.
2. Materials and Methods
2.1. Animals
All studies with mice followed NIH guidelines and were approved by the Stony Brook University IACUC committee. For these studies we used Tg-5x FAD mice, a model for AD-like parenchymal amyloid pathology. Tg- 5xFAD mice co-express human APP695 and human presenilin 1 with five familial mutations (APP K670N/ M671L + I716V + V717I and PS1 M146L + L286V) and develop early-onset Aβ accumulation and fibrillar Aβ plaques in brain starting at about two months of age [21]. Tg-5xFAD mice and NOS3−/− mice were obtained from Jackson Laboratories. Heterozygous Tg-5xFAD mice were bred with NOS3−/− to obtain cohorts of heterozgous bigenic Tg-5xFAD/NOS3+/− mice. These mice were subsequently bred to heterozygous NOS3+/− mice to obtain heterozygous Tg-5xFAD mice and bigenic heterozygous Tg-5xFAD/NOS3−/− mice all on the same background. The presence of the Tg-5xFAD transgenes and absence of NOS3 were confirmed by PCR genotyping.
2.2. Tissue Collection
Prior to termination blood was collected from each mouse by retro-orbital bleeding to obtain plasma samples. The mice were perfused with cold phosphate-buffered saline (PBS), the brains removed and dissected through the mid-sagittal plane. One hemisphere was snap frozen in liquid nitrogen, pulverized on dry ice and divided into three equivalent aliquots for biochemical and molecular analyses. The other hemisphere was placed in 70% ethanol, followed by xylene treatment and embedding in paraffin for immunohistochemical and histological analyses.
2.3. Mouse Plasma Isolation
Mouse blood was collected by retro-orbital bleed from anesthetized mice. Blood was collected in one tenth volume of 3.8% sodium citrate to prevent coagulation. Blood was centrifuged at 8000× g for 5 min at room temperature to remove platelets and cellular components. Plasma samples were stored at −80˚C until analysis.
2.4. ELISA Measurement of Aβ Peptides
Soluble pools of Aβ40 and Aβ42 were determined by using specific ELISAs on carbonate extracted mouse forebrain tissue and subsequently the insoluble Aβ40 and Aβ42 levels were determined by ELISA of guanidine lysates of the insoluble pellets resulting from the carbonate extracted brain tissue as described [22,23]. For plasma Aβ analysis, 70 µl of sample was incubated with 35 µl of 5 M guanidine-HCl for 30 min at room temperature. The treated plasma samples were then diluted to a volume of 350 µl in a buffer of 200 mM Tris-HCl, pH 6.8 containing 0.25% bovine serum albumin. In the sandwich ELISAs Aβ40 and Aβ42 were captured using their respective carboxyl-terminal specific antibodies m2G3 and m21F12 and biotinylated m3D6, specific for human Aβ, was used for detection [22]. Each brain lysate or plasma sample was measured in triplicate and compared to linear standard curves generated with known concentrations of human Aβ using a Spectramax M2 plate reader (Molecular Devices, Sunnyvale, CA).
2.5. Immunohistochemical Procedures
For immunolabeling, sections were cut from paraffinembedded mouse brain hemispheres in the sagittal plane at 10 µm using a microtome, placed on a flotation water bath at 45˚C and then placed onto glass slides. Paraffin was removed from the sections by washing with xylene and the tissue sections were rehydrated in decreasing concentrations of ethanol. Antigen retrieval was performed by treating the tissue sections with proteinase K (0.2 mg/ml) for 10 min at 22˚C. Deposited Aβ was detected using an affinity purified rabbit polyclonal antibody directed to residues 1 - 28 of human Aβ (1:250; [24]). Primary antibody was detected with HRP-conjugated anti-rabbit secondary antibody (1:1000; GE Healthcare, Buckinghamshire, UK) and visualized with a stable diaminobenzidine solution as substrate (Spring Bioscience, Fremont, CA). Sections were counterstained with hematoxylin. Cerebral vessels were identified using a rabbit polyclonal antibody to collagen type IV (1:100; Research Diagnostics, Flanders, New Jersey) and Alexa Fluor 594-conjugated secondary antibody (1:1000; Invitrogen, Eugene, Oregon). Thioflavin-S staining for fibrillar amyloid was performed as described previously [25,26].
2.6. Immunoblotting
Aliquots of pulverized mouse forebrain were homogenized in 50 mM Tris-HCl (pH 7.5) containing 150 mM NaCl, 1% SDS, 0.5% NP-40, 5 mM EDTA and proteinase inhibitor cocktail (Roche Biochemicals, Indianapolis, IN, USA). The tissue homogenates were clarified by centrifugation at 14,000× g for 10 min. Protein concentrations of the resulting supernatants was determined using the BCA Protein Assay kit (Pierce, Rockford, IL, USA). 35 μg of total protein from each sample was electrophoresed in either SDS 10% polyacrylamide gels under nonreducing conditions and the proteins were transfered onto Hybond nitrocellulose membranes (Amersham, Arlington Heights, IL, USA). Unoccupied sites on the membranes were blocked overnight with 5% non-fat milk in PBS with 0.05% Tween—20. The following antibodies were used for detection of target proteins: human APP was detected using the human APP-specific N-terminal monoclonal antibody P2-1 as described previously (1:1000; [26]); BACE1 was detected using a rabbit monoclonal antibody (1:1000; Cell Signaling Technology, Danvers, MA, USA); APP C-terminal fragments were detected using an affinity purified rabbit polyclonal antibody directed to APP695 residues 677 - 695 (1:1000); and β-tubulin was detected using a rabbit polyclonal antibody (1:1000; Abcam, Cambridge, MA, USA). Quantitation of protein bands was performed using a VersaDoc Imaging System (BioRad, Hercules, CA) and the manufacturer’s Quantity Oneton software.
2.7. Stereological Measurements
Total Aβ deposition and amyloid burden in the regions of the fronto-temporal cortex and subiculum were quantified on the same set of systematically sampled immunostained sections and thioflavin-S stained sections, respectively, using NIH Image J 1.32 software. The numbers of thioflavin-S labeled surface meningeal blood vessels in the same fields as above were determined using the stereological principles as described as described [27,28].
2.8. Statistical Analysis
Data are presented as mean and standard deviation of the mean of n = six or more mice per group. The t test was performed to determine the statistical difference between the groups of Tg-5xFAD mice and Tg-5xFAD/NOS3−/− mice. p < 0.05 was considered as significant.
3. Results
3.1. Deletion of NOS3 Increases Parenchymal Aβ Accumulation in the Brains of Tg-5xFAD Mice
To investigate the effect of NOS3 on cerebral Aβ pathology we bred Tg-5xFAD mice, a model of AD-like parenchymal Aβ pathology, with NOS3−/− mice. The absence of NOS3 in bigenic Tg-5xFAD/NOS3−/− mice was confirmed at the genetic level by PCR genotyping (data not shown) and at the protein level by immunoblotting brain samples to confirm the absence of NOS3 protein (Figure 1). It was previously reported that NOS3−/− mice exhibit a significant increase in blood pressure compared to wild-type mice due to loss of endothelial NO and vascular relaxation [29]. Accordingly, we found that bigenic Tg-5xFAD/NOS3−/− had elevated blood pressure measurements compared to Tg-5xFAD mice (186.3 ± 7.4 mmHg vs 154.3 ± 18.3 mmHg systolic blood pressure and 152.2 ± 10.7 mmHg vs 120.5 ± 16.0 mmHg diastolic blood pressure, respectively) further confirming the lack of NOS3 in the bigenic animals.
Groups of Tg-5xFAD mice and bigenic Tg-5x FAD/NOS3−/− mice were aged to six months, a point where there is marked parenchymal Aβ deposition in this APP transgenic line [21]. ELISA analysis was performed to determine the levels of soluble and insoluble Aβ40 and Aβ42 in brain homogenates prepared from each line. As shown in Figure 2, there were increased levels of both soluble and insoluble Aβ40 and Aβ42 in the Tg-5x

Figure 1. Lack of NOS3 in Tg-5xFAD/NOS3−/− mice. Brain homogenates prepared from Tg-5xFAD mice and Tg-5x FAD/NOS3−/− mice were subjected to immunoblotting with an antibody specific to NOS3. The results confirm that Tg-5xFAD/NOS3−/− mice do not express NOS3 protein.
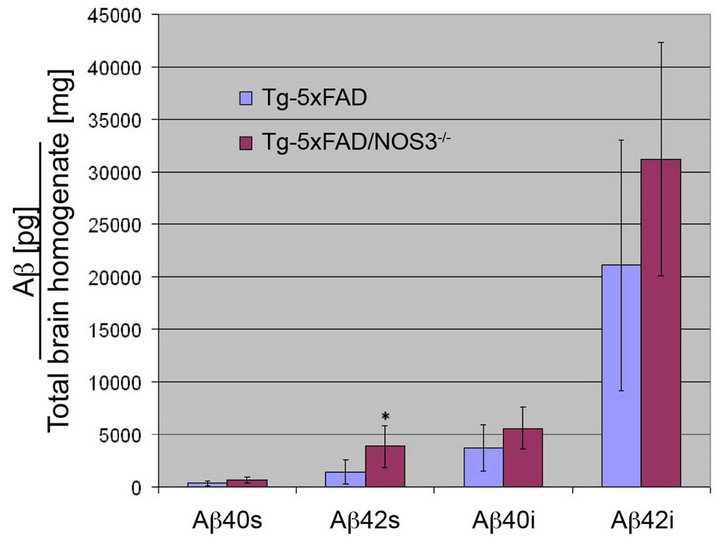
Figure 2. The absence of NOS3 increases Aβ levels in Tg-5xFAD mice. ELISA measurements for soluble and insoluble forms of Aβ40 and Aβ42 peptides were performed on homogenates prepared from forebrain hemispheres from Tg-5xFAD and Tg-5x FAD/NOS3−/− mice. The results show that the levels of Aβ40 and Aβ42 peptides were generally higher in the absence of NOS3. ELISA data shown are mean ± SD of 6 mice per group. *p < 0.05.
FAD/NOS3−/− mice compared with Tg-5xFAD littermate controls.
To determine the cerebral distribution of Aβ deposition we performed immunostaining on brain sections prepared from Tg-5xFAD and Tg-5xFAD/ NOS3−/− mice. As shown in Figure 3, Tg-5xFAD mice and Tg-5x FAD/NOS3−/− mice exhibited a similar pattern of parenchymal Aβ deposition with abundant plaques in the cortex and subiculum. Quantitative image analysis confirmed that there was increased Aβ deposition in the Tg-5xFAD/NOS3−/− mice compared with Tg-5xFAD littermates, consistent with the ELISA results obtained in Figure 2.
To differentiate between Aβ deposition and fibrillar amyloid deposition we performed thioflavin S staining of brain sections. Figure 4 shows representative images of fibrillar amyloid deposition in the cortex and subiculum of Tg-5xFAD and Tg-5xFAD/NOS3−/− mouse brains. Quantitation of the thioflavin S staining indicated that there was significantly higher amounts of fibrillar amy-

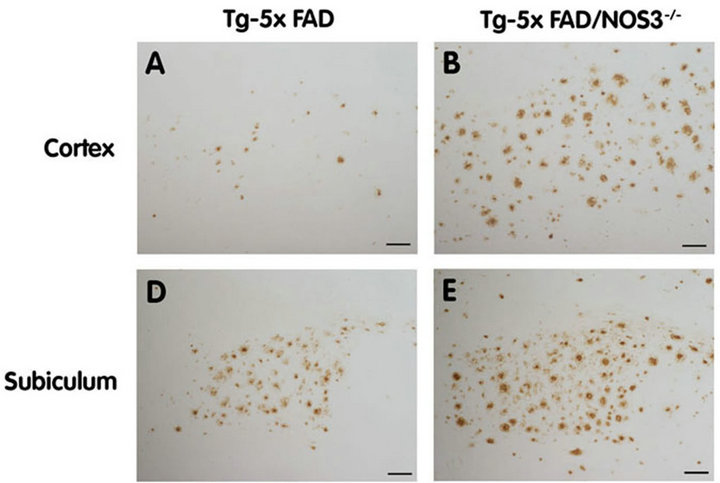
Figure 3. The absence of NOS3 increases parenchymal Aβ deposition in Tg-5xFAD mice. Human Aβ deposits were detected by immunostaining (brown) in the cortex and subiculum regions of Tg-5xFAD and Tg-5xFAD/NOS3−/− mouse cortex (A) (B) respectively and subiculum (D) (E) respectively. Scale bars = 100 μm. Quantitative image analysis of Aβ deposition in Tg-5xFAD and Tg-5xFAD/NOS3−/− mouse cortex (C) and subiculum (F) showed elevated levels of Aβ in the absence of NOS3. Data presented are the mean ± SD of 6 mice per group. *p < 0.001.
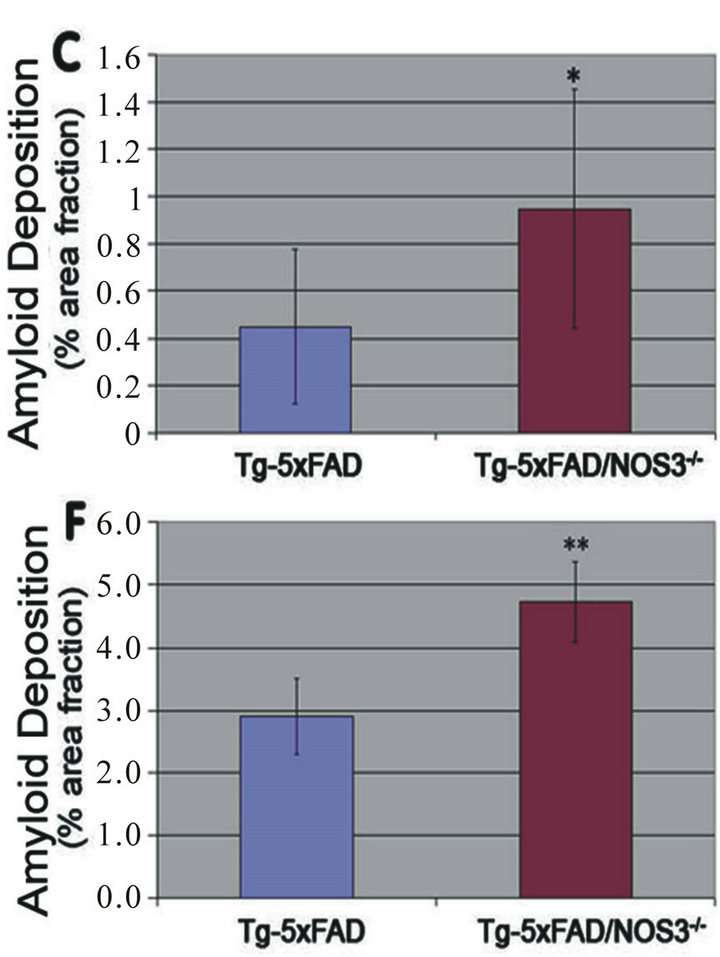
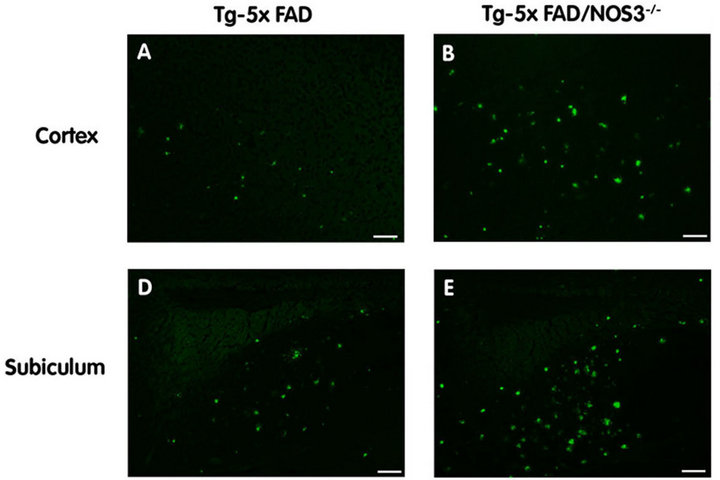
Figure 4. The absence of NOS3 increases parenchymal amyloid deposition in Tg-5xFAD mice. Fibrillar amyloid was detected using thioflavin-S (green) in the cortex and subiculum regions of Tg-5xFAD and Tg-5xFAD/NOS3−/− mouse cortex (A) (B) respectively and subiculum (D) (E) respectively. Scale bars = 100 μm. Quantitative image analysis of fibrillar amyloid deposition in Tg-5xFAD and Tg-5xFAD/NOS3−/− mouse cortex (C) and subiculum (F) showed elevated levels of in the absence of NOS3. Data presented are the mean ± SD of 6 mice per group. *p < 0.03, **p < 0.0005.
loid in both the cortex and subiculum of Tg-5xFAD/ NOS3−/− mice compared to Tg-5xFAD littermates.
Although CAA is not a prominent phenotype in the Tg-5xFAD mice, they do develop some meningeal surface vessel CAA with age. Since NOS3 is expressed in endothelial cells and affects vascular function it may have an influence on CAA. Therefore, we performed double labeling experiments on the mouse brain sections where we immunolabeled for collagen IV to identify cerebral vessels and stained with thioflavin S to identify fibrillar amyloid. As shown in Figure 5, some large meningeal vessel amyloid was observed in both Tg-5x FAD and Tg-5xFAD/NOS3−/− mice that was somewhat elevated in the latter group. Together, these findings show that the absence of NOS3 increases the levels of cerebral Aβ peptides, parenchymal Aβ deposition and fibrillar amyloid in six months old Tg-5xFAD mice.
3.2. APP Production and Metabolism Are Not Affected in the Tg-5xFAD/NOS3−/− Mice
Since Aβ and fibrillar amyloid levels were elevated in the Tg-5xFAD/NOS3−/− mice we next determined if this was a result of increased APP production and/or metabolism. It was previously reported that the levels of endogenous mouse APP are elevated in NOS3−/− mice [20]. However, as shown in Figure 6, quantitative immunoblotting analysis showed no difference in the levels of transgene encoded human APP between Tg-5xFAD and Tg-5xFAD/ NOS3−/− mice.
Similarly, it was reported that endogenous BACE 1 was increased in NOS3−/− mice [20]. Thus, elevated BACE 1 levels could enhance amyloidogenic processing of APP causing increased production of Aβ peptides. However, quantitative immunoblotting analysis showed there was no appreciable change in the levels of BACE 1 between Tg-5xFAD and Tg-5xFAD/NOS3−/− mice suggesting that increased processing of APP does not occur in the Tg-5xFAD/NOS3−/− mice. This contention was further supported by quantitative measures of the C99/ C83 APP C-terminal fragments again showing no differences between Tg-5xFAD and Tg-5xFAD/NOS3−/− mice (Figure 6). Together, these results suggest that the increased Aβ and parenchymal fibrillar amyloid levels in Tg-5xFAD/NOS3−/− mice is not due to elevated APP or BACE expression or altered amyloidogenic processing of APP.
3.3. Tg-5xFAD/NOS3−/− Mice Exhibit Increased Levels of Plasma Aβ
An important clearance route of cerebral Aβ is across the blood-brain barrier into the circulation [29]. Since the absence of NOS3 occurs in endothelial cells at the blood-brain barrier it is possible that this efflux pathway is altered in Tg-5xFAD/NOS3−/− mice resulting in the increased Aβ in brain. However, ELISA measurements of plasma Aβ40 and Aβ42 showed that Tg-5xFAD/ NOS3−/− mice have significantly higher amounts of plasma Aβ compared to Tg-5xFAD mice (Figure 7). These higher plasma Aβ levels mirror the higher cerebral Aβ levels in the absence of NOS3 suggesting that Aβ clearance into the circulation is likely not impaired.
4. Discussion
The Aβ cascade hypothesis proposes that the accumulation of Aβ in the brain promotes deleterious downstream consequences such as neuroinflammation, hyperphosphorylation of tau and neuronal death, which in turn causes the cognitive deficits seen in AD [2,3]. Therefore, identifying factors that influence the accumulation of Aβ in brain is important for understanding mechanisms that might underlie the initiation of this pathological process. In the present study, we show that the loss of NOS3 increases cerebral Aβ levels and the amount of deposited fibrillar amyloid in Tg-5xFAD mice, a model of AD-like parenchymal amyloid pathology. Due to the presence of the FAD mutations in Tg-5xFAD mice, Aβ42 is the predominant species of the peptide that is produced and ac-

Figure 5. The absence of NOS3 increases surface meningeal vessel CAA in Tg-5xFAD mice. CAA was detected using thioflavin-S (green) to identify fibrillar amyloid and an antibody to collagen IV (red) to identify cerebral blood vessels. Representative images of surface vessel CAA in Tg-5xFAD mice (A) and Tg-5xFAD/NOS3−/− mice (B). (C) Quantitation of the percent of surface meningeal vessels affected with CAA in Tg-5xFAD mice and Tg-5xFAD/NOS3−/− mice. Data presented are the mean ± SD of 6 mice per group.
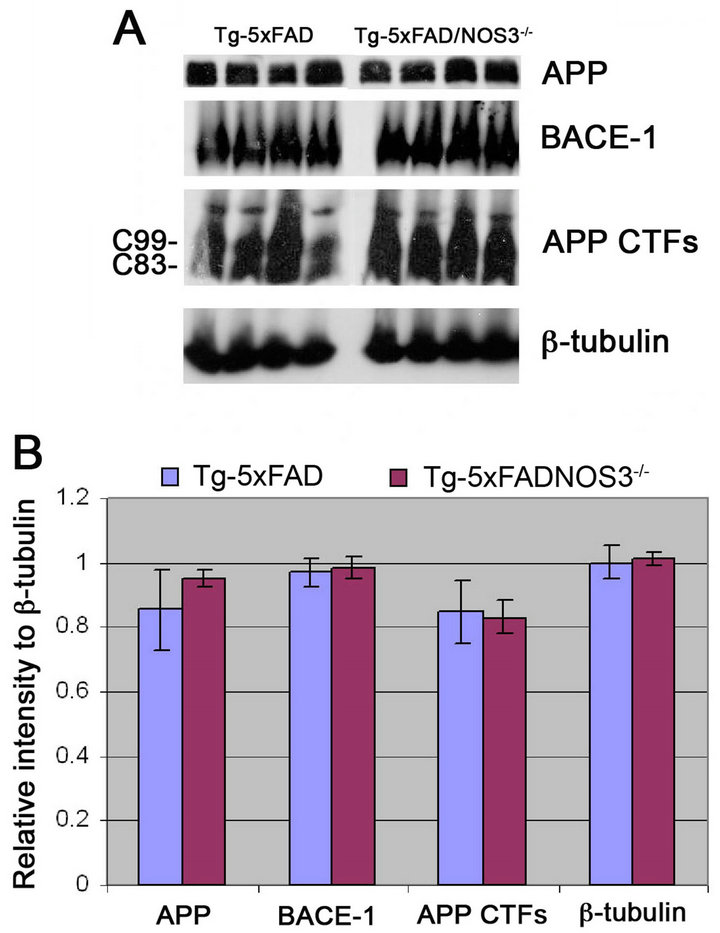
Figure 6. The absence of NOS3 does not alter APP expression and processing in Tg-5xFAD mice. (A) Representative quantitative immunoblots for detecting APP, BACE-1, APP CTFs and β-tubulin in forebrain homogenates prepared from Tg-5xFAD mice and Tg-5xFAD/NOS3−/− mice; (B) Quantitation of protein levels revealed there was no difference between Tg-5xFAD mice and Tg-5xFAD/NOS3−/− mice. Data presented are the mean ± SD of 6 mice per group.
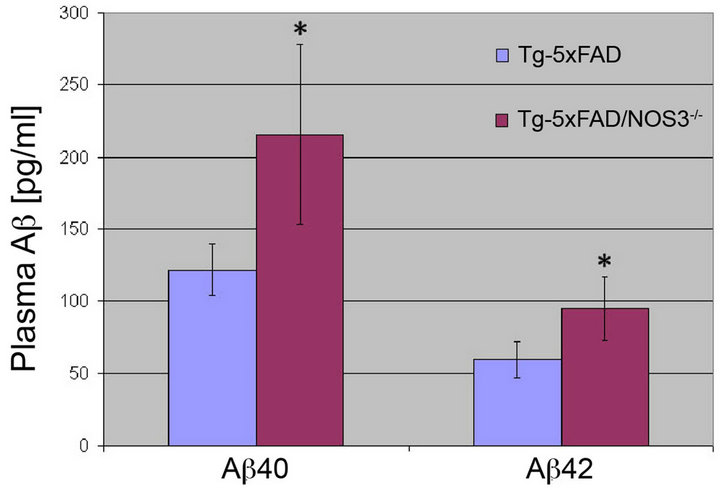
Figure 7. The absence of NOS3 increases plasma Aβ levels in Tg-5xFAD mice. ELISA measurements for Aβ40 and Aβ42 peptides were performed on plasma samples collected from Tg-5xFAD and Tg-5xFAD/NOS3−/− mice. The results show that the plasma levels of Aβ40 and Aβ42 peptides were significantly elevated in the absence of NOS3. ELISA data shown are mean ± SD of 6 mice per group. *p < 0.05.
cumulates in brain. Notably, although increases in both Aβ40 and Aβ42 occur in the absence of NOS3 the latter remains the predominant form of Aβ that accumulates in brain of Tg-5xFAD mice. Since Aβ42 is elevated and more fibrillogenic this also explains why there are significant increases in parenchymal fibrillar amyloid deposition in the absence of NOS3.
Austin et al. [20] reported that in NOS3−/− mice the levels of endogenous mouse APP and the APP processing enzyme BACE1 were increased. However, when we performed quantitative measures of transgene encoded human APP and endogenous mouse BACE1 we found no appreciable differences between Tg-5xFAD mice and bigenic Tg-5xFAD/NOS3−/− mice suggesting that increased expression and processing of APP was not altered in the absence of NOS3. This was further supported by the lack of changes in the amount of APP C-terminal fragments that result from β-secretase processing (C83) and β-secretase processing (C99) of APP. The lack of changes in APP or BACE1 expression observed in Tg-5xFAD/NOS3−/− mice could be due to several different reasons. For example, in the study of NOS3−/− mice by Austin et al. [20] they measured endogenous mouse APP where in our study we measured transgene encoded human APP that is under transcriptional control of the thy1 promoter element [21]. Perhaps the effect of increased APP expression in the absence of NOS3 is restricted to the activity of the endogenous mouse APP promoter element. The absence of increased BACE1 expression in bigenic Tg-5xFAD/NOS3−/− mice could be due to the presence of FAD mutations in APP and PS or different background strains of the mice. Overall though, the increased levels of cerebral Aβ and parenchymal amyloid deposition do not appear to result from enhanced APP expression and amyloidogenic processing.
The accumulation of Aβ in brain can result from an increase in production or decrease in one or more clearance mechanisms. The present findings argue that deletion of NOS3 does not appear to increase production of Aβ in Tg-5xFAD mice, suggesting that clearance pathways may be impaired. One prominent clearance route for cerebral Aβ peptides is transport across the endothelial blood-brain barrier into the circulation [28]. Since NOS3 is expressed in the endothelium and its production of NO influences vascular function it is possible that Aβ efflux into blood could be impaired in bigenic Tg- 5xFADx/NOS3−/− mice. However, the plasma levels of Aβ40 and Aβ42 were significantly higher in the bigenic Tg-5xFAD/NOS3−/− mice compared to the Tg-5xFAD mice reflecting the elevated brain levels in the former. Thus, the efflux of Aβ out of brain into the circulation does not appear to be altered in the absence of NOS3. This could suggest that other clearance mechanisms are altered in the absence of NOS3. For example, there are a number of endogenous Aβ degrading enzymes in brain including neprilysin, insulin degrading enzyme and matrix metalloproteinases [31-33]. It is possible that the absence of NOS3 reduces the levels or activities of one or more of these enzymes, resulting in increased Aβ levels. Alternatively, there could be changes in Aβ binding proteins that facilitate the assembly and deposition of Aβ. For example, the β4 isoform of apolipoprotein E can decrease the age of onset, increase the severity and promote cerebral deposition of fibrillar amyloid [34]. Other factors that can influence cerebral Aβ deposition include apolipoprotein J, otherwise known as clusterin [23], apolipoprotein A-1 [35,36], β1-anti-chymotrypsin [37] and transthyretin [38]. In the future it will be important to determine if in the absence of NOS3 Aβ-degrading enzymes or Aβ binding proteins are altered in a manner that promotes the accumulation and deposition of Aβ in brain.
In conclusion, our results suggest that NOS3 plays a neuroprotective role in the development of Aβ pathologies. The present findings with NOS3 deletion are similar to the previous studies of NOS2 deletion [18,19] suggesting that reduced NOS activity and NO levels in brain enhance amyloid-associated pathologies in human APP transgenic mice.
5. Acknowledgements
This research was supported by a gift from the Cowles Charitable Trust and National Institutes of Health Grant NS55118 (to WEVN). Antibody reagents for the ELISA analysis of Aβ were generously provided by Lilly Research Laboratories.
REFERENCES
- Alzheimer’s Association, “Alzheimer’s Disease Facts and Figures,” Alzheimer’s and Dementia: The Journal of the Alzheimer’s Association, Vol. 8, No. 2, 2012, pp. 131-168. doi:10.1016/j.jalz.2012.02.004
- D. J. Selkoe, “Alzheimer’s Disease: Genes, Proteins and Therapy,” Physiological Reviews, Vol. 8, 2001, pp. 741- 766.
- J. Hardyand D. J. Selkoe, “The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics,” Science, Vol. 297, No. 5580, 2002, pp. 353-356. doi:10.1126/science.1072994
- A. A. Rensink, R. M. de Waal, B. Kremer and M. M. Verbeek “Pathogenesis of Cerebral Amyloid Angiopathy,” Brain Research Brain Research Reviews, Vol. 43, No. 2, 2003, pp. 207-223. doi:10.1016/j.brainresrev.2003.08.001
- J. Attems, K. Jellinger, D. R. Thal and W. Van Nostrand, “Sporadic Cerebral Amyloid Angiopathy,” Neuropathology Applied Neurobiology, Vol. 37, No. 1, 2011, pp. 75- 93. doi:10.1111/j.1365-2990.2010.01137.x
- J. Kang, et al. “The Precursor of Alzheimer's Disease Amyloid A4 Protein Resembles a Cell-Surface Receptor,” Nature, Vol. 325, 1987, pp. 733-736. doi:10.1038/325733a0
- R. E. Tanzi, et al., “Amyloid ß Protein Gene: cDNA, mRNA Distribution and Genetic Linkage near the Alzheimer’s Locus,” Science, Vol. 235, No. 4791, 1987, pp. 880-884. doi:10.1126/science.2949367
- R. Vassar, et al., “ß-Secretase Cleavage of the Alzheimer’s Precursor Protein by the Transmembrane Aspartic Protease BACE,” Science, Vol. 286, No. 5440, 1999, pp. 735-741. doi:10.1126/science.286.5440.735
- B. De Strooper, B., et al., “Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein,” Nature, Vol. 391, 1998, pp. 387-390. doi:10.1038/34910
- M. S. Wolfe, et al. “Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and a Secretase Activity,” Nature, Vol. 398, 1999, pp. 513- 517. doi:10.1038/19077
- K. D. Kroncke, C. V. Fehsel and V. Kolb-Bachofen, “Implications of Inducible Nitric Oxide Synthase Expression and Enzyme Activity,” Antioxidant Redox & Signaling, Vol. 2, No. 3, 2000, pp. 585-605. doi:10.1089/15230860050192341
- D. A. Wink, et al. “Mechanisms of the Antioxidant Effects of Nitric Oxide,” Antioxidants Redox & Signaling, Vol. 3, No. 2, 2001, pp. 203-213. doi:10.1089/152308601300185179
- W. A. Pryor and G. L. Squadrito, “The Chemistry of Peroxynitrite: A Product from the Reaction of Nitric Oxide with Superoxide,” American Journal of Physiology, Vol. 268, 1995, pp. 699-722.
- P. Pacher, J. S. Beckman and L. Liaudet, “Nitric Oxide and Peroxynitrite in Health and Disease,” Physiological Reviews, Vol. 87, No. 1, 2007, pp. 315-424. doi:10.1152/physrev.00029.2006
- C. Iadecola, “Bright and Dark Sides of Nitric Oxide in Ischemic Brain Injury,” Trends in Neuroscience, Vol. 20, No. 3, 1997, pp. 132-139. doi:10.1016/S0166-2236(96)10074-6
- I. N. Mungrue, D. S. Bredt, D. J. Stewart and M. Husain, “From Molecules to Mammals: What’s NOS Got to Do with It?” Acta Physiologica Scandinavica, Vol. 179, No. 2, 2003, pp. 123-35. doi:10.1046/j.1365-201X.2003.01182.x
- U. Förstermann and W. C. Sessa, “Nitric Oxide Synthases: Regulation and Function,” European Heart Journal, Vol. 33, No. 7, 2012, pp. 829-837. doi:10.1093/eurheartj/ehr304
- C. A. Colton, et al., “NO Synthase 2 (NOS2) Deletion Promotes Multiple Pathologies in a Mouse Model of Alzheimer’s Disease,” Proceedings of the National Academy of Sciences USA, Vol. 103, No. 34, 2006, pp. 12867- 12872. doi:10.1073/pnas.0601075103
- D. M. Wilcock, et al., “Progression of Amyloid Pathology to Alzheimer’s Disease Pathology in an Amyloid Precursor Protein Transgenic Mouse Model by Removal of Nitric Oxide Synthase 2,” Journal of Neuroscience, Vol. 28, 2008, pp. 1537-1545. doi:10.1523/JNEUROSCI.1090-04.2004
- S. A. Austin, A. V. Santhanam and Z. S. Katusic, “Endothelial Nitric Oxide Modulates Expression and Processing of Amyloid Precursor Protein,” Circulation Research, Vol. 107, 2010, pp. 1498-1502. doi:10.1161/CIRCRESAHA.110.233080
- H. Oakley, et al., “Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation,” Journal of Neuroscience, Vol. 26, No. 40, 2006, pp. 10129-10140. doi:10.1523/JNEUROSCI.1202-06.2006
- K. Johnson-Wood, et al., “Amyloid Precursor Protein Processing and Aß42 Deposition in a Transgenic Mouse Model of Alzheimer’s Disease,” Proceedings of the National Academy of Sciences USA, Vol. 94, No. 4, 1997, pp. 1550-1555. doi:10.1073/pnas.94.4.1550
- R. B. DeMattos, et al., “Clusterin Promotes Amyloid Plaque Formation and Is Critical for Neuritic Toxicity in a Mouse Model of Alzheimer’s Disease,” Proceedings of the National Academy of Sciences USA, Vol. 99, No. 16, 2002, pp. 10843-10848. doi:10.1073/pnas.162228299
- J. Davis-Salinas, S. M. Saparito-Irwin, F. M. Donovan, D. D. Cunningham and W. E. Van Nostrand, “Thrombin Receptor Activation Induces Secretion and Nonamyloidogenic Processing of Amyloid ß-Protein Precursor,” Journal of Biological Chemistry, Vol. 269, 1994, pp. 22623-22627.
- D. W. Dickson, et al., “Ubiquitin Immunoelectron Microscopy of Dystrophic Neurites in Cerebellar Senile Plaques of Alzheimer’s Disease,” Acta Neuropathologica, Vol. 79, 1990, pp. 486-493.
- J. Davis, et al., “Early-Onset and Robust Cerebral Microvascular Accumulation of Amyloid ß-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant form of Amyloid ß-Protein Precursor,” Journal of Biological Chemistry, Vol. 279, 2004, pp. 20296-20306. doi:10.1074/jbc.M312946200
- J. M. Long, et al., “Stereological Estimation of Total Microglia Number in Mouse Hippocampus,” Journal of Neuroscience Methods, Vol. 84, No. 1-2, 1998, pp. 101- 108. doi:10.1016/S0165-0270(98)00100-9
- J. Miao, et al., “Cerebral Microvascular Amyloid ß-Protein Deposition Induces Vascular Degeneration and Neuroinflammation in Transgenic Mice Expressing Human Vasculotropic Mutant Amyloid ß-Protein Precursor,” American Journal of Pathology, Vol. 167, No. 2, 2005, pp. 505-515. doi:10.1016/S0002-9440(10)62993-8
- E. G. Shesely, et al., “Elevated Blood Pressures in Mice Lacking Endothelial Nitric Oxide Synthase,” Proceedings of the National Academy of Sciences USA, Vol. 93, No. 23, 1996, pp. 13176-13181. doi:10.1073/pnas.93.23.13176
- R. Deane, R. D. Bell, A. Sagare and B. V. Zlokovic, “Clearance of Amyloid-Beta Peptide across the BloodBrain Barrier: Implication for Therapies in Alzheimer’s Disease,” CNS Neurological Disorders Drug Targets, Vol. 8, 2009, pp. 16-30. doi:10.2174/187152709787601867
- M. Carpentier, Y. Robitaille, L. DesGroseillers, G. Boileau and M. Marcinkiewicz, “Declining Expression of Neprilysin in Alzheimer Disease Vasculature: Possible Involvement in Cerebral Amyloid Angiopathy,” Journal Neuropathology and Experimental Neurology, Vol. 61, 2002, pp. 849-856.
- A. Perez, L. Morelli, J. C. Cresto and E. M. Castano, “Degradation of Soluble Amyloid Beta-Peptides 1 - 40, 1 - 42 and the Dutch Variant 1 - 40 Q by Insulin Degrading Enzyme from Alzheimer Disease and Control Brains,” Neurochemical Research, Vol. 25, No. 2, 2000, pp. 247- 255. doi:10.1023/A:1007527721160
- J. R. Backstrom, G. P. Lim, M. J. Cullen and Z. A. Tokes, “Matrix Metalloproteinase-9 (MMP-9) Is Synthesized in Neurons of the Human Hippocampus and Is Capable of Degrading the Amyloid-Beta Peptide (1 - 40),” Journal of Neuroscience, Vol. 16, 1996, pp. 7910-7919.
- C. C. Liu, T. Kanekiyo, H. Xu and G. Bu, “Apolipoprotein E and Alzheimer’s Disease: Risk, Mechanisms and Therapy,” Nature Review Neurology, Vol. 9, No. 184, 2013, pp. 106-118. doi:10.1038/nrneurol.2013.32
- I. Lefterov, et al., “Apolipoprotein A-1 Deficiency Increases Cerebral Amyloid Angiopathy and Cognitive Deficits in APP/PS1dE9 Mice,” Journal of Biological Chemistry, Vol. 285, 2010, pp. 36945-36957. doi:10.1074/jbc.M110.127738
- T. L. Lewis, et al., “Overexpression of Human Apolipoprotein A-I Preserves Cognition and Attenuates Neuroinflammation and Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer’s Disease,” Journal of Biological Chemistry, Vol. 285, 2010, pp. 36958-36968. doi:10.1074/jbc.M110.127829
- H. Potter, I. M. Wefes and L. N. Nilsson, “The Inflammation-Induced Pathological Chaperones ACT and apoE Are Necessary Catalysts of Alzheimer Amyloid Formation,” Neurobiology of Aging, Vol. 22, No. 6, 2001, pp. 923-930. doi:10.1016/S0197-4580(01)00308-6
- J.N. Buxbaum, et al., “Transthyretin Protects Alzheimer’s Mice from the Behavioral and Biochemical Effects of Abeta Toxicity,” Proceedings of the National Academy of Sciences USA, Vol. 105, No. 7, 2008, pp. 2681-2686. doi:10.1073/pnas.0712197105
NOTES
*Corresponding author.