American Journal of Analytical Chemistry
Vol. 3 No. 2 (2012) , Article ID: 17285 , 7 pages DOI:10.4236/ajac.2012.32017
Fast Determination of Benzodiazepines in Human Urine via Liquid-Liquid Extraction with Low Temperature Partitioning and LC-HRMS
1Departamento de Química, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
2Departamento de Química, Universidade Federal de Viçosa, Viçosa, Brazil
3Departamento de Química, Universidade Federal de Ouro Preto, Ouro Preto, Brazil
Email: *clesia@qui.ufmg.br
Received November 2, 2011; revised December 19, 2011; accepted December 27, 2011
Keywords: Benzodiazepines; Urine; LC-HRMS; Extraction Procedure; Low-Temperature Partitioning
ABSTRACT
A simple and high-throughput method to simultaneously determine selected benzodia-zepines (i.e., diazepam, lorazepam, clonazepam, and bromazepam) in urine was developed and validated. The entire methodology consisted of the application of an innovative extraction/cleanup procedure, namely liquid-liquid extraction with low-temperature partitioning (LLE-LTP), and analysis by liquid chromatography combined with high-resolution mass spectrometry (LC-HRMS). The LLE-LTP procedure was optimized via factorial design and by evaluating crucial variables, specifically the freezing mode (either slow or fast), the urine/acetonitrile volume ratio, and the sample ionic strength. The benzodiazepines were quantified using matrix-matched calibration curves where the following parameters were assessed by validation protocol: in general, linearity range of 17 - 200 µg·L–1 (r > 0.9957); limits of detection lower than 5 µg·L–1; relative standard deviations (RSD) lower than 12.5%; and accuracy ranging from 72.3% to 117%. To test this procedure’s performance, the method was applied to determine the content of diazepam in actual urine samples. The validation results obtained for the method demonstrated that the present methodology could be potentially applied in proficient laboratories as a routine approach for determining benzodiazepines compounds content in urine.
1. Introduction
Benzodiazepines are the most widely prescribed sedative and hypnotic drugs that are used and are readily available worldwide. Their main pharmacological actions produce hypnotic, antianxiety, muscle relaxing, and anti-convulsive effects. Benzodiazepines may also induce anterograde amnesia at therapeutic doses with increasing risks at higher dosages [1]. Moreover, the misuse of these drugs can lead to intoxication and even dependence. Benzodiazepines and related drugs have also been associated with cases of crime, homicide, suicide, robbery, and sexual assault, which certainly represent a matter of major concern [2,3]. Benzodiazepines are extensively metabolized. Acid and enzyme hydrolysis of the urine specimens to convert these analytes to their free forms could be used to improve detection [3-5]. However, acidcatalyzed hydrolysis procedures used to de-conjugate benzodiazepine glucuronides during urine drug testing are known to decompose some benzodiazepines into common benzophenones [6,7]. Recently, the literature has reported that hydrolysis methods employing β-glucuronidase may produce artifacts [8]. Moreover, these procedures are time consuming, varying from 1 to 18 hours [9].
Sensitive, fast, and low-cost analytical methods are increasingly required in forensic and clinical toxicology to identify and quantify the most common benzodiazepines in different biological matrices. Urine has been the most widely used biological matrix for the determination of benzodiazepines and metabolites in drug abuse testing and toxicology. Compared to other matrices, urine exhibits several advantages, particularly its ease of collection. Because of that, most of the analytical procedures have been designed to use this matrix [10]. Among such procedures, two techniques have risen to the fore: immunoassay [11] and chromatography (gas chromatography or liquid chromatography) [4,5,12]. However, whereas gas chromatography (GC) usually requires derivatization steps that demand an inconveniently long analysis time [13], liquid chromatography (LC) does not require these steps. Furthermore, by combining a liquid chromatographer to a time-of-flight mass spectrometer (TOFMS) bearing an electrospray ionization (ESI) source, an instrument of superior performance (LC-ESI-TOFMS) is obtained. These instruments have proven to be extremely effective in the analyses of abused drugs, including benzodiazepines, not only for urinalysis but also for a number of other biological samples [14].
Different procedures that are used to extract drugs from biological fluids for analysis by LC-MS and to remove impurities contained in human body fluids have been described, including LLE (liquid-liquid extraction) [12], SPE (solid phase extraction) [4,15,16], and SPME (solid phase micro-extraction) [17]. Recently an innovative methodology, called liquid-liquid extraction with low temperature partitioning (LLE-LTP), has demonstrated excellent performance in analysis of organic contaminants in different matrices, such as water [18] and food [19]. This simple extraction procedure consists of adding a small amount of an organic solvent (usually acetonitrile) to a given aqueous sample (urine in this case) and refrigerating the resulting mixture at –20˚C for approximately 1 hour. Under these conditions, the aqueous phase solidifies, whereas the liquid supernatant, consisting mainly of the organic solvent and solubilized analytes, can be promptly isolated and subsequently analyzed. The main advantage of this procedure is that only one step is typically all that is required to obtain a clean extract.
The purpose of the present study was therefore to develop and validate a simple and fast method based on an unprecedented application of the LLE-LTP procedure and to determine the most common benzodiazepine drugs (i.e., diazepam, clonazepam, lorazepam, and bromazepam) in urine without having to go through a hydrolysis step. The extracts obtained upon the LLE-LTP procedure were analyzed in an LC-ESI-IT-TOFMS instrument, which comprises a liquid chromatographer coupled to a high-resolution mass spectrometer containing an electrospray ionization source and two sequentially arranged analyzers (i.e., an ion trap and a time-of-flight mass spectrometer).
2. Experimental
2.1. Reagents and Chemicals
Diazepam, clonazepam, lorazepam, bromazepam, and chlordiazepoxide (internal standard—IS) were all purchased from Sigma-Aldrich (St. Louis, USA). Figure 1 shows the structures of benzodiazepines evaluated in this work and of the internal standard. Stock solutions were prepared by dissolving 10 mg of a given analyte in 10 mL of methanol (Merck, Darmstadt, Germany) and were stored at –20˚C. Working solutions of these analytes were prepared by successive dilutions with acetonitrile
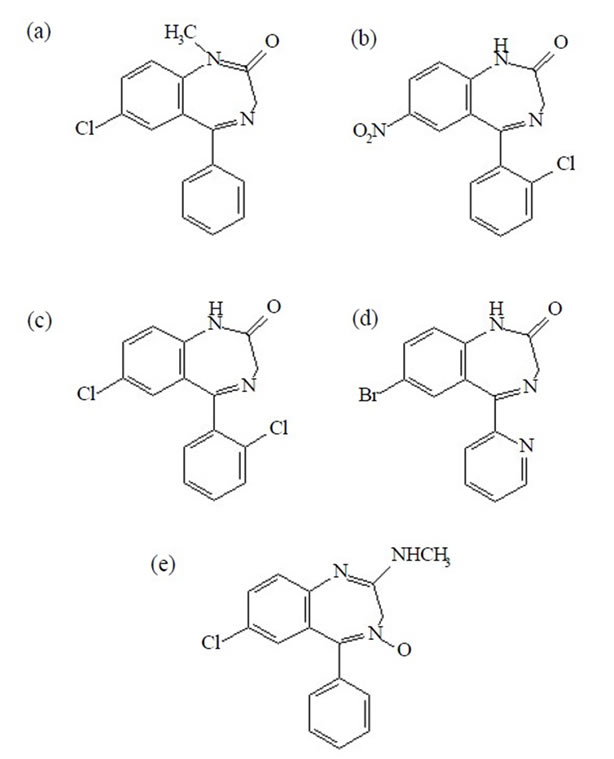
Figure 1. Structures of benzodiazepines evaluated in this work: (a) Diazepam; (b) Clonazepam; (c) Lorazepam; (d) Bromazepam; (e) Chlordiazepoxide (IS).
(Merck, Darmstadt, Germany). Other reagents used were formic acid (Synth, Diadema, SP, Brazil), ammonia (Synth, Diadema, SP, Brazil), and sodium chloride (Merck, Darmstadt, Germany). Deionized water obtained from a Milli-Q system (Millipore DirectQ 3, France) was used to prepare solutions and to perform dilutions. A solution of ammonium formate (2.0 mmol·L–1) was obtained by diluting 0.105 mL of ammonia (25 % m·v–1) and 0.154 mL of pure formic acid to a final volume of 1000 mL with deionized water.
2.2. Samples
A pool of blank human urine samples, collected from ten healthy volunteers in respective flasks, was used for the optimization and validation experiments. To evaluate the method’s overall performance, urine samples were collected from two benzodiazepine users. All of these samples were stored immediately on receipt at 4˚C. The LC analyses were performed within three days of the initial sample storage.
2.3. Instrumentation
LC analyses were conducted on a Shimadzu Prominence system that had been equipped with a high-pressure binary solvent delivery system (LC-20AD) and a SIL 20AC autosampler. The injection volume was 5 μL, and the chromatographic separation was performed on an Agilent R × C18 column (3.0 μm × 150 × 2.1 mm) maintained at room temperature. Flow rate was 0.2 mL·min–1, and the mobile phase comprised mixtures at distinct proportions of an aqueous solution of ammonium formate 2.0 mmol·L–1 (A) and acetonitrile (B). Chromatographic separation was carried out using a gradient method according to the following regimen: 30% B to 50% B in 7.5 min; hold at 50% B for 1.5 min; 50% to 100% B in 4 min; reduce to 30% B for 0.5 min and hold for another 4.5 min. The total run time was 18 min. Mass spectrometry detection was performed using a Shimadzu LC-ESI-IT-TOFMS instrument working at high-resolution and high mass accuracy (<5 ppm) under the following conditions: ESI ionization at +4.5 KV and nebulizer gas at 1.5 L·min–1, curved desorption line (CDL) interface at 200˚C, and drying gas at 100 kPa; octapole ion accumulation time of 100 ms. Full scan mass spectra from m/z 100 to 1000 were acquired with a scan time of 0.2 s. Resolution was at least 10,000 FWHM (Full Width at Half Maximum) at 500 m/z. The ion chromatogram was divided into four segments: 0 to 3.4 min, 3.5 to 8.1 min, 8.2 to 11 min and 11.1 to 18 min). High resolution scan mass spectra were recorded in all segments and a selected ion monitoring (SIM) step was realized, where the ion trap selected m/z of 300.0889 and 316.0080 (segment 2), m/z of 321.0192 and 316.0483 (segment 3) and m/z of 285.0789 (segment 4). Therefore, the TOF ion chromatogram for bromazepam, clordiazepoxide (IS), lorazepam, clonazepam and diazepam was obtained at 316.0080, 300.0898, 321.0192, 316.0483 and 285.0789 m/z, respecttively (Table 1).
2.4. Optimization of the LLE-LTP Procedure and LC-ESI-IT-TOFMS Analyses
The LLE-LTP procedure was optimized by evaluating the following variables: 1) type of freezing (1 h in a freezer at approximately –20˚C or 8 s in liquid nitrogen); 2) ionic strength of the solution (adjusted by the addition of NaCl at concentrations of 0 or 0.2 mol·L–1); and 3) urine/acetonitrile volume ratio (1:1 or 2:1). Aliquots (0.5 mL or 1.0 mL) of blank human urine samples were pipetted into 1.5 mL vials and enriched with the solutions of the benzodiazepines (i.e., diazepam, clonazepam, lorazepam, and bromazepam) to reach a final concentration of 100 μg·L–1 for each compound and with 150 μg·L–1 chlordiazepoxide (IS). The ionic strength of each solution was adjusted by adding the aforementioned amounts of NaCl followed by adding 0.5 mL of acetonitrile. The resulting mixtures were vortex-mixed for 30 s and then placed in a freezer for 1 h or submitted to freezing in liquid nitrogen for 8 s. The organic extracts were then separated from the frozen aqueous phase. In this procedure, around 0.2 mL of each organic extract was transferred to another vial with a micropipette, and 5 μL was injected into the LC-ESI-IT-TOFMS system. After acquisition, extracted-ion chromatograms were generated for each analyte by selecting the specific [M + H]+ ions (see Table 1). Extraction mass window widths that ranged from 10 to 200 mDa were employed.
2.5. Method Validation
Validation was conducted to assess the following parameters: selectivity, linearity, extraction recovery, limit of detection (LOD), limit of quantification (LOQ), accuracy, and precision. Matrix-matched (MM) calibration curves were used. For this, aliquots of blank urine samples were submitted to the LLE-LTP procedure, and the extracts were spiked with the benzodiazepine solutions to generate final concentrations of each compound at concentrations of 0, 1, 5, 10, 30, 50, 100, 150 and 200 μg·L–1. Analyses at each concentration level were performed in triplicate, and the MM calibration curves were constructed by plotting the chromatographic peak-area versus the nominal concentration of a given analyte. Linearity was investigated in a concentration range of 0 - 200 μg·L–1 and was evaluated by the coefficient of correlation (R).
To determine the extraction recovery, human urine samples (0.5 mL) were spiked with each benzodiazepine in the concentration of 100 µg·L–1 (n = 3) and submitted to the extracting procedure. Another set of samples were prepared extracting 0.5 mL aliquots of human urine and then spiking the extract with the same amount of benzodiazepines. The recovery was calculated by comparing the areas obtained before and after extraction and it was expressed as percentage of the amount extracted.
The limits of detection and quantification were estimated at signal-to-noise ratios of 3 and 10, respectively.
Precision and accuracy were evaluated by spiking human urine samples with the benzodiazepine compounds at concentrations of 30, 100 and 150 μg·L–1 (n = 5). The results obtained for precision and accuracy were expressed as relative standard deviation (RSD, %) and recovery (%), respectively.
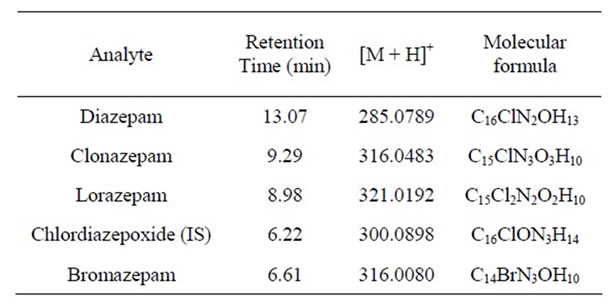
Table 1. Chromatographic and mass spectrometric parameters for each benzodiazepine compound.
2.6. Method Application
After validation, the method was applied to actual urine samples collected from two subjects undergoing benzodiazepine treatment. The optimized conditions obtained for the LLE-LTP procedure and LC-ESI-IT-TOFMS analysis were employed.
3. Results and Discussion
3.1. LC-ESI-IT-TOFMS Analyses
The development of a sensitive and selective chromatographic methodology to simultaneously determine four benzodiazepine compounds (i.e., diazepam, lorazepam, clonazepam, and bromazepam) in urine required extensive studies to obtain the composition of the mobile phase and the gradient program that yielded optimal results. This assessment is of central importance because the acquisition of sharp and intense chromatographic peaks may improve overall method accuracy. A gradient of acetonetrile/ammonium formate was therefore selected and used to verify the efficiency of the developed chromatographic method. Figure 2 shows the resulting extracted-ion chromatograms for all the analytes, obtained in segmented mode. The unequivocal determination of all analytes was facilitated by the use of a mass analyzer (TOF) with high-resolution capabilities. The ability to determine the m/z of the protonated benzodiazepines within 5 parts per million (i.e., mass assignment to four decimal places) enabled the assignment of a unique elemental formula for each compound based on the mass sufficiency of the constituent atoms (Table 1).
3.2. Extraction Method
The LLE-LTP procedure was optimized to maximize the extraction rates. To accomplish this, factorial designs were constructed, and three variables were evaluated: 1) type of freezing (1 h in a refrigerator or 8 s in liquid nitrogen); 2) ionic strength of the solution (adjusted by the addition of NaCl at 0 or 0.2 mol·L–1) and 3) urine/acetonitrile volume ratio (1:1 or 2:1). As a urine/acetonitrile volume ratio of 2:1 (1 mL of urine to 0.5 mL of acetonetrile) did not promote phase separation, a volume ratio of
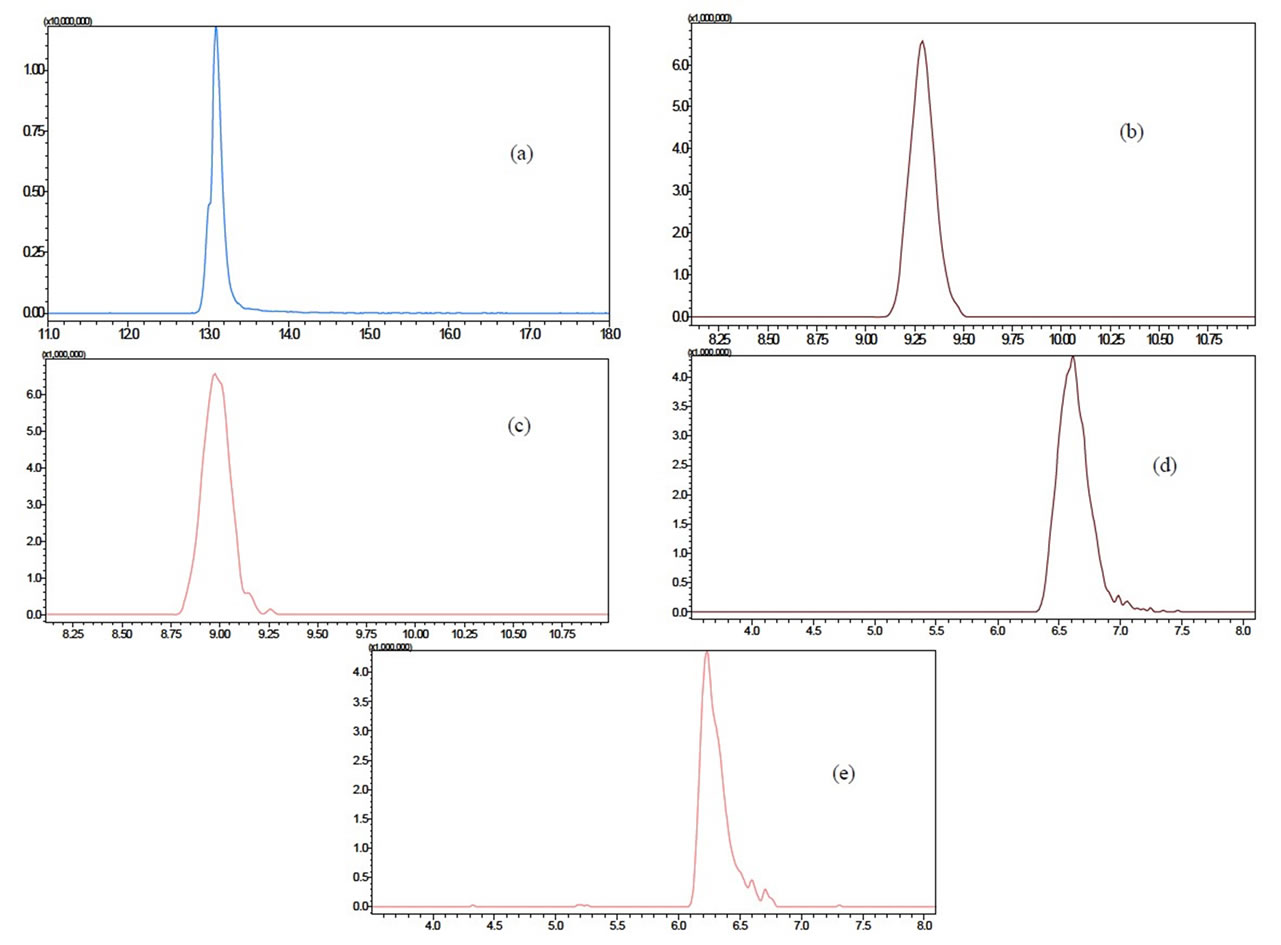
Figure 2. Extracted-ion chromatograms of a solution at 150 µg·L–1 containing the following benzodiazepines: (a) Diazepam, (b) Clonazepam; (c) Lorazepam; (d) Bromazepam; and (e) Chlordiazepoxide (IS).
1:1 was selected as the optimized value for this variable. To verify the influence of the other two variables, i.e., the type of freezing (variable 1) and the ionic strength (variable 2) a 22 factorial design was planned. The results indicated that variable 1 had a significant and negative effect. This result means that higher recoveries were obtained when the slower freezing method was applied. A longer freezing time is relevant as it probably allows the analytes to migrate more efficiently from the aqueous into the organic phase. Finally, variable 2 presented a positive effect; therefore, the increasing of the ionic strength of the urine samples (by the addition of NaCl 0.2 mol·L–1) improved the recovery rates for all benzodiazepines.
The extraction method optimized present advantages: easy to perform, low consume of acetonitrile (0.5 mL/ sample) and high-throughput (200 samples can be extracted simultaneously in 2 hours).
The optimized conditions were used to validate the entire method as further discussed below.
3.3. Method Validation
To validate the method, the following parameters were evaluated: selectivity, linearity, extraction recovery, LOD, LOQ, precision, and accuracy.
3.3.1. Selectivity
The method was shown to be selective when no peaks (>LOQ) were detected in known blank samples of urine at the retention times of the analytes (Table 1). The application of the LLE-LTP procedure, which obtained clean extracts, and the use of a high-resolution mass analyzer (TOF), which served to differentiate between compounds with slightly different masses, allowed the development of a method with high selectivity, and negligible background interference.
3.3.2. Linearity, Limits of Detection and Quantification
The suppression effect and matrix effect were previously assayed. An ion suppression effect was not present. The matrix effect was observed when comparing the slopes of two calibration curves by an F test and Student’s t test. The first curve was prepared by diluting a working solution containing all benzodiazepines in pure acetonitrile. The second calibration curve was prepared by diluting the working solution in an extract of the blank matrix. For this reason, all the calibration curves used in this work were prepared in the matrix extract. Table 2 shows the correlation coefficients (R), the slopes, and the intercepts of the MM calibration curves for all analytes at the concentration range evaluated. The correlation coefficients, curve range from LOQ to 200 µg·L–1, were satisfactory for all compounds, varying from 0.9957 for diazepam to 0.9998 for clonazepam. With the data provided by the lowest concentration level evaluated, the limits of detection and limits of quantification were estimated. The LODs ranged from 1 to 5 μg·L–1, whereas the LOQs ranged from 3.3 to 17 μg·L–1. The LODs and LOQs for diazepam, lorazepam, clonazepam, and bromazepam that were obtained from the present method were lower than previously reported results [4]. Finally, it should be emphasized that the present method is fully adequate to be applied to cases of clinical and forensic toxicology when the concentrations of benzodiazepines in urine reach values considerably higher than the LOQs obtained in this study.
3.3.3. Extraction Recovery, Precision and Accuracy
Recoveries of benzodiazepines ranged from 72.4% to 100.4% employing proposed method. Results are showed in Table 3.
The method’s precision ranged from 6.3% to 12.5% for the lowest concentration evaluated (30 μg·L–1), 4.1% to 8.6% for the middle concentration (100 μg·L–1), and 2.6% to 8.0% for the highest concentration (150 μg·L–1). These results are summarized in Table 4. According to Peters and Maurer [20], acceptable precision parameters for a given method must be below 15%. These results thus indicated that the present method is suitably precise. The accuracy for the different benzodiazepines in urine ranged from 72.9% to 117%. In general, the accuracy obtained in this work is suitably better than the values obtained from previous methods developed for a simultaneous detection of benzodiazepines [4] as well as lorazepam [13].
Acid and enzymatic hydrolyses of the urine specimens are used to convert the analytes to their free forms and to improve detection [21], but these procedures present a number of drawbacks, such as producing artifacts and
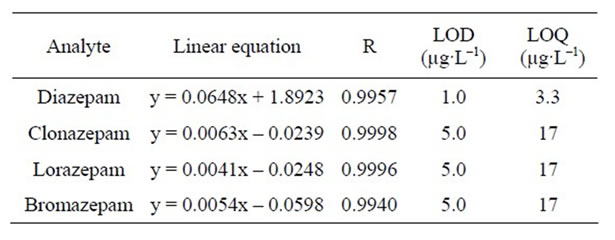
Table 2. Linear equation, correlation coefficient, limit of detection, and limit of quantification for the benzodiazepines studied.
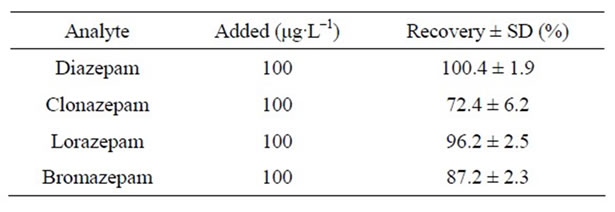
Table 3. Extraction recovery for the four benzodiazepines from human urine samples (n = 3).
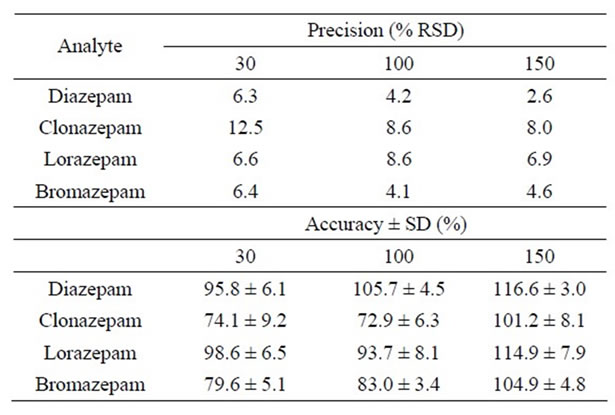
Table 4. Precision and accuracy for each analyte obtained from urine samples spiked at three concentration levels: 30, 100, and 150 µg·L−1.
consuming time [6-9]. In this work, a hydrolysis reaction was not produced; nevertheless, the proposed method presented characteristics suitable to clinical and forensic applications involving the use of benzodiazepines, where the concentration of the unchanged compound is appropriate. Demonstrating the applicability of this method, it was employed to analyze actual samples.
3.4. Method Application
The optimized method was applied to analyze urine samples collected from two subjects undergoing diazepam treatment. For the first subject, the concentration of diazepam was determined as 41 ± 3 μg·L–1. For the second subject, diazepam was quantified in a concentration of 105 ± 5 μg·L–1.
4. Conclusions
In this work a simple and high-throughput method was developed and validated to analyze benzodiazepines in urine samples. The method employed an innovative procedure, LLE-LTP, followed by analysis via liquid chromatography combined with high-resolution mass spectrometry (HRMS). The extraction method proposed not required sophisticated materials, the reagent consumption is minimal and approximately 200 samples can be extracted simultaneously in 2 hours. Acceptable LOQs, precision, and accuracy were obtained. Furthermore, this method was applied for the first time to the determination of diazepam in actual urine samples. Finally, these results clearly demonstrate that the proposed methodology is simple, rapid, and sensitive enough to be used as a routine procedure to determine the presence of benzodiazepines in urine.
5. Acknowledgements
The authors are grateful to Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) for financial support and research fellowships.
REFERENCES
- P. Xiang, Q. Sun, B. Shen, P. Chen, W. Liu and M. Shen, “Segmental Hair Analysis Using Liquid Chromatography-Tandem Mass Spectrometry after a Single Dose of Benzodiazepines,” Forensic Science International, Vol. 204, No. 1-3, 2011, pp. 19-26. doi:10.1016/j.forsciint.2010.04.046
- T. Ishida, K. Kudo, M. Hayashida and N. Ikeda, “Rapid and Quantitative Screening Method for 43 Benzodiazepines and Their Metabolites, Zolpidem and Zopiclone in Human Plasma by Liquid Chromatography/Mass Spectrometry with a Small Particle Column,” Journal of Chromatography B, Vol. 877, 2009, No. 25, pp. 2652- 2657.
- M. Hayashida, M. Takino, M. Terada, E. Kurisaki, K. Kudo and Y. Ohno, “Time-of-Flight Mass Spectrometry (TOF-MS) Exact Mass Database for Benzodiazepine Screening,” Legal Medicine, Vol. 11, Suppl. 11, 2009, pp. S423-S425. doi:10.1016/j.legalmed.2009.02.078
- P. Adamowicz and M. Kała, “Simultaneous Screening for and Determination of 128 Date-Rape Drugs in Urine by Gas Chromatography-Electron Ionization-Mass Spectrometry,” Forensic Science International, Vol. 198, No. 1-3, 2010, pp. 39-45. doi:10.1016/j.forsciint.2010.02.012
- S. Kinani, S. Bouchonnet, N. Milan and I. Ricordel, “A Sensitive and Selective Method for the Detection of Diazepam and Its Main Metabolites in Urine by Gas Chromatography-Tandem Mass Spectrometry,” Journal Chromatography A, Vol. 1141, No. 1, 2007, pp. 131-137. doi:10.1016/j.chroma.2006.12.008
- H. Maurer and K. Pfleger, “Identification and Differentiation of Benzodiazepines and Their Metabolites in Urine by Computerized Gas-Chromatography Mass-Spectrometry,” Journal Chromatography A, Vol. 422, No. 1, 1987, pp. 85-101.
- M. A. ElSohly, S. Feng, S. J. Salamone and R. Wu, “Sensitive GC-MS Procedure for the Analysis of Flunitrazepam and Its Metabolites in Urine,” Journal Analytical Toxicology, Vol. 21, No. 2, 1997, pp. 335-340.
- S. Fu, J. Lewis, H. Wang, J. Keegan and M. Dawson, “A Novel Reductive Transformation of Oxazepam to Nordiazepam Observed during Enzymatic Hydrolysis,” Journal Analytical Toxicology, Vol. 34, No. 2, 2010, pp. 243-251.
- S. Fu, A. Molnar, P. Bowron, J. Lewis and H. Wang, “Reduction of Temazepam to Diazepam and Lorazepam to Delorazepam during Enzymatic Hydrolysis,” Analytical and Bioanalytical Chemical, Vol. 400, No. 1, 2011, pp. 153-164. doi:10.1007/s00216-011-4723-y
- M. Augsburger, L. Rivier and P. Mangin, “Comparison of Different Immunoassays and GC-MS Screening of Benzodiazepines in Urine,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 18, No. 4-5, 1998, pp. 681-687. doi:10.1016/S0731-7085(98)00266-0
- K. M. Kirschbaum, F. Musshoff, A. Wilbert, J. Röhrich and B. Madea, “Direct ELISA Kits as a Sensitive and Selective Screening Method for Abstinence Control in Urine,” Forensic Science International, Vol. 207, No. 1-3, 2011, pp. 66-69. doi:10.1016/j.forsciint.2010.09.002
- K. B. Borges, E. F. Freire, I. Martins and M. E. P. B. De Siqueira, “Simultaneous Determination of Multibenzodiazepines by HPLC/UV: Investigation of Liquid-Liquid and Solid-Phase Extractions in Human Plasma,” Talanta, Vol. 78, No. 1, 2009, pp. 233-241. doi:10.1016/j.talanta.2008.11.003
- J. C. Eichhorst, M. L. Etter, N. Rousseaux and D. C. Lehotay, “Drugs of Abuse Testing by Tandem Mass Spectrometry: A Rapid, Simple Method to Replace Immunoassays,” Clinical Biochemistry, Vol. 42, No. 15, 2009, pp. 1531-1542. doi:10.1016/j.clinbiochem.2009.07.019
- D. Borrey, E. Meyer, W. Lambert, C. Van Peteghem and A. P. De Leenheer, “Simultaneous Determination of Fifteen Low-Dosed Benzodiazepines in Human Urine by Solid-Phase Extraction and Gas Chromatography-Mass Spectrometry,” Journal of Chromatography B, Vol. 765, No. 2, 2001, pp. 187-197. doi:10.1016/S0378-4347(01)00419-4
- M. K. K. Nielsen, S. S. Johansen, P. W. Dalsgaard and K. Linnet, “Simultaneous Screening and Quantification of 52 Common Pharmaceuticals and Drugs of Abuse in Hair Using UPLC-TOF-MS,” Forensic Science International, Vol. 196, No. 1-3, 2010, pp. 85-92. doi:10.1016/j.forsciint.2009.12.027
- W. Cheng, T. Yau, M. Wong, L. Chan and V. K. Mok, “A High-Throughput Urinalysis of Abused Drugs Based on a SPE-LC-MS/MS Method Coupled with an In-House Developed Post-Analysis Data Treatment System,” Forensic Science International, Vol. 162, No. 1-3, 2006, pp. 95-107. doi:10.1016/j.forsciint.2006.04.019
- I. Marchi, J. Schappler, J. Veuthey and S. Rudaz, “Development and Validation of a Liquid ChromatographyAtmospheric Pressure Photoionization-Mass Spectrometry Method for the Quantification of Alprazolam, Flunitrazepam, and Their Main Metabolites in Haemolysed Blood,” Journal of Chromatography B, Vol. 877, 2009, No. 23, pp. 2275-2283.
- S. M. Goulart, R. D. Alves, A. A. Neves, J. H. de Queiroz, T. C. de Assis and M. E. L. R. de Queiroz, “Optimization and Validation of Liquid-Liquid Extraction with Low Temperature Partitioning for Determination of Carbamates in Water,” Analytica Chimica Acta, Vol. 671, No. 1-2, 2010, pp. 41-47. doi:10.1016/j.aca.2010.05.003
- S. M. Goulart, M. E. L. R. Queiroz, A. A. Neves and J. H. Queiroz, “Low-Temperature Clean-Up Method for the Determination of Pyrethroids in Milk Using Gas Chromatography with Electron Capture Detection,” Talanta, Vol. 75, 2008, No. 5, pp. 1320-1323.
- F. T. Peters and H. H. Maurer, “Bioanalytical Method Validation and Its Implications for Forensic and Clinical Toxicology—A Review,” Accreditation and Quality Assurance, Vol. 7, No. 11, 2002, pp. 441-449. doi:10.1007/s00769-002-0516-5
- D. S. Ming and J. Heathcote, “Rapid and Accurate UPLC/MS/MS Method for the Determination of Benzodiazepines in Human Urine,” Journal of Chromatography B, Vol. 879, No. 5-6, 2011, pp. 421-428.
NOTES
*Corresponding author.