Open Journal of Medicinal Chemistry
Vol.2 No.4(2012), Article ID:26238,10 pages DOI:10.4236/ojmc.2012.24017
Computer-Aided Drug Design: An Innovative Tool for Modeling
Medicinal Chemistry Research Laboratory, JSPM’s Charak College of Pharmacy & Research, Pune, India
Email: *rishiantre@gmail.com
Received October 1, 2012; revised November 5, 2012; accepted November 15, 2012
Keywords: CADD; HTS; Software for General Purpose Molecular Modeling; SBDD
ABSTRACT
Strategies for CADD vary depending on the extent of structural and other information available regarding the target (enzyme/receptor) and the ligands. Computer-aided drug design (CADD) is an exciting and diverse discipline where various aspects of applied and basic research merge and stimulate each other. In the early stage of a drug discovery process, researchers may be faced with little or no structure activity relationship (SAR) information. The process by which a new drug is brought to market stage is referred to by a number of names most commonly as the development chain or “pipeline” and consists of a number of distinct stages. To design a rational drug, we must firstly find out which proteins can be the drug targets in pathogenesis. In present review we reported a brief history of CADD, DNA as target, receptor theory, structure optimization, structure-based drug design, virtual high-throughput screening (vHTS), graph machines.
1. Introduction
All the world’s major pharmaceutical and biotechnology companies use computational design tools. At their lowest level the contributions represent the replacement of crude mechanical models by displays of structure which are a much more accurate reflection of molecular reality capable of demonstrating motion and solvent effects [1-3]. Beyond this, theoretical calculations permit the computation of binding free energies and other relevant molecular properties. The theoretical tools include empirical molecular mechanics, quantum mechanics and, more recently, statistical mechanics. This latest advance has permitted explicit solvent effects to be incorporated. Underpinning all this work is the availability of high quality computer graphics, largely supported on workstations [1-5].
Two distinct categories of research are clearly distinguishable:
1) Crystallography, NMR or homology modelling. A detailed molecular structure of the target macromolecule, the drug receptor, is known from x-ray.
2) Variable activity of otherwise similar molecules.
The target receptor binding site has properties which can only be inferred from a knowledge of the both these types of approach will now be considered and illustrated with some recent examples.
DNA as Target
The sequencing of the human genome represents one of the major scientific endeavours of this century. A major aspect of the utilization of this information will be the provision of small molecules which will recognize selected sequences, perhaps with the goal of switching off particular genes as in cancer chemotherapy. For some time, antibiotics such as netropsin have been known to bind preferentially to sequences rich in A-T pairs. A variant based on this research has been to try to design a bioreductive ligand based upon netropsin [1]. The idea of bioreductive anti-cancer agents, statts with the fact that tumours receive less blood and hence less oxygen than normal tissue.
A second starting point for sequence selective ligands is an organometallic molecule with chiral properties. The propeller-like ruthenium tris-phenanthroline complexes do show differential binding between A-T and G-C sequences [2] and moreover may exhibit a preference for purine 3’, 5’ pyrimidine sites in DNA [3]. Perhaps the most intriguin starting point for a molecule upon which to build nucleic acid selectivity is the ubiquitous spermine. It has been proposed [4] that spermine can bind to DNA in a cross-groove manner, with relatively non-specific interactions between the positive nitrogens of the spermine and the negatively-charged phosphate backbone.
The Ras family of protooncogenes (N-Ras, H-Ras and K-Ras) codes for small proteins of 189 amino acids with molecular weight 21 kDa protein [5]. Ras proteins are localized in the inner plasma membrane and are involved in the transduction of external stimuli to effect molecule Raf serine/threonine kinase [6]. These proteins bind GDP/ GTP and possess intrinsic GTPase activity allowing inactivation following signal transduction in the normal cellular environment [7]. Activation of point mutations in the Ras is one of the most frequent genetic alterations associated with human cancers [8]. Approximately 90% of these activating mutations occur in codons 12 and 59, identifying these codons as hot-spot targets [9]. A particular genetic alteration has been identified in a significant percentage of bladder tumors; this mutation changes a single amino acid in the H-Ras protein. Specifically, the mutation replaces the amino acid glycine with the amino acid valine at position 12 (RasG12V) [10]. The mutant form of RasA59T, which is known to undergo autophosphorylation on Thr-59 [11], shows a very strong signal that argues against the occurrence of a covalently bound phosphate [8]. As a result of these mutational changes, the mutated Ras-p21 has a structure that disables its ability to bind with GTPase activating protein (GAP) and creation of an autophosphorylation site [12], thus keeping the Ras-p21 in the GTP-bound, activated state contributing to a malignant cell phenotype . Drug target discovery involves the identification and early validation of disease-associated targets. Mutations occurring in the Ras gene(s) lead to uncontrolled cell growth and proliferation. In general, 30% of human tumor occurs through mutation in Ras gene. In colorectal and pancreatic cancers, the occurrence of mutation in Ras is 50% - 90% [5]. When we consider treatments for cancer, they depend on the types and stages of cancer development. Chemotherapy, targeted therapies, surgery, radiation therapy, biological therapy, and hormonal therapy are the various treatments that currently exist.
2. A Brief History of CADD [9]
1900: The receptor and lock-and-key concepts P. Ehrich (1909) and E. Fisher (1894);
1970s: Quantitative structure-activity relationships (QSAR), Limitations: 2-Dimensional, retrospective analysis;
1980s: Beginning of CADD Molecular Biology, X-ray crystallography, multi-dimensional NMR Molecular modeling, computer graphics;
1990s: Human genome Bioinformatics, Combinatorial chemistry, High-throughput screening.
2.1. How Does CADD Work? [13]
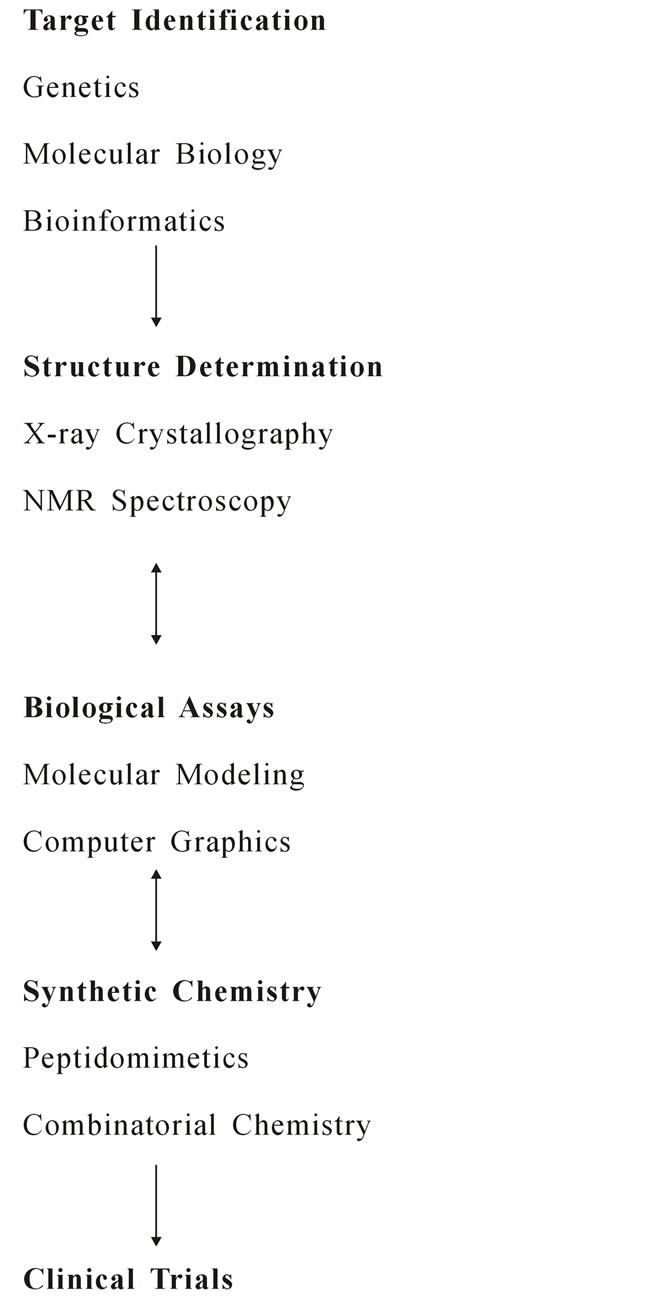
2.2. Software for Molecular Modeling
General purpose molecular modeling (large & small molecules)
—molecular mechanics, dynamics and multifunctional programs;
Quantum Chemistry calculations (small molecules)
—molecular orbital or quantum mechanical calculation;
Database of molecular structures (large & small molecules)
—software for storage and retrieval of molecular structure data;
Molecular graphics (large & small molecules)
—programs to visualize molecules QSAR (small molecules).
2.3. Software for General Purpose Molecular Modeling [13]
For workstations, minicomputers, and supercomputers (SGI, Sun, Cray, etc.)
AMBER—Peter Kollman and coworkers, UCSF.
Computer assisted model building, energy minimization, molecular dynamics, and free energy perturbation calculations.
Midas Plus—UCSF Computer Graphics Laboratory.
CHARMM—Martin Karplus and cowrokers, Harvard.
QUANTA/CHARMm—Molecular Simulations Inc. (MSI) molecular/drug design, QSAR, quantum chemistry.
X-ray & NMR data analysis Insight/DISCOVER— Biosym, Inc. Now MSI and Biosym became Accelrys Inc.
SYBYL—Tripos, Inc.
ECEPP—Harold Scheraga and coworkers, Cornell MM3—Norman Allinger and coworkers, Georgia For personal computers (Apple, Compaq, IBM, etc.)
Alchemy III—Tripos, Inc.
Structure building and manipulation, SYBYL energy minimization, molecular display, conformational searching Chem3D Pro—CambridgeSoft Corp.
Desktop Molecular Modeller—Oxford Elec. Publishing Molecular Modeling Pro—WindowChem Software Energy minimization, QSAR (surface area, volume, logP), etc.
PC MODEL—Serena Software.
2.4. Optimization [14]
The second step of drug discovery involves the modification of the hits in order to improve the biological properties of the compound by changing its pharmacophore. Using QSAR to modify lead compounds would be less tedious then having to physically synthesize the compounds. Moreover, such in silico methods could theoretically help to modify the compounds to exhibit the most potency, most selectivity, best pharmacokinetics and least toxicity. QSAR involves mainly physical chemistry and molecular docking tools that lead to tabulated data and first and second order equations. There are many theories, being the most relevant Hansch’s analysis that involves Hammett electronic parameters, Esteric parameters and logP parameters [15].
2.5. Receptor Theory
A receptor [16], in the biochemistry context, is a/are protein molecule(s), found in either the plasma membrane or the cytoplasm of a cell, to which one or more specific kinds of signaling molecules may attach. A molecule which attaches to a receptor is called a ligand, and may be a peptide or other small molecule, such as a neurotransmitter, a hormone, a pharmaceutical drug, or a toxin. Each kind of receptor can bind only certain ligand shapes. Each cell typically has many receptors, of many different kinds.
An agonist is a drug that binds to a receptor of a cell and triggers a response by the cell. An agonist often mimics the action of a naturally occurring substance. An agonist produces an action. An antagonist blocks an action of an agonist. Endogenous (such as hormones and neurotransmitters) or exogenous (such as drugs) agonists and antagonists, either stimulate or inhibit a biological response in receptors [17].
3. CADD Strategies in the Drug Discovery Process
Strategies for CADD vary depending on the extent of structural and other information available regarding the target (enzyme/receptor) and the ligands. “Direct” and “indirect” design are the two major modeling strategies currently used in the drug design process. In the indirect approach the design is based on comparative analysis of the structural features of known active and inactive compounds. In the direct design the three-dimensional features of the target (enzyme/receptor) are directly considered.
3.1. CADD in Lead Generation
3D Structure of the Protein Unknown
In the early stage of a drug discovery process, researchers may be faced with little or no structure activity relationship (SAR) information. At this point, assay development and screening should be undertaken immediately by the high-throughput screening (HTS) group [18], and chemists should immediately follow up on any screening leads or other sources of initial information. The compounds screened could be commercially available, natural products, collections of in-house synthesized compounds or emerge from combinatorial libraries. Computational chemists can, however, help in the choice of the compounds to be selected for HTS.
Instead of performing random screening, a set of compounds presenting diversity in their physicochemical properties can be selected to find leads. The aim of these analyses is to select and test fewer compounds, whilst gaining as much information as possible about the dataset [19]. Any reduction of the number of compounds to be tested, while only reducing the amount of redundancy within a database without introducing any voids, should have an important impact on research efficiency and the costs associated [20]. Recently, the use of rational design to maximize the structural diversity of database, for lead findings and refinements, was investigated. Hierarchical clustering and maximum dissimilarity methods were compared to a random approach in order to study their efficiency for the diversity enhancement of three-dimension databases. The investigations were done using twodimensional fingerprints as a validated molecular descriptor and the performance of rational selection methods vs. random approach has been compared [18,19,21-23].
The first step to derive a new lead, also called seconddary lead, will be to study the stereoelectronic properties of the selected primary leads Table 1. The primary leads should be selected among a set of compounds showing a large variety in chemical structures, and interact with the same target via the same binding mechanism. By comparison of the stereoelectronic properties of primary leads, a pharmacophore is defined. A pharmacophore model is a spatial arrangement of atoms or functional groups believed to be responsible for biological activity [24]. In this model the rest of the molecule acts as a skeleton to hold the groups in the right place. Typically, the derived pharmacophores consist, generally, of 3 - 5 features, and the distances between them (angles and other geometric measures are sometimes used).
3.2. Structure-Based Drug Design
Within many of the rational drug design projects in the group, computer-aided methods, such as virtual screening and de novo design techniques, play an important role. NMR spectroscopy in conjunction with molecular modeling and other spectroscopic methods allows investtigations to be made into molecular mechanisms of ligand-target recognition at the atomic level [25]. This information is a necessary component in the design of novel therapeutics and in prediction of interactions of drugs with the targets. Also over the years, the group hasstudied details of binding of ligands to the minor groove of DNA, such as Hoechst 33,258, or to tRNA [26]. NMR methods are also used by the group to study interactions of proteins with ligands. There is 300 MHz instrumentation in the school, and the group has shared usage of 500 MHz high-field instruments housed in the Department of Chemistry. The group collaborates extensively with Professor Gareth Morris, inventor and pioneer of many modern NMR techniques, thereby bringing
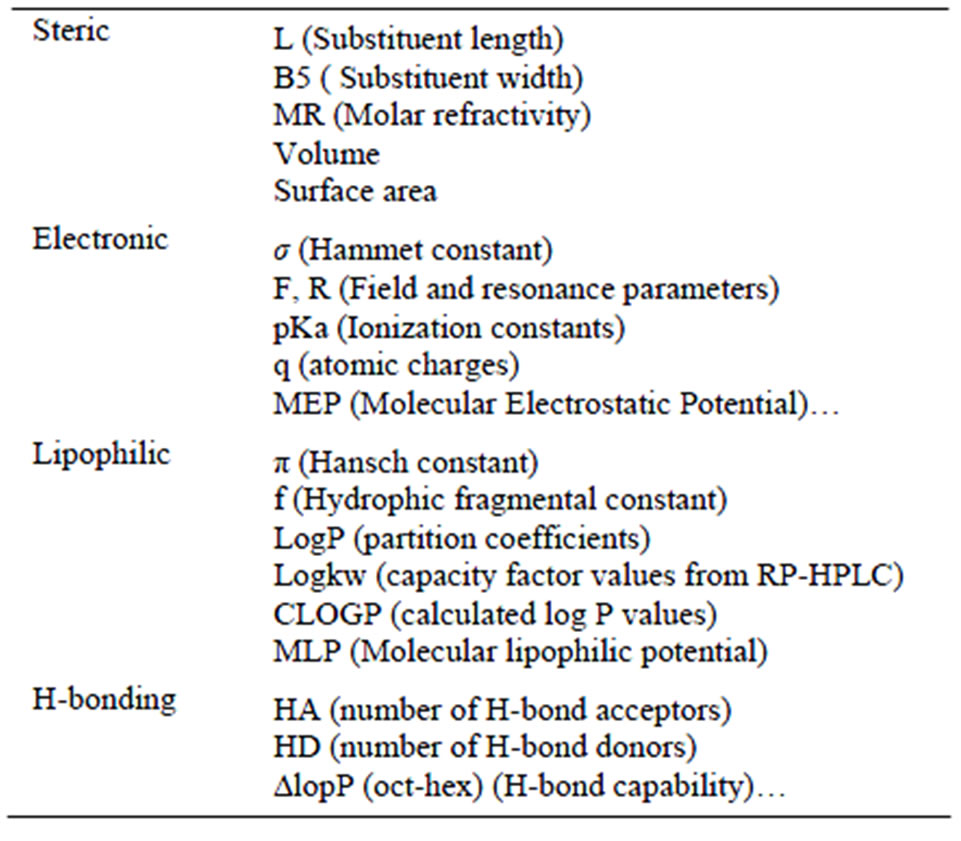
Table 1. Main stereoelectronic properties used in CADD [13].
novel techniques to bear on red biological problems [27].
3.3. Bioinformatics in Computer-Aided Drug Design
A few years ago, the National Institutes of Health (NIH) created the Biomedical Information Science and Technology Initiative (BISTI) to examine the current state of bioinformatics in the United States. BISTI’s working definition of bioinformatics included its use in biomedical research, in particular for drug discovery and development programs. Bioinformatics was seen as an emergin how drugs are found, brought to clinical trials and eventually released to the marketplace [28].
Computer-Aided Drug Design (CADD) is a specialized discipline that uses computational methods to simulate drug-receptor interactions. CADD methods are heavily dependent on bioinformatics tools, application and on the support side of the hub, information technology, information management, software applications, databases and computational resources all provide the infrastructure for bioinformatics. On the scientific side of the hub, bioinformatics methods are used extensively in molecular biology, genomics, proteomics, other emerging areas (i.e. metabolomics, transcriptomics) and in CADD research. There are several key areas where bioinformatics supports CADD research Figure 1 [29].
3.4. Virtual High-Throughput Screening (vHTS)
Pharmaceutical companies are always searching for new leads to develop into drug compounds. One search method is virtual high-throughput screening. In vHTS, protein targets are screened against databases of small molecule compounds to see which molecules bind
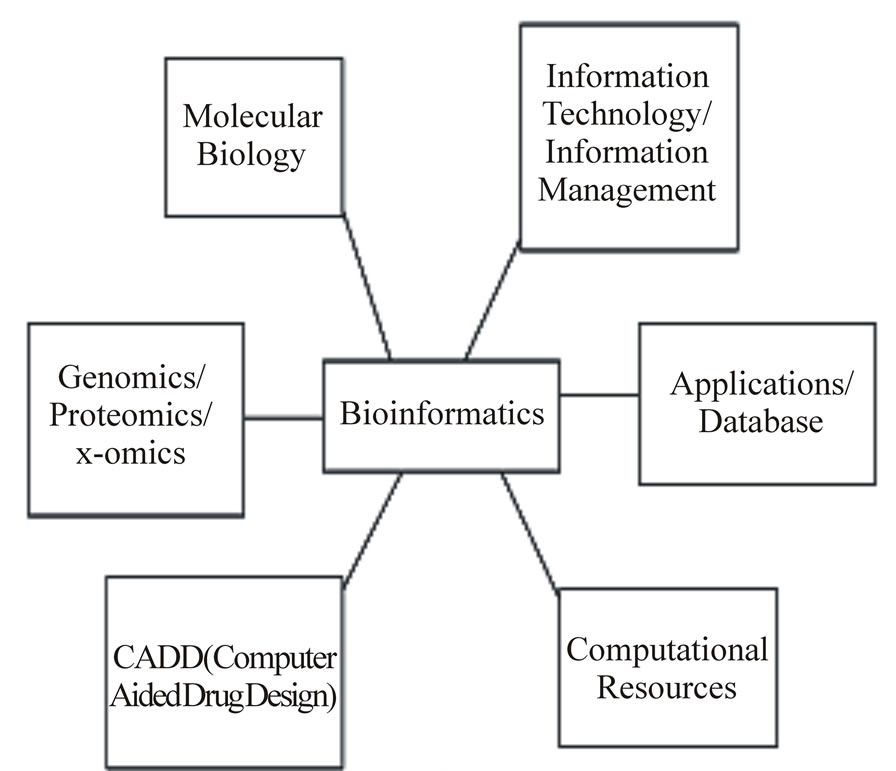
Figure 1. Bioinformatics in computer-aided drug design.
strongly to the target. If there is a “hit” with a particular compound, it can be extracted from the database for further testing. With today’s computational resources, several million compounds can be screened in a few days on sufficiently large clustered computers. Pursuing a handful of promising leads for further development can save researchers considerable time and expense. ZINC is a good example of a vHTS compound library [30].
4. Serendipity in Drug Research
Accidental discoveries always played an important role in science [31], especially in the search for new drugs [32-36]. Even if we do not count the traditional evaluation of plants, animal toxins and minerals for therapeutic potential, in ancient history, and the more or less systematic screening of synthetic compounds in our century, the number of serendipitous findings in drug history is legion. “Ein glücklicher Zufall hat uns ein Präparat in die Hand gespielt” (a lucky accident played a new drug in our hands) are the first words of a publication which describes the fortunate discovery of the fever-reducing activity of acetanilide. Erroneously this compound was clinically tested, instead of naphthalene that should have been investigated as an intestinal worm-killing agent [32-36].
The two best known examples of serendipitous findings are the discovery of the antibiotic effect of a certain Penicillium strain by Sir Alexander Fleming, which led to the development of penicillin and its synthetic derivatives, and the discovery that Chlordiazepoxide, which resulted from an unexpected chemical rearrangement, is a potent tranquillizer. Five years later, he prepared the compound once again; this time he experienced hallucinations, after accidental intake or inhalation of minute amounts of this highly potent compound [31,34,36]. Acetylsalicylic acid was originally designed as a prodrug of salicylic acid to treat headache, fever and rheumatic diseases. Much later it turned out to be an irreversible cyclooxygenase inhibitor, preventing blood coagulation by the inhibition of thrombocyte aggregation. A careful inspection of the reasons for the observed cytotoxic effect led to the surprising result that it was due to the action of ammonium and chloride ions on the platinum electrode, forming Cisplatin in the electrolysis medium [34]. Cyclosporin was developed because of its antifungal activity. Sandoz was already going to stop the program when the compound turned out to be an immunosuppressant, highly valuable to prevent the rejection of organ transplants. The story of the anticoagulants Dicoumarol and Warfarin is full of serendipitous findings. First, cattle bleeded to death after they were fed with moldy hay. The toxicagent Dicoumarol was isolated and introduced into human therapy. Because of its narrow therapeutic range and its frequent side effects it was abandoned after a short period. The Wisconsin Alumni Research Foundation developed the Dicoumarol analog Warfarin as a rat poison. New clinical trials started when a US army cadet unsuccessfully attempted to commit suicide. Warfarin is now the drug of choice to protect against stroke and other acute thrombotic events [37]. Recently it was recognized as a valuable lead for the development of potent HIV protease inhibitors. Application of a water-soluble salt, lithium urate, led to the serendipitous discovery of the beneficial effect of lithium salts [31,34,37]. Phenylbutazone and Valproic acid were designed as solubility enhancers for other drugs. However, both compounds turned out to be valuable drugs on their own. Phenolphthalein was discovered to be a potent laxative when it was tested as a possible marker to label cheap Hungarian wines. The three most important artificial sweeteners, saccharine, cyclamate and aspartame, were also serendipitous discoveries. Chemists experienced the sweet taste when licking their fingers or smoking a cigarette [38]. An important discovery in receptor research was also a case of serendipity. The second messenger cyclic AMP was discovered in 1957, adenylate cyclase in 1958. Fluoride ions activated adenylate cyclase but the mechanism of this surprising stimulation of enzymatic activity could not be explained for the next 24 years. Adenylate cyclase originally consisted of two components, the cyclase and a regulatory unit, the G protein. Fluoride activation of the G protein was observed in disposable glass tubes or in the presence of tap water, but not with distilled water in plastic tubes [39]. Further systematic investigation of these confusing results made clear that fluoride ions activate the G proteins only in the presence of minute amounts of aluminum ions. Whereas a GDP-G protein complex is inactive, the GTPG protein or GDP-fluoroaluminate-G protein complexes activate adenylate cyclase: It was supposed that the fluoroaluminate ion mimics the outer phosphate group of GTP [39].
5. Graph Machines
Definitions and notations for handling acyclic graphs, and the construction of graph machines from general graphs (possibly cyclic). It is shown that the training and model selection methods developed for vector machines can be extended to graph machines.
5.1. Handling Directed Acyclic Graphs
Definitions: We consider the mapping from a set of acyclic graphs G to a set of realvalued numbers. Graph machines and their applications to computer-aided drug design for each acyclic graph Gi of G, a parameterized function gi Rn R is constructed, which is intended 1) to encode the structure of the graph [40].
5.2. Model Selection
Similarly to vector machines, usual model selection techniques such as hold-out, K-fold cross-validation, leaveone-out, can be applied to recursive networks and to graph machines. In the present section, we show how virtual leave-one-out, a powerful method for estimating the generalization capability of a vector machine, can be extended to graph machines.
5.3. Graph Machines for the Prediction of Properties and/or Activities of Molecules
The prediction of the physico-chemical properties and pharmaceutical activities of molecules is a critical task in the drug industry for shortening the development times and costs. Typically, one synthesized molecule out of 10,000 becomes a commercial drug, and the development time of a new drug is approximately 10 years. Therefore, predicting the activity of a hitherto non-existent molecule may lead to tremendous savings in terms of development time and cost. Hence, over the past few years, QSPR/ QSAR has become a major field of research in the chemical industry. In a typical QSAR/QSPR scenario, a database of measured properties or activities of molecules is available, and it is desired to infer, from those data, the property/activity of molecules that have not yet been synthesized.
5.4. Encoding the Molecules
Molecules are usually described in databases in a representation called SMILES (Simplified Molecular Input Line Entry System), which provides a description of the graph structure of the molecule as a character string. In the applications described here, the functions gi were generated from the SMILES files of the molecules by the following procedure: The molecules, described by these files, were converted into labeled graphs by the association of each non-hydrogen atom to a node, and each bond to an edge. The nodes were also assigned labels describeing the atoms they were related to (e.g. their nature, their degree or stereoisomery). Then the adjacency matrices associated to those labeled graphs were generated. In the subsequent step, the matrices were cast into a canonical form, by an algorithm that ranks the nodes according to criteria such as their degree, the fact that they belong to a cycle [41]. This canonical form allowed the choice of the root nodes, and the conversion of the graphs into directed acyclic graphs.
5.5. Predicting the Boiling Points of Halogenated Hydrocarbons
The volatility of halogenated hydrocarbons is an important property, because those compounds are widely used in the industry, for example as solvents, anaesthetics, blowing agents, and end up in the environment, where they can damage the ozone layer or be greenhouse gases. The volatility of a molecule can be assessed by its boiling point, a property measured only for a small proportion of possible halogenated hydrocarbons. We studied a data set of 543 haloalkanes, whose boiling points were previously predicted by Multi Linear Regression (MLR) [41,42]. This regression required the computation of numerous molecular descriptors, including arithmetic descriptors, topological indices, geometrical indices, and counts of substructures and fragments. The best feature subset was then selected, and generally comprised 6 or 7 descriptors.
To provide a comparison with the results obtained by this method, we used the same training and test sets [43]. They feature 507 and 36 halo alkanes respectively, whose boiling points range from −128˚C to 249˚C. In order to select the number of neurons required by the complexity of the problem, we first performed 10-fold crossvalidation on the 507 examples of the training set. The results suggested the use of neural networks with 4 hidden neurons. We then trained the selected graph machines, and predicted the boiling points of the test set molecules. The results of this study are shown in Table 2, where they are compared to the results obtained by [43] on the same sets, using a 7-regressor MLR model.
5.6. Predicting the Anti-HIV Activity of TIBO Derivatives
TIBO (Tetrahydroimidazobenzodiazepinone) derivatives are a family of chemicals with a potential anti-HIV activeity. They belong to the category of non-nucleoside inhibitors, which block the reverse transcriptase of the retrovirus and prevent its duplication. We studied a data set of 73 of those compounds, whose activity was previously modeled with several QSAR methods, including conventional neural networks [44], multi-linear regression, comparative molecular field analysis (CoMFA) [45], and the substructural molecular fragments (SMF) method. The latter approach is based on the representation of the molecules with graphs, which are split into fragments,
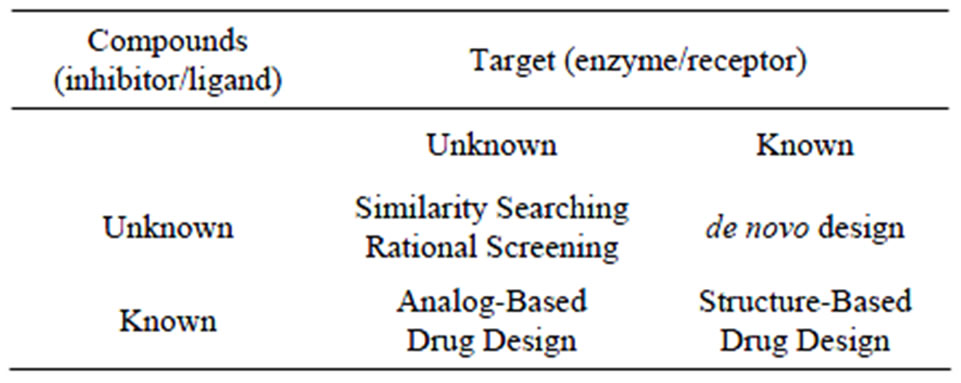
Table 2. Four major cases in CADD also known as “direct” and “indirect design when the structure of the target is respectively known or unknown [18].
whose contribution to the modeled activity is then computed with linear or non-linear regression. Those fragments are either atom-bond sequences, or “augmented atoms”, defined as atoms with their nearest neighbors. In order to compare the prediction abilities of graph machines to the performances of the SMF method , the data set was split into a training and a test set of 66 and 7 examples respectively, exactly as in. The activity is expressed as log (1/IC50) where IC50 is the concentration leading to the inhibition of 50% of the HIV-1 reverse transcriptase enzyme. Since some compounds of the set are stereoisomers, a label that described the enantiomer (R or S) of the atoms was added when necessary. We first performed 6-fold cross-validation on the training set with node functions having up to five hidden neurons to select the complexity of the model [46].
6. Drug Discovery Process
The process by which a new drug is brought to market stage is referred to by a number of names most commonly as the development chain or “pipeline” and consists of a number of distinct stages. Broadly it can be grouped under two stages Preclinical and the Clinical. Subsequently, the drug goes through many phases of clinical development in humans. In the clinical phase, the drug is administered to human volunteers to determine:
• The passage of the drug through the body-from the time it is taken to the time it is excreted;
• The effect of the drug on the body;
• Its effectiveness on the disease being targeted;
• Undesirable side effects of the drug.
6.1. Pitfall in Current Drug Discovery Process the Productivity Gap
A recent US Government Accountability Office (GAO) report found that Pharma R & D spending grew by 147% between 1993 and 2004 while the overall number of New drug applications (NDAs) submitted to the FDA increased only 38% and, worse still, the number of NDAs submitted for the presumably more innovative New molecular entities (NMEs) increased by only 7% in that time.
The attrition rate is unacceptably high. Only 1 out of 12 drugs entering clinical trials become a new drug. A particular worry or the pharmaceutical industry is that, despite a variety of approaches being used for R & D, attrition rates remain high during drug development. There are a number of factors attributed to the high attrition rates observed, but the number of active substances with poor pharmacological properties has been cited as a major concern. These are active substances that lack appropriate bioavailability, exhibit poor pharmacokinetics or cause adverse events and will therefore need to be withdrawn from development. It is estimated that these types of failures represent approximately 50% of all failures in drug development.
6.2. Need for an Alternative Tool
From the above facts and figures it is evident that there is an urge for an alternative tool that would not only shorten the R & D time cycle but also reduce the ever increasing cost involved in the drug discovery process. There is a general perception that applied sciences have not kept pace with the advances of basic sciences [48].
6.3. Impact of Technology
The process of finding a drug molecule that attaches itself to the target protein in the body has now moved from the lab to the computer [49]. The words in silico drug design and computer aided drug design are almost synonymous. In the post genomic era, computer aided drug design (CADD) has considerably extended its range of applications, spanning almost all stages in the drug discovery pipeline, from target identification to lead discovery, from lead optimization to preclinical or clinical trials Figure 2 [50].
6.4. In Silico Drug Discovery Process Comprises of 3 Stages
Stage 1 It involves Identification of a therapeutic target and building a heterogeneous small molecule library to be tested against it. This is followed by the development of a virtual screening protocol initialized by either docking of small molecules from the library or building these structures in the active site by employing De novo design methods.
Stage 2 These selected hits are checked for specificity by docking at binding sites of other known drug targets.
Stage 3 These selected hits are subjected to detail in silico ADMET profiling studies and those molecules that pass these studies are termed as leads.
6.5. Target Identification and Validation in Silico
Target identification and validation is the first key stage in the drug discovery pipeline. However, identification and validation of drug able targets from among thousands of candidate macromolecules is still a challenging task. Numerous technologies for addressing the targets have been developed recently. Genomic and proteomic approaches are the major tools for target identification. For example, a proteomic approach for identification of binding proteins for a given small molecule involvescomparison of the protein expression profiles for a given cell or tissue in the presence or absence of the given molecule.
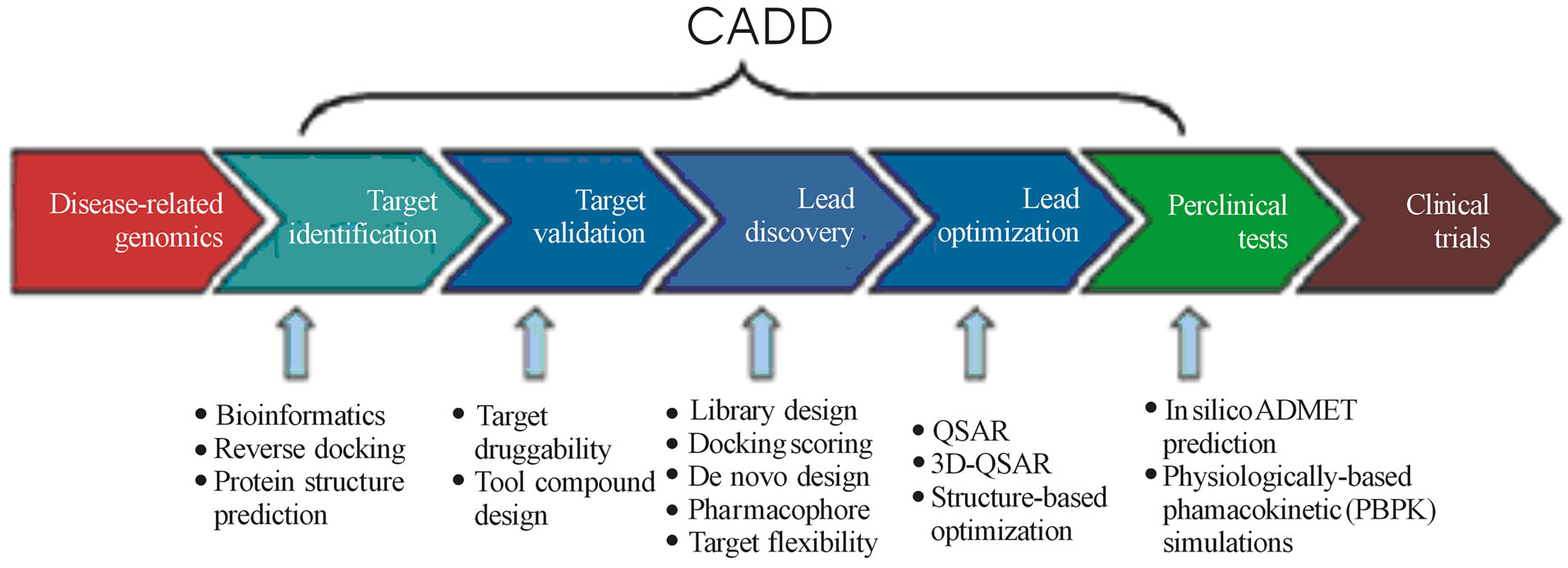
Figure 2. It shows the applications of CADD to the various stages of drug development [47].
6.6. In Silico ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) Prediction [51]
Studies indicate that poor pharmacokinetics and toxicity are the most important causes of costly late stage failures in drug development and it has become widely appreciated that these areas should be considered as early as possible in the drug discovery process. Combinatorial chemistry and high throughput screening have significantly increased the number of compounds for which early data on absorption, distribution, metabolism, excretion (ADME) and toxicity (T) are needed. With use of in silico tools it is possible to model the most relevant pharmacokinetic, metabolic and toxicity endpoints, thereby accelerating the drug discovery process.
6.7. In Silico Prediction of Drug Safety [52]
There is considerable interest in computational models to predict drug safety in drug discovery and development. Significant adverse toxicological findings for a drug in late-stage clinical trials or post marketing can cause enormous financial losses and place patients at risk. The earlier such molecules are identified and the drug development process halted the better.
There are tools to predict toxicities like 1) Genotoxicity;
2) Liver toxicity;
3) CYP450 inhibition; and 4) Cardiotoxicity.
6.8. In Silico Prediction of Drug-Drug Interactions [53]
Recently, metabolic drug-drug interactions (MDDI) have raised some high profile problems in drug development resulting in restricted use, withdrawal or non approval by regulatory agencies. The use of in vitro technologies to evaluate the potential for MDDI has become routine in the drug development process. Nevertheless, in the absence of an integrated approach, their interpretation and value remains the subject of debate, and the vital distinction between a useful “simulation” and a precise “prediction” is not often appreciated. Various in silico software are now available for the simulation of MDDI. One such software is SIMCYP.
6.9. Virtual Screening [54]
Virtual screening involves the docking of selected lead molecules against the biological target. This is followed by a scoring pattern. There is a number of software available for this. Some are commercially available and some are free to use.
7. Conclusion
The successful stories of CADD application in drug discovery in recent years have demonstrated the potential value of CADD in drug development. CADD approaches can provide valuable information for target identification and validation, lead selection, small-molecular screening and optimization. In particular, those sub disciplines of CADD have demonstrated promising application for design of drug. The latest technological advances (QSAR/ QSPR, structure-based design, combinatorial library design, chemoinformatics & bioinformatics); the growing number of chemical and biological databases; and an explosion in currently available software tools are providing a much improved basis for the design of ligands and inhibitors with desired specificity. In future our review will helpful for design of drug with minimal side effect and high potency.
REFERENCES
- W. G. Richards, “Computer-Aided Drug Design,” Pure and Applied Chemistry, Vol. 66, No. 8, 1994, pp. 1589- 1596. doi:10.1351/pac199466081589
- I. S. Haworth, C. Burt, F. Gago, C. A. Reynolds and W. G. Richards, “A Prototype Bioreductive DNA Groove Binding Ligand,” Anticancer Drug Design, Vol. 6, No. 1, 1991, pp. 59-61.
- I. S. Haworth, A. H. Elcock, A. Rodger and W. G. Richards, “Sequence Selective Binding to the DNA Major Groove: Tris (1,lO-phenanhline) Metal Complexes Binding to Poly(dG-dC) and Poly(dA-dT),” Journal of Biomolecular Structure and Dynamics, Vol. 9, No. 1, 1991, pp. 23-29. doi:10.1080/07391102.1991.10507891
- I. S. Haworth, A. H. Elcock, A. Rodger and W. G. Richards, “A Binding Mode of A-[tris(l,lO-phenanthrohe)ruthenium(II)]2+ Exhibiting Preference for Purine-3’,5’-Pyrimidine Sites of DNA,” Journal of Biomolecular Structure and Dynamics, Vol. 9, No. 1, 1991, pp. 553-569. doi:10.1080/07391102.1991.10507936
- M. Jayakanthan, G. Wadhwa, T. M. Mohan, L. Arul, P. Balasubramanian and D. Sundar, “Computer-Aided Drug Design for Cancer-Causing H-Ras p21 Mutant Protein,” Letters in Drug Design and Discovery, Vol. 6, No. 1, 2009, pp. 14-20. doi:10.2174/157018009787158526
- D. A. Spandidos and N. M. Wilkie, “Malignant Transformation of Early Passage Rodent Cells by a Single Mutated Human Oncogene,” Nature, Vol. 310, 1984, pp. 469- 475.
- A. Valencia, P. Chardin, A. Wittinghofer and C. Sander, “The Ras Protein Family: Evolutionary Tree and Role of Conserved Amino Acids,” Biochemistry, Vol. 30, No. 19, 1991, pp. 4637-4648. doi:10.1021/bi00233a001
- M. Barbacid, “Ras Genes,” Annual Review of Biochemistry, Vol. 56, 1987, pp. 779-827. doi:10.1146/annurev.bi.56.070187.004023
- R. Khosravi-Far and C. J. Der, “The Ras Signal Transduction Pathway,” Cancer and Metastasis Reviews, Vol. 13, No. 1, 1994, pp. 67-89. doi:10.1007/BF00690419
- A. Russo, V. Bazan, V. Agnese, V. Rodolico and N. Gebbia, “Prognostic and Predictive Factors in Colorectal Cancer: Kirsten Ras in CRC (RASCAL) and TP53CRC Collaborative Studies,” Annals of Oncology, Vol. 4, Suppl 4, 2005, pp. 44-49.
- G. Oxford and D. Theodorescu, “The Role of Ras Superfamily Proteins in Bladder Cancer Progression,” Journal of Urology, Vol. 170, No. 5, 2003, pp. 1987-1993. doi:10.1097/01.ju.0000088670.02905.78
- J. John, M. Frech and A. Wittinghofer, “Biochemical Properties of Haras Encoded p21 Mutants and Mechanism of the Autophosphorylation Reaction,” The Journal of Biological Chemistry, Vol. 263, 1988, pp. 11792-11799.
- H. Kiaris and D. A. Spandidos, “Mutations of Ras Genes in Human Tumors,” International Journal of Oncology, Vol. 7, No. 3, 1995, pp. 413-421.
- L. George and J. H. Ching, “VPython Application To The Computer-Aided Drug Design Problem,” The Python Papers Monograph, Vol. 2, 2010, pp. 1-5.
- D. G. Sprous, J. Zhang, L. Zhang, Z. Wang and M. A. Tepper, “Kinase Inhibitor Recognition by Use of a Multivariable QSAR Model,” Journal of Molecular Graphics and Modelling, Vol. 24, No. 4, 2006, pp. 278-295. doi:10.1016/j.jmgm.2005.09.004
- C. D. Selassie, “Burger’s Medicinal Chemistry, Drug Discovery and Development,” 6th Edition, John Wiley & Sons, Inc., New York, 2003.
- Structure-Activity Relationships (QSAR), “A Review, Combinatorial Chemistry & High Throughput Screening,” Vol. 9, 2006, pp. 213-228.
- K. R. Oldenburg, “Annual Report in Medicinal Chemistry,” J. A. Bristol, Ed., Academic Press, London, Vol. 33, 1998, pp. 301-307.
- W. H. Moos, G. D. Green and M. R. Pavia, “Chapter 33. Recent Advances in the Generation of Molecular Diversity,” Annual Reports in Medicinal Chemistry, Vol. 28, 1993, pp. 315-324. doi:10.1016/S0065-7743(08)60903-3
- F. Ooms, “Molecular Modeling and Computer Aided Drug Design. Examples of their Applications in Medicinal Chemistry,” Current Medicinal Chemistry, Vol. 7, No. 2, 2000, pp. 141-158. doi:10.2174/0929867003375317
- J. Kuhlman, International Journal of Clinical Pharmacology and Therapeutics, Vol. 35, 1997, pp. 541-552.
- R. G. Halliday, S. R. Walker and C. E. Lumley, Journal of Pharmaceutical Medicine, Vol. 2, 1992, pp. 139-154.
- A. K. Ghose and J. J. Wendoloski, “Perspective in Drug Discovery and Design,” Kluwer/Escom, Vol. 9-11, 1998, pp. 253-271.
- D. J. Abraham and G. E. Kellogg, “3D-QSAR in Drug Design,” H. Kubinyi, Ed., Escom, Leiden, Vol. 1, 1993, pp. 506-522.
- D. Scherer, P. Dubois and B. Sherwood, “VPython: 3D Interactive Scientific Graphics for Students,” Computing in Science and Engineering, Vol. 2, No. 5, 2000, pp. 56-62.
- J. Irwin, D. M. Lorber, S. L. McGovern, B. Wei and B. K. Shoichet, “Docking and Drug Discovery,” Computational Nanoscience and Nanotechnology, Vol. 2, 2002, pp. 50-51.
- C. A. Taft, V. B. da Silva and C. H. T de P. da Silva, “Current Topics in Computer-Aided Drug Design,” Journal of Pharmaceutical Sciences, Vol. 97, No. 3, 2008, pp. 1089-1098. doi:10.1002/jps.21293
- D. Bernard, A. Coop and A. D. MacKerell Jr., “Computer-Aided Drug Design: Structure-Activity Relationships of Delta Opioid Ligands,” Drug Design Reviews, Vol. 2, No. 4, 2005, pp. 277-291. doi:10.2174/1567269054087596
- W. G. Richards, “Computer-Aided Drug Design,” Pure and Applied Chemistry, Vol. 66, No. 8, 1994, pp. 1589- 1598. doi:10.1351/pac199466081589
- D. S. Park, J. M. Kim, Y. B. Lee and C. H. Ahn, “QSID Tool: A New Three-Dimensional QSAR Environmental Tool,” Journal of Computer-Aided Drug Design, Vol. 22, No. 12, 2008, pp. 873-883.
- H. Kubinyi, “Chance Favors the Prepared Mind—From Serendipity to Rational Drug Design,” Journal of Receptor and Signal Transduction Research, Vol. 19, No. 1-4, 1999, pp. 15-39. doi:10.3109/10799899909036635
- M. von Itzstein, W. Y. Wu, G. B. Kok, M. S. Pegg, J. C. Dyason, B. Jin, T. V. Phan, M. L. Smythe, H. F. White, S. W. Oliver, P. M. Colman, J. N. Varghese, D. M. Ryan, J. M. Woods, R. C. Bethell, V. J. Hotham, J. M. Cameron and C. R. Penn, “Rational Design of Potent SialidaseBased Inhibitors of Influenza Virus Replication,” Nature, Vol. 363, No. 6428, 1993, pp. 418-423. doi:10.1038/363418a0
- J. Greer, J. W. Erickson, J. J. Baldwin and M. D. Varney, “Application of the Three-Dimensional Structures of Protein Target Molecules in Structure-Based Drug Design,” Journal of Medicinal Chemistry, Vol. 37, No. 8, 1994, pp. 1035-1054. doi:10.1021/jm00034a001
- J. P. Vacca and J. H. Condra, “Clinically Effective HIV-1 Protease Inhibitors,” Drug Discovery Today, Vol. 2, No. 1, 1997, pp. 6-18. doi:10.1016/S1359-6446(97)01053-2
- P. C. Sternweis and A. G. Gilman, “Aluminum: A Requirement for Activation of the Regulatory Component of Adenylate Cyclase by Fluoride,” Proceedings of the National Academy of Sciences USA, Vol. 79, No. 16, 1982, pp. 4888-4891. doi:10.1073/pnas.79.16.4888
- J. Sondek, D. G. Lambright, J. P. Noel, H. E. Hamm and P. B. Sigler, “GTPase Mechanism of Gproteins from the 1.7-Å Crystal Structure of Transducin α.GDP.AlF4-,” Nature, Vol. 372, 1994, pp. 276-279. doi:10.1038/372276a0
- G. De Stevens, “Serendipity and Structured Research in Drug Discovery,” Progress in Drug Research, Vol. 30, 1986, pp. 189-203.
- P. J. Goodford, “Drug Design by the Method of Receptor Fit,” Journal of Medicinal Chemistry, Vol. 27, No. 5, 1984, pp. 557-564. doi:10.1021/jm00371a001
- J. P. Vacca and J. H. Condra, “Clinically Effective HIV-1 Protease Inhibitors,” Drug Discovery Today, Vol. 2, No. 1, 1997, pp. 6-18. doi:10.1016/S1359-6446(97)01053-2
- A. Goulon, A. Duprat and G. Dreyfus, “Graph Machines and Their Applications to Computer-Aided Drug Design: A New Approach to Learning from Structured Data,” Lecture Notes in Computer Science, Vol. 4135, 2006, pp. 1-19.
- C. Jochum and J. Gasteiger, “Canonical Numbering and Constitutional Symmetry,” Journal of Chemical Information and Computer Sciences, Vol. 17, No. 2, 1977, pp. 113-117. doi:10.1021/ci60010a014
- A. T. Balaban, S. C. Basak, T. Colburn and G. D. Grunwald, “Correlation between Structureand Normal Boiling Points of Haloalkanes C1-C4 Using Neural Networks,” Journal of Chemical Information and Computer Sciences, Vol. 34, No. 5, 1994, pp. 1118-1121. doi:10.1021/ci00021a016
- C. Rucker, M. Meringer and A. Kerber, “QSPR Using MOLGEN-QSPR: The Example of Haloalkane Boiling Points,” Journal of Chemical Information and Computer Sciences, Vol. 44, No. 6, 2004, pp. 2070-2076. doi:10.1021/ci049802u
- L. Douali, D. Villemin and D. Cherqaoui, “Exploring QSAR of Non-Nucleoside Reverse Transcriptase Inhibitors by Neural Networks: TIBO Derivatives,” International Journal of Molecular Sciences, Vol. 5, No. 2, 2004, pp. 48-55. doi:10.3390/i5020048
- J. Huuskonen, “QSAR Modeling with the Electrotopological State, TIBO Derivatives,” Journal of Chemical Information and Computer Sciences, Vol. 41, No. 2, 2001, pp. 425-429. doi:10.1021/ci0001435
- Z. Zhou and J. D. Madura, “CoMFA 3D-QSAR Analysis of HIV-1 RT Nonnucleoside Inhibitors, TIBO Derivatives Based on Docking Conformation and Alignment,” Journal of Chemical Information and Computer Sciences, Vol. 44, No. 6, 2004, pp. 2167-2178. doi:10.1021/ci049893v
- B. Hammer, “Recurrent Networks for Structured Data— A Unifying Approach and its Properties,” Cognitive Systems Research, Vol. 3, No. 2, 2002, pp. 145-165. doi:10.1016/S1389-0417(01)00056-0
- C. Jochum and J. Gasteiger, “Canonical Numbering and Constitutional Symmetry,” Journal of Chemical Information and Computer Sciences, Vol. 17, No. 2, 1977, pp. 113-117. doi:10.1021/ci60010a014
- A. T. Balaban, S. C. Basak, T. Colburn and G. D. Grunwald, “Correlation between Structure and Normal Boiling Points of Haloalkanes C1-C4 Using Neural Networks,” Journal of Chemical Information and Computer Sciences, Vol. 34, No. 5, 1994, pp. 1118-1121. doi:10.1021/ci00021a016
- C. Rucker, M. Meringer and A. Kerber, “QSPR Using MOLGEN-QSPR: The Example of Haloalkane Boiling Points,” Journal of Chemical Information and Computer Sciences, Vol. 44, No. 6, 2004, pp. 2070-2076. doi:10.1021/ci049802u
- J. Augen, “The Evolving Role of Information Technology in the Drug Discovery Process,” Drug Discovery Today, Vol. 7, Suppl 5, 2002, pp. 275-282.
- H. Van de waterbeemd and E. Gifford, “ADMET in Silico Modelling, towards Prediction Paradise?” Nature Reviews Drug Discovery, Vol. 2, No. 3, 2003, pp. 192-204. doi:10.1038/nrd1032
- W. J. Egan, G. Zlokarnik and P. D. J. Grootenhuis, “In Silico Prediction of Drug Safety: Despite Progress There Is Abundant Room for Improvement,” Drug Discovery Today: Technologies, Vol. 1, Suppl 4, 2004, pp. 381-387.
- A. Rostami-Hodjegan and G. Tucker, “‘In Silico’ Simulations to Assess the ‘in Vivo’ Consequences of ‘in Vitro’ Metabolic Drug-Drug Interactions,” Drug Discovery Today: Technologies, Vol. 1, Suppl 4, 2004, pp. 441-448.
NOTES
*Corresponding author.