Open Journal of Rheumatology and Autoimmune Diseases
Vol.3 No.1(2013), Article ID:27908,5 pages DOI:10.4236/ojra.2013.31005
Sicca to Lymphoma: Sjogren Syndrome
![]()
Department of Internal Medicine and Rheumatology, “Sf. Maria” Clinical Hospital, University of Medicine and Pharmacy “Carol Davila”, Bucharest, Romania.
Email: lauraheretiu@yahoo.com, d.predet@yahoo.com
Received August 7th, 2012; revised September 12th, 2012; accepted September 27th, 2012
Keywords: Autoimmune Disease; Prognosis Factors; Non Hodgkin Lymphoma
ABSTRACT
Sjogren Syndrome (SS) is an autoimmune disease characterized by a lymphocytic infiltration of exocrine glands. This abnormality leads to the destruction of these glands with the clinical expression of the sicca syndrome (dry eyes and dry mouth). Usually Sjogren Syndrome follows a non-aggressive course, but patients suffering from this disease have a very high risk of developing malignant lymphoma (MALT). The evolution from benign lymphocytic infiltration characteristic of Sjogren Syndrome to malignant lymphoma (usually MALT subtype) is a multistep process [1-3]. We present the case of 35 years old woman admitted to “Sf. Maria” Clinical Hospital—Bucharest, Romania, in the Rheumatology Department in May 2011, representative for the diagnosis difficulties as well as for the unfortunate evolution. She was diagnosed with primary Sjogren Syndrome in 2004. The first symptoms of the disease, dry eyes and dry mouth, appeared in 2000 but they were undermined by some hematological disorders, as the patient had a low white blood cell count at the biological evaluation. Four years later, when the diagnosis of Sjogren Syndrome was set, she already had some negative prognosis factors. In the end, the disease had an unfavorable outcome, as the patient developed nonHodgkin malignant lymphoma for an unfortunate evolution.
1. Introduction
Sjogren Syndrome (SS) is an autoimmune disease characterized by a lymphocytic infiltration of exocrine glands leading to a progressive destruction of these glands and resulting in their dysfunction. The clinical expression of this abnormality is the sicca syndrome (dry eyes and dry mouth). Sjogren Syndrome generally follows a non-aggressive course, but development of malignant lymphoma may occur. Patients with Sjogren Syndrome have an increased risk of developing lymphoma of up to 44 times when compared with the general population. In most cases with this complication, there is a low grade B cell lymphoma of the mucosa associated lymphoid tissue (MALT). The evolution from benign lymphocytic infiltration characteristic of Sjogren Syndrome to malignant lymphoma is a multistep process: polyclonal gammopathy, monoclonal gammopathy, MALT lymphoma and malignant lymphoma [1-3].
2. Case Report
We present the case of a 35 years old woman diagnosed with Sjogren Syndrome in 2004.
The onset of the disease was in 2000, when the first symptoms appeared (dry eyes, dry mouth and fatigue). The symptoms were undermined by biological evaluation: low white blood cell count- 3600/mmc (normal range 4000 - 9000/mmc), modified electrophoresis with low albumin 49.2% (normal range 52% - 66%) and high gamma globulins 24.2% (normal range 12% - 22%), low level of complement (C3 and C4), positive RF and antinuclear antibodies but with normal value of the serum proteins (7.86 g/dl). At that moment there weren’t sufficient diagnosis criteria for an autoimune disease. The biological profile indicated towards a hematological disorder, therefore a bone marrow biopsy was performed. The result showed only a benign lymphocytic infiltration.
In 2004, based on the persistent symptoms and on the biological abnormalities (dry eyes, dry mouth, inflammatory syndrome, high levels of anti R0 (SSA), LA (SSB) antibodies, positive Schirmer test), the patient was diagnosed with Sjogren Syndrome and she started treatment with Plaquenil and corticosteroids.
In December 2004, the patient had an agressive episode of arthralgia and fatigue. Upon examination of blood samples, the patient showed a very low white blood cell count 1800/mmc (normal range 4000 - 8000/mc), anemia Hb 10.45 g/dl, (normal range 12 - 14 g/dl), very high levels of RF 270 u/ml, (normal range <50 u/ml), hipergammaglobulinemia and inflammatory syndrome with C reactive protein of 50 mg/dl (normal range 3 - 10 mg/dl) and ESR 17 mm/h (normal range 2 - 20 mm/H). Considering the low white blood cell level and the anemia, the possibility of developing a blood neoplasia was again taken into account. Therefore, a second bone marrow biopsy was performed and it revealed a benign aspect of a 10% lymfoplasmocitic infiltrate. At that moment, the biological and clinical follow up was considered as being a relapse of the high activity of the Sjogren Syndrome.
In 2005, next to the clinical and biological findings, consistent since the previous evaluation, some small laterocervical masses were found upon the clinical examination. Anti RO (SSA), Anti LA (SSB) antibodies and RF were positive and the complement level was low. The protein electrophoresis did not show any modifications from the previous one. The new clinical elements suggested that the disease progressed; therefore the next step that was taken was conducting a salivary gland biopsy. The biopsy revealed diffuse infiltrations with portioned ductal and acinar atrophy and nodules, as well as benign epithelial lymphatic lesions. This was a sign of the high activity of the disease. A third bone marrow biopsy was also performed and it revealed a 10% lymfoplasmocitic infiltration (benign aspect).
During the same year some purpuric lesions appeared on the legs. The biopsy of the lesions highlighted the diagnosis of leucocytoclasic vasculitis.
Between 2005-2011, the patient constantly presented low white blood cell levels, discontinuous inflammatory syndrome, high levels of anti RO (SSA) and anti LA (SSB) antibodies. The immune activity maintained with low C3 and C4, and high levels or G immunoglobulin. During this time the patient carried one pregnancy to term without any complications, and was under treatment with corticosteroids and Plaquenil (discontinued during the pregnancy)
In 2011, she was admitted to our hospital as she had developed new lateral cervical masses, increased in dimension and swollen. She also accused the presence of arthralgia in the small joints of the upper limbs and vasculitic lesions on the lower limbs. The clinical examination revealed the presence of non-painful lateral cervical masses (2 - 3 cm in dimensions) (see Figure 1), as well as purpura on the lower limbs (see Figure 2).
The laboratory exams showed moderate inflammation (ESR 47 mm/hnormal range 2 - 20 mm/h), positive anti RO (SSA) antibodies and positive antinuclear antibodies and negative anti LA (SSB) antibodies. The immune electrophoresis showed an increased level on the immunoglobulin M fraction, as well as a high level of cryoglobulins. The white blood cell count was normal. The tests for hepatitis B and C were also negative. The electrophoresis aspect was modified from the last one,

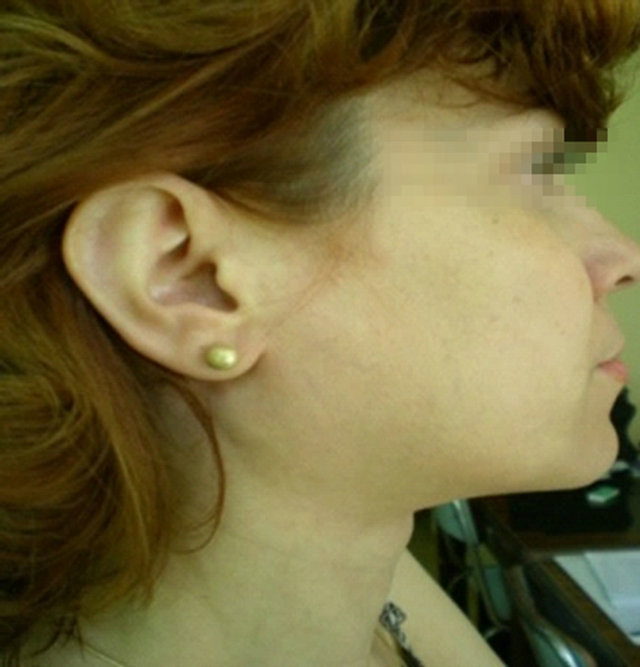
Figure 1. Lateral cervical masses.
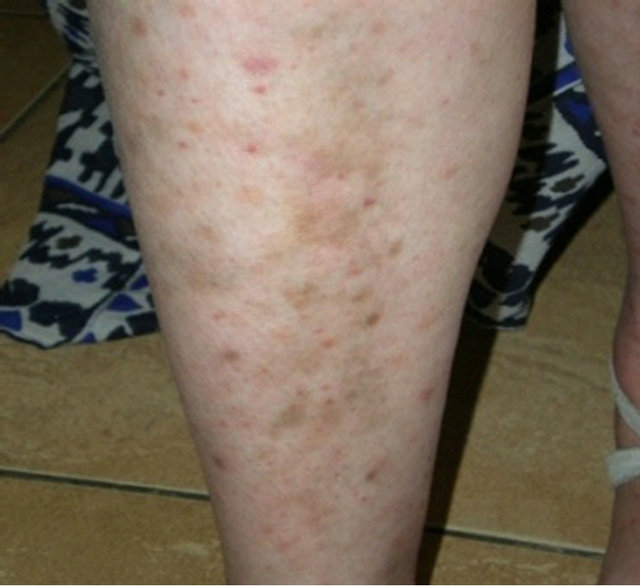
Figure 2. Purpura on the lower limbs.
showing that there was a new disturbance in the immune balance. In order to complete the investigations and taking into account the clinical context, we performed the biopsy of the lateral cervical masses. The biopsy result, with histological and immunohistochemical evaluation revealed the presence of severe small B cell infiltration (marginal type) with plasmocitic infiltration and Kappa chains secretion, with high proliferation index (see Figure 3). The diagnosis was of a diffuse Non-Hodgkin MALT lymphoma, with B type small cell. After we established the diagnosis, we directed the patient towards a hematology unit.
In the hematology unit, some tests were taken in order to establish the spread of the disease:
• The bone marrow biopsy showed only a reactive lymphocytic infiltration (2% - 3%).
• The thoraco-abdominal CT showed small lymph nodes under and over the diaphragm.
The 2 test established stage III non Hodgkin lymphoma and the patient started the treatment with Rituximab (6 monthly courses combined with Vincristine). The course of the disease was favorable with the regression of the adenopathies and with the maintenance of the hematological parameters within normal range. After finishing the treatment with Rituximab, she started again the treatment with low dose of corticoteroids, which she also follows also in the present, with a good clinical and biological evolution.
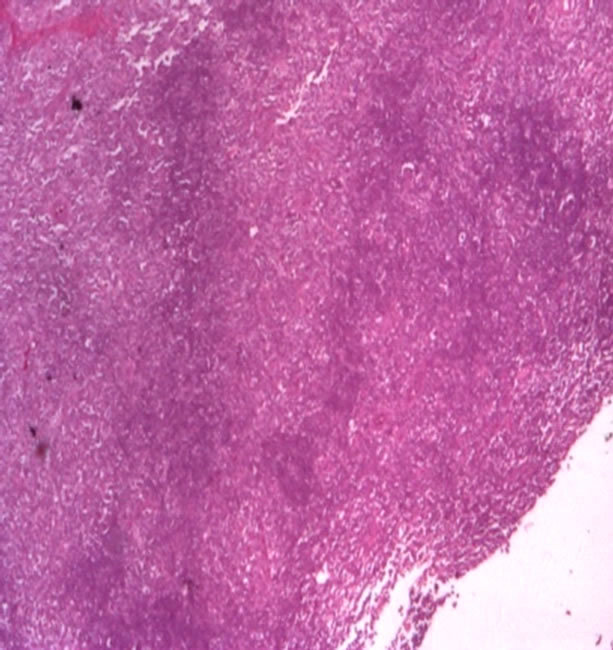 (a)
(a) (b)
(b)
Figure 3. (a) Severe lymphocytic small B cell infiltration (ME); (b) Lymphocites with lambda fraction.
3. Discussions
Sjogren Syndrome (SS) is an autoimmune disease characterized by a lymphocytic infiltration of exocrine glands leading to a progressive destruction of the architecture of these glands and resulting in their dysfunction. The clinical expression of this abnormality is usually the sicca syndrome (dry eyes and dry mouth) but the disease onset may also be followed by symptoms showing the involvement of other internal organs (lung, liver, kidneys, pancreas, pharynx, esophagus, heart) and skin [1,2,4].
The etiology and pathogenesis is not very clear: there is a combination of environmental factors and genetic predisposition (HLAB8/HLADR3, type-1 interferon-regulated genes). There is also a combination between immunologic and nonimmunologic mechanisms. Some studies show the persistence of CMV and EBV viral fragments in the salivary glands of patients with Sjogren Syndrome. Also, retroviruses such as HIV can cause symptoms very similar to Sjogren Syndrome. Moreover, a small study showed an improvement of clinical manifestations in patients with Sjogren Syndrome treated with Zidovudine. The cells infiltrating the glands (Tand Bcells, dendritic cells) and their interactions, have multiple functions: periductal infiltration and fibrosis, cell-mediated destruction of glandular elements, secretion of cytokines, production of autoantibodies to the ribonucleoprotein particles SS-A/Ro and SS-B/La and other that interfere with muscarinic receptors and secretion of metalloproteinases (MMPs). Nonimmune mechanisms include local and systemic androgen deficiency and autonomic nervous system dysfunction [5-7].
For the positive diagnosis of primary Sjogren Syndrome, 4 of 6 from the diagnosis criteria are required (see Table 1). Either criterion number 5 or criterion number 6 must be included [5,8].
*Our patient had 4 of the 6 diagnosis criteria for primary Sjogren Syndrome: dry eyes, dry mouth, persistent high levels of anti RO (SSA), LA (SSB) antibodies and positive Schirmer test.
There are 2 types of Sjogren Syndrome: primary and secondary (which accompanies other autoimmune disease: SLE, Rheumatoid Arthritis, Biliary cirrhosis, etc).
Table 1. Revised international classification criteria for Sjögren’s Syndrome [8].

For the positive diagnosis of secondary Sjogren Syndrome, symptoms of oral or ocular dryness in addition to criterion 3, 4, or 5 are required, all in the presence of a connective-tissue disease (see Table 2) [7,8].
• *Exclusion criteria:
• Past head and neck radiation treatment
• Hepatitis C infection
• Acquired immunodeficiency disease
• Pre-existing lymphoma
• Sarcoidosis
• Graft versus host disease
• Use of anticholinergic drugs (since a time shorter than 4-fold the half-life of the drug) [8]
At the onset of the disease, the patient had sicca syndrome (dry eyes, dry mouth). These symptoms were initially not sufficient neither for the diagnosis of Sjogren Syndrome nor for other autoimmune disease (such as SLE), therefore the biological profile rather pointed towards a hematological disorder (low white blood cell
Table 2. Revised rules for Sjogren Syndrome classification [8].

count) developed on an autoimmune field (positive RF and anti-nuclear antibodies). From that moment on, that was a negative prognosis factor.
The delay of the diagnosis is not unusual as the symptoms which appear at the onset of the disease are usually undermined by other abnormalities. When the diagnosis of Sjogren Syndome was established (4 years after the onset of the first symptoms) the patient had already had some negative biological abnormalities that predicted its evolution towards lymphoma. In most cases Sjogren Syndrome has a less aggressive course but about 5 percent of people with this disease develop malignant hematological disorders. [8] This unfortunate evolution happens when the autoimmune disorder is aggressive. We must take into account that at the onset of the sicca syndrome there was an aggressive immune disorder. This hypothesis is sustained by the aspect of the electrophoresis with high gamma globulins, low level of complement, positive RF anti-nuclear antibodies. There were no apparent lymph nodes present (clinically) but the presence of molecular abnormalities in the lymphatic organs couldn’t be excluded.
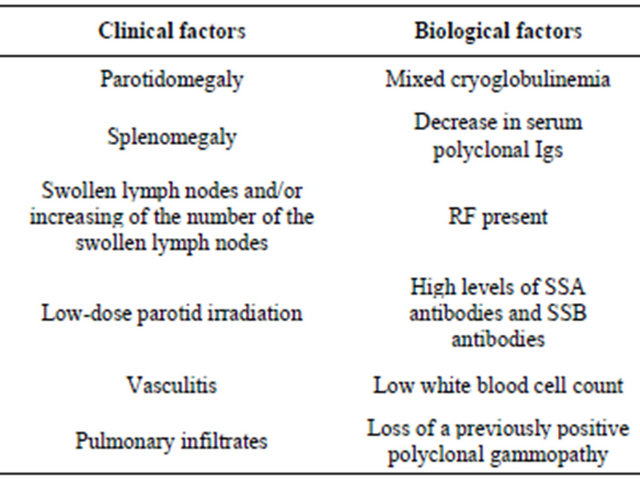
There were a few negative prognosis factors for the developing of the lymphoma (see Table 3). In our case some of these prognosis factors developed at the onset of the disease (high levels of rheumatoid factor, anti RO (SSA) and anti LA (SSB) antibodies, low levels of white blood cells, low C3 and C4 ), while others appeared during the disease follow up (vasculitis, adenopathy, lateral cervical masses, cryoglobulines). Even if the lateral cervical mass biopsy and the bone marrow biopsy had a benign aspect at that moment, these new elements correlated with the biological findings predicted a negative evolution of the disease. This is why attentive clinical examination and periodical evaluation of the negative prognosis factors are needed in patients with Sjogren Syndrome. [9]
The data given above suggests that the development of
Table 3. Negative prognosis factors of Sjogren Syndrome [9].
malignant lymphoma in Sjogren Syndrome patients is a multistep process: polyclonal gammopathy, monoclonal gammopathy, MALT lymphoma and malignant lymphoma. This process involves interactions between epithelial cells, T cells, and B cells and the adhesion molecules. B cells activation, together with oncogenic alterations, including p53 inactivation (by anti p53 antibodies) and bcl- 2 activation (by rearrangement), as well as the presence of EB virus DNA and HTLV specific genes may play important roles in B cell monoclonal proliferation and malignant transformation. The association between the virus presence and lymphoma needs further studies as it is not well established [3,10].
The most common symptom of lymphoma is a painless swelling of the lymph nodes in the neck and/or underarm but there are other symptoms that must be taken into account: fever, unexplained fatigue, nausea, early satiety. They might be a possible sign of extra salivary dissemination. The mucosa-associated lymphoid tissue (MALT) lymphoma, the most common histological subtype of non-Hodgkin lymphoma in Sjogren Syndrome, most commonly develops in the salivary glands but it can develop in any non-lymphoid tissue stomach, nasopharynx, skin, liver, and lungs. Performing labial small gland biopsies is considered to be one of the most important tools in early diagnosis of MALT lymphomas derived from Sjogren Syndrome [1,9].
Our case is highly representative for the clinical and biological profile as well as for the negative course and negative prognosis factors. Any clinical modification of the salivary glands or of the ganglions accompanied by biological alterations should be attentively examined as they might be the sign of a malignant evolution.
• 4. Conclusions
• Sjogren Syndrome is an autoimmune disease with a multifactorial etiology.
• The diagnosis of Sjogren Syndrome is often delayed by other biological or clinical manifestations.
• The disease usually follows a benign course. About 5 percent of the people diagnosed with this disease develop hematologic malignancies.
• There are a few negative prognosis factors important for the negative course of the disease: high levels of rheumatoid factor and anti RO (SSA), and anti LA (SSB) antibodies, low C3 and C4 vasculitis, adenopathy, lateral cervical masses, cryoglobulines, parotidomegaly, splenomegaly, vasculitis, pulmonary infiltrates, mixed cryoglobulinemia, low blood white cell count.
• In patients with negative prognosis factors, a very attentive follow up is mandatory.
Our case is highly representative both for the delay of the diagnosis, the negative prognosisi factors and the negative course of the diasease
REFERENCES
- N. M. Van Mello, S. R. Pillemer, P. P. Tak, et al., “B Cell MALT Lymphoma Diagnosed by Labial Minor Salivarygland Biopsy in Patients Screened for Sjo¨Gren’s Syndrome,” Annals of Rheumatic Disease, Vol. 64, No. 3, 2005, pp. 471-473. doi:10.1136/ard.2004.022707
- R. Ionescu, et al., “Esentialul in Reumatologie,” Cap.19, 2nd Revised Edition, Daniela Opris, Amaltea, Bucharest, 2007, pp. 270-279.
- B. Royer , D. C. Hatem and J. Sibilia, “Lymphomas in Patients with Sjögren’s Syndrome Are Marginal Zone BCell Neoplasms, Arise in Diverse Extranodal and Nodal Sites, and Are Not Associated with Viruses,” Blood, 15, Vol. 90, No. 2, 1997, pp. 766-775.
- A. V. Miller, S. K. M. Ranatunga, M. L. Francis, et al., Medscape Reference (Durgs, Diseases &Procedures), Last Updated 31 October 2011.
- M. Garcia Carrasco, S. Fuentes Alexandro and R. O. Escarcega, “Pathophysiology of Sjögren’s Syndrome,” Archives of Medical Research, Vol. 37, No. 8, 2006, pp. 921-932. doi:10.1016/j.arcmed.2006.08.002
- J. Maggie, “A Short Review of the Pathogenesis of Sjogren Syndrome,” Larche Autoimmunity Reviews, Vol. 5, No. 2, 2006, pp. 132-135.
- P. N. Nikolay and G. I. Gabor, “Pathogenesis of Sjogren’s Syndrome,” Current Opinion in Rheumatology, Vol. 21, No. 5, 2009, pp. 465-470.
- C. Vitali, S. Bombaridieri. R. Jonsson, et al., “Classification Criteria for Sjögren’s Syndrome: A Revised Version of the European Criteria Proposed by the AmericanEuropean Consensus Group,” Annals of Rheumatic Disease, Vol. 61, No. 6, 2002, pp. 554-558. doi:10.1136/ard.61.6.554
- P. B. Zeron, M. Ramos-Casals, A. Bove., et al., Predicting Adverse Outcomes in Primary Sjögren’s Syndrome: Identification of Prognostic Factors,” Rheumatology, Vol. 46, No. 8, 2007, pp. 1359-1362.
- Y. Masaki and S. Sugai, “Lymphoproliferative Disorders in Sjögren’s Syndrome,” Autoimmunity Reviews, Vol. 3, No. 3, 2004, pp. 175-182.