Open Journal of Ecology
Vol.04 No.17(2014), Article ID:52763,22 pages
10.4236/oje.2014.417091
Impact of Natural Selection on Lignin and Cellulose Candidate Genes in a Natural Population of Eucalyptus urophylla
Létizia Camus-Kulandaivelu1*, Bénédicte Favreau1, Saneyoshi Ueno1,2, Jonathan Przybyla1, Jean-Marc Bouvet1
1Cirad-Bios Department, AGAP Research Unit 108 “Adaptation and Breeding of Tropical and Mediterranean Plants”, International Campus of Baillarguet, Montpellier, France
2Tree Genetics Laboratory, Department of Forest Genetics, Forestry and Forest Products Research Institute, Tsukuba, Japan
Email: *letizia.camus-kulandaivelu@cirad.fr
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 August 2014; revised 6 September 2014; accepted 21 September 2014
ABSTRACT
Wood plays a major role in land ecosystems and in human activity. Better understanding the genetic basis and evolutionary implication of wood variability are thus key issues with both ecological and economical implications. The present paper addresses the question of the extending and the nature of natural selection on wood related genes in Eucalyptus urophylla, a tropical tree species with key economical importance. We conducted a genetic study on an E. urophylla population from Timor Island using a set of 17 SSR characterized on a main sample of 43 individuals and six candidate genes sequenced on a subset of 18 individuals. The candidate genes include three cellulose synthase genes (EuCesA1, EuCesA2 and EuCesA3), and three genes involved in lignin synthesis (EuCAD2, EuC4H1 and EuC4H2). Based on SSR data, the investigated population appeared to have no structure and have undergone past population expansion. Accounting for this demographic history, we were able to draw neutral expectation for polymorphism distribution on candidate genes and to determine their potential selective status. We hence identified two gene portions exhibiting unexpected polymorphism pattern, consistent with natural selection imprint.
Keywords:
Eucalyptus urophylla, Adaptation, Demography, Approximate Bayesian Computation, Candidate Genes

1. Introduction
Wood is a structural tissue resulting from the secondary growth of vascular cambium in tree stems and roots. It is a natural composite of cellulose fibbers embedded in a lignin matrix which provides trees with physical support, water and nutriment transport between roots and leaves and nutriment storage. Wood is quantitatively and qualitatively a key component of land ecosystems and its properties further determine biomass decomposition rates, playing a key role in carbon and nitrogen cycle [1] [2] . It is also a very important resource for humans, being used to produce paper, charcoal, furniture and many other facilities. Wood first evidence dates back from early Devonian, 407 million years ago [3] and, since then, its chemical and physical properties have highly evolved. This variation exhibits complex patterns, which globally appear to be the results of functional constraints, phylogenetic conservatism and environmental adaptation [4] . Focusing at intra-specific level, wood traits exhibit important variability with high heritability [5] , raising the question of their particular role in species adaptation and the selective forces acting on them. Wood is a complex structure with thousands of genes involved [6] - [12] but an increasing number of studies have managed to pinpoint gene SNPs associated to wood property variability in natural populations of several tree genus, including Eucalyptus and allied species [13] - [17] . On the other hand, the question of the natural selection pressures acting on wood related genes has been rarely addressed, in particular at intra-specific level. Wood related genes are generally considered to be under purifying or balancing selection, but only few examples have been reported so far [18] [19] .
The present paper attends to decipher the selective forces acting on six full length wood synthesis candidate genes in Eucalyptus urophylla S T Blake. E. urophylla is the only Eucalyptus species not naturally found in Australia and has a relatively restricted area, the Lesser Sunda Islands where it occurs as disjunct populations across seven islands, with main stands in Timor and Wetar [20] . E. urophylla is one of the main Eucalyptus species used for industrial plantations in tropical regions and it is also described to be one of the most morphologically variable species among Eucalyptus [21] , being thus a very good model to study adaptation. E. urophylla grows from sea level up to 3000 m and displays dramatic phenotypic differences as for growth and shape as well as for fruit size and bark morphology [20] [22] .
We focus here on a set of lignin (EuC4H1, EuC4H2 and EuCAD2) and cellulose synthesis (EuCesA1, EuCesA2 and EuCesA3) gene set for which association with wood related traits has been already investigated in E. urophylla [17] . Cellulose synthesis (CesA) genes encode components of an enzyme complex embedded in the cellular membrane [23] and characterized by a “rosette” like structure [24] . This enzyme complex catalyzes the last biochemical reaction of the cellulose synthesis and synthesizes six β-1,4-glucan chains that cocrystallize to form a 36-chain microfibril [25] . While CesA genes can be expressed either in primary tissues or in secondary tissues [26] , EuCesA1, EuCesA2 and EuCesA3 were chosen because in E. globulus Labill., a species closely related to E. urophylla, the three orthologous CesA genes are specifically and strongly expressed in wood [27] . C4H and CAD2 are two enzymes that play an important role at the beginning and the end of the lignin biosynthesis pathway. Cinnamate 4-hydroxylase (C4H) belongs to the core reactions of phenylpropanoid metabolism. It catalyzes the hydroxylation of cinnamic acid to 4-hydroxycinnamate (or p-Coumaric acid), which is the second step of the phenylpropanoid pathway [28] . Evidences suggest that C4H has been duplicated before monocot and dicot separation and can be found in a single or several copies in plants [28] - [30] . Finally the CAD2 enzyme belongs to the specific lignin pathway. The Cinnamoyl CoA Reductase (CCR) converts hydroxycinnamoyl CoA esters to their corresponding aldehydes, which are converted in the monomeric precursors of lignin by the Cynnamyl Alcohol Dehydrogenase (CAD2) [28] .
In order to characterize the selective forces acting on E. urophylla wood gene at intra-specific level, we sequenced the six candidate genes on 18 individuals from Timor Island and investigated the imprint of natural selection on their polymorphism pattern. Since demographic events can generate polymorphism pattern similar to what is expected under selection at the whole genome scale, it is important to account for it in order to avoid false positives. However, the demographic history of E. urophylla is not well known. Previous studies showed no [22] or weak population structure [31] at species range level. Evidence for bottleneck was specifically investigated and was found on chloroplast markers [32] but not on nuclear SSR data. To clarify demographic history of our E. urophylla sampled region, we used a set of 43 individuals genotyped for 17 SSR markers to evaluate the fit of several simple demographic scenarios using Approximate Bayesian Computation (ABC) framework. ABC is a flexible class of Monte-Carlo algorithms used to perform model-based inference [33] and relying on summary statistics of the data, making it much less computationally demanding than full likelihood relying approaches. The demographic history being elucidated on SSR data, the results served as a basis to build an ad-hoc expected distribution for sequence polymorphism pattern accounting for demographic history under selective neutrality. We used this ad-hoc distribution along with other neutrality test to evaluate selective status of the candidates.
2. Material and Methods
2.1. Sampling and DNA Extraction
To perform our study, we have selected samples from the East Timor (Figure 1 and Table 1). No endangered or protected species were involved in this study. Seeds of Eucalyptus urophylla were collected between in 1973 and preserved in the CIRAD Genetic Forestry Laboratory. At the time of the sampling, East Timor was a Portuguese territory and sampling authorization was obtained from the military governor (SUPDOC). As GPS technology was not available at the time of the sampling, coordinates were estimated using a 1/50,000 topographic map. The seeds had been stored in a cold room at 4˚C and 30% humidity. In 2003, some seeds from this collection were planted. The plantlets were maintained in humid tropical nursery at the following conditions: 28˚C day temperature, 25˚C night temperature, 70% humidity, pH4 substrate. Leaves were collected on 4 months old plantlets, dried in silica gel and stored at room temperature until DNA extraction. To perform our study, we have selected samples from the East of Timor Island (Figure 1 and Table 1). We concentrate on Timor Island because the E. urophylla stands growing there are bigger and likely more stable over time than in other islands [20] . DNA was extracted from 43 individuals according to a modified protocol adapted from Gawel & Jarret [34] and Saghai- Maroof [35] . Each individual is coming from a different mother tree. All the 43 genotypes were used for the microsatellite analysis while 18 individuals were used for gene sequencing and gene polymorphism studies.
2.2. Microsatellite Genotyping
A total of 17 microsatellite loci were used for this study. They were all previously described: EMB18 [36] ; EMB22, EMB27, EMB30, EMB32, EMB33, EMB37, EMB38, EMB42, EMB44, EMB47, EMB52, EMB56, EMB63 [37] ; FMRA1, FMRA4, FMRA5 [38] . They were selected according to several criteria: specificity of the amplification, high polymorphism level, belonging to different linkage groups. The genotyping was per-

Figure 1. Sample location map. E. urophylla provenances sampled in this study are materialized on the map by pins associated with their number, as given in Table 1. Detailed geographical information about provenances is presented in Table 1. These geographic coordinates were obtained using a 1/50,000 topographic map. Map is similar to Open Street Map (http://www.openstreetmap.org) but not identical and is therefore for representative use only.

Table 1. Geographical data of the sampled individuals.
Num: location number reported on Figure 1 map; Microsatellites: number of individuals used for the microsatellite analysis; Gene: number of individuals used for the gene sequence analysis; Lat, Long: latitude, longitude; Alt: altitude (expressed in meters). Latitude and longitude were estimated with a 1/50,000 topographic map.
formed on the 43 individual set and 3 DNA controls with known allele size were added to each 96 well plates. The PCR amplification was performed by multiplexing 2 primers. Five microliters of QIAGEN multiplex mix, 0.08 µM of both forward primer with 5’-tail-end M13 (CACGACGTTGTAAAACGAC), 0.10 µM of both reverse primer, 0.10 µM IRDye fluorescent-labelled M13-primer (700 or 800 nm) and 5.0 ng of genomic template DNA. A touchdown cycling programme was used: 95˚C for 15 min, 67˚C for 1.5 min, 72˚C for 1 min, followed by eight cycles of 94˚C for 30 s, 65˚C for 1.5 min with 2˚C decrease at each cycle, 72˚C for 1 min then 24 cycles at 94˚C for 30 s, 51˚C for 1.5 min, 72˚C for 1 min, and a final extension of 60˚C for 30 min. Amplified fragments were analysed at 700 and 800 nm by electrophoresis on an IR2-DNA analyzer (LI-COR 4200 sequencer) at the Montpellier Languedoc-Roussillon Genopole genotyping platform. Allele scoring was done with SAGA software (LI-COR).
2.3. Candidate Gene Sequencing and Sequence Analysis
The E. urophylla genome has not been sequenced yet and at the time of the experiments, the E. grandis W. Hill ex Maiden sequence was not released. In that context, primer design was based on gene sequences from other species found in databases. Primers were designed in order to get the whole sequence of the candidate genes (Table S1). Details of primer design for each gene are given in Supplementary Online Material. PCRs were performed in 10 mL reaction mixtures with 10 mM incubation buffer 1X CORE Kit Q-Biogene (Tampon CORE Kit Q-Biogene, MP Biomedical), 0.30 µM forward and reverse primers, 1.2 units of Taq DNA polymerase (Invitrogen) and 15 ng of template DNA. We used the following PCR program for the amplification: 94˚C for 4 min; then 35 cycles (40 cycles for CesAs) of 94˚C for 30 sec, an annealing step for 1 min at the primer’s optimized annealing temperature for 1 min and 72˚C for 1 min (2 min for C4Hs), followed by a final extension at 72˚C for 5 min. PCR products were loaded on agarose gels for electrophoresis and the quantity of PCR product was estimated by image analysis with the freeware Image J v1.45b (Wayne Rasband, NIH). One hundred ng of primer product were sent to High-Throughput Genomics Unit (HTGU, Department of Genome Sciences, Univ. of Washington, Seattle) for direct sequencing. For each gene, the overlapping sequences obtained after direct PCR sequencing were assembled and aligned with Codon Code Aligner software (Licor. 2002-2007 ver. 3.5.2), and manually edited. For heterozygote sequences due the presence of INDELs, the sequences of the two alleles were either deducted manually or with INDELLIGENT software [39] .
2.4. Genetic Structure Analysis
While Tripiana et al. [22] found no population structure on a E. urophylla sample encompassing our studied area, we verified the absence of any cryptic population structure on our 43 individual SSR data set by using Structure software (version 2.3.3) [40] [41] . We used sample group information [41] and an admixture model with correlated allele frequencies. 12 independent runs were performed with 100,000 MCMC repetitions and a 100,000 burn-in period for K = 1 to K = 4. The most likely number of clusters was determined using the log probability of data.
2.5. Determining the Demographic History of E. urophylla in Timor Island Using Microsatellites Data
We used the Approximate Bayesian Computation (ABC) approach as implemented in DIYABC software [42] , [43] to determine the demographic scenario that may have shaped E. urophylla diversity pattern observed on SSR data. ABC uses coalescence tool to generate thousands of simulated data having a similar configuration then observed one in terms of individual number, marker type and number and relies on summary statistics to summarize and compare simulated and observed data. Simulations are launched from a set of user-defined demographic scenarios with parameters sampled from prior distributions. DIYABC software provides a full set of tools that we used to simulate SSR data and to perform scenario choice, parameter evaluation and several steps of model validation.
Based on Tripiana et al. [22] results, we considered a single panmictic population and investigated five simple scenarios accordingly: SNM Standard Neutral Model (constant population size), BOT: bottleneck, EXP: demographic expansion, EXPBOT: recent expansion and past bottleneck and BOTEXP: recent bottleneck and past expansion. Cycles of population bottlenecks and expansions are possible in E. urophylla due to the volcanic characteristics of Sunda Islands and motivate the exploration of scenario EXPBOT and BOTEXP. These five scenarios are detailed in Figure 2, along with parameter description and imposed constraints. Since we have only scarce information about E. urophylla tree, we gave the five scenarios equal prior probability and used non-informative parameter priors, i.e. sampled from uniform distribution (Table S2). For each of these five scenarios, we sampled 100,000 simulated data sets whose corresponding information (parameters and summary statistics) was stored in the so-called reference table used in all further analysis except model checking. We recorded mean allele number, mean genetic diversity [44] , mean allele size variance and mean Garza-Williamson’s M [45] as summary statistics. Scenario posterior probabilities were calculated using a local logistic regression procedure proceeding on the 10% closest simulated points and that, shortly saying, gives more weight to those closer to the observed data. We then performed a model checking step to verify if the scenario with highest probability fits properly the observed data. As advised by Cornuet et al. [43] who recommended not to use for model checking the same summary statistics as for model choice, we constructed a new reference table with same parameter priors than previously but with recording only mean allele number and mean allele size variance as summary statistics and used mean genetic diversity [44] and mean Garza-Williamson’s M [45] for model checking. We simulated then 1000 additional data sets using the parameter posteriors of the scenario with highest probability and compared their summary statistic with those of the observed data. The scenario with highest probability is considered as suitable if the summary statistics of the observed data are included in the 95% confidence interval of the summary statistic’s posterior predictive distribution. Parameter posterior distributions were estimated only for the scenario with highest probability using the 10% closest simulated points with a local regression procedure and a logit transformation of the parameters. Confidence in scenario choice was finally evaluated by calculating power and alpha risk associated with choosing the scenario with highest probability. Alpha risk was evaluated by generating 1000 additional simulated sets for each of the four rejected scenario using their respective prior and by assessing the proportion wrongly attributed to the scenario with highest probability based on the reference table. Power was calculated by generating 1000 additional simulated data sets for the scenario with highest probability and by calculating the proportion correctly assigned using the reference table. Confidence in parameter estimates was evaluated by comparing the known parameters of a set of 1000 newly simulated data set obtained for the most likely scenario to those estimated using the current reference table. As measure of parameter estimation error and bias, we calculated the average relative bias and the factor 2, i.e. the proportion of pseudo observed data set for which the point estimate is at least half and at most twice the true value [42] .


Figure 2. Demographic scenarios investigated with ABC analysis. Events are considered backward in time, from present to past. Constraints on the parameters are indicated along with each scenario along with the detail of parameters used with DIYABC. Scenario “SNM” (Standart Neutral Model) considers a population of constant size N. Scenario “BOT” considers a population of current size Nb having experienced a sudden bottleneck event tb generations ago, from ancestral population size N1. Scenario “EXP” considers a population of current size Ne having experienced a sudden expansion event Te generations ago from ancestral population size N2. Scenario EXPBOT considers a population of current size
2.6. Nucleotide Diversity Analysis on Candidate Genes
The SNPs and INDELs were identified by comparison of the 18 genotypes. Polymorphism estimation and neutrality tests were performed with DNAsp software [46] . For all the analyses, the missing data and the INDELs were excluded in all the subsets. Nucleotide diversity on a base pair basis was estimated by ΘW from the number of polymorphic segregating (S) sites [47] and by π [44] from the number of pairwise mismatches. These two site frequency spectrum related statistics are two estimators of 4Nμ, with N effective population size and μ the mutation rate per nucleotide and per generation. They are complementary since ΘW is not sensitive to allelic frequencies and π is. To determine a possible non-equilibrium status of the studied genes, we measured Tajima’s D [48] . Tajima’s D quantifies the difference between π and Θw. A significant negative value of Tajima’s D can indicate an excess of rare variants as expected under positive selection. It can also reflect demographic event such as population expansion. A significant positive Tajima’s D value, at the opposite, indicates an excess of high frequency variants as expected under balancing selection or diversifying selection. It can also reflect population structure or bottleneck events. We calculated the rate of synonymous (dS) and non-synonymous (dN) substitution according to Nei’s [44] method. The  ratio should provide insights into the long-term selective pressures on a full gene or a particular gene region and allows us to identify purifying selection
ratio should provide insights into the long-term selective pressures on a full gene or a particular gene region and allows us to identify purifying selection  and positive selection
and positive selection . As the later criteria for positive selection is overly stringent because positive selection often only acts on functional domain [49] we also performed a sliding window analysis with window length of 100 bp and step of 25 bp. We finally performed Hudson Kreitman and Aguade (HKA) test [50] and McDonald & Kreitman (MDK) test [51] with publicly available sequences of E. pillularis Smith for CesA1 (NCBI sequence AB591266.1), CesA3 (NCBI sequence AB591273.1) and CAD2 (NCBI sequence AB591254.1) genes as outgroup sequences. HKA and MDK neutrality tests are based on polymorphism to divergence comparison and are much less sensitive to demographic history than neutrality tests based on site frequency spectrum.
. As the later criteria for positive selection is overly stringent because positive selection often only acts on functional domain [49] we also performed a sliding window analysis with window length of 100 bp and step of 25 bp. We finally performed Hudson Kreitman and Aguade (HKA) test [50] and McDonald & Kreitman (MDK) test [51] with publicly available sequences of E. pillularis Smith for CesA1 (NCBI sequence AB591266.1), CesA3 (NCBI sequence AB591273.1) and CAD2 (NCBI sequence AB591254.1) genes as outgroup sequences. HKA and MDK neutrality tests are based on polymorphism to divergence comparison and are much less sensitive to demographic history than neutrality tests based on site frequency spectrum.
2.7. Determining Selective Status of Candidate Genes Accounting for Demographic History
Demographic history strongly impact sequence polymorphism pattern and it is necessary to account for it while searching for selection imprint on candidate genes. To achieve that aim, we build the expected null distribution of several summary statistics for sequence data, conditional to the most likely demographic scenario inferred from the SSR data set, and used it to assess neutrality status of the candidate genes. We built this conditional distribution and detected outlier sequences using EGGLIB Python/C++ library and its ABC commands [52] . This library provides tools to easily perform coalescent simulations from inferred parameter posteriors and determine outlier status of candidate loci. Since DYIABC and EGGLIB do not use the same scenario parameters and that SSR and SNPs have different mutation rate and pattern, it is not possible to use the parameter posterior distributions inferred from SSR with DIYABC in our candidate gene analysis. We thus first calibrate scenario parameters for sequence data by running a new ABC analysis using EGGLIB, with candidate gene sequences as observed data and investigating only the most likely scenario inferred from SSR data. We simulated 106 data sets, using uninformative priors and accounting for gene recombination. As summary statistics, we recorded average ΘW and the relative frequency of 4 classes of minor allele frequency i.e. overall proportion of all polymorphic sites from all loci with minor allele frequency: below 0.125, between 0.125 and 0.25, between 0.25 and 0.375 and under 0.375. Parameter posterior distribution was established through simple rejection procedure [53] , keeping the 1% simulated data points closest to the observed data and applying a log transformation. To further proceed with posterior based coalescent simulations, we transformed the obtained continuous parameter posterior distributions into a single discrete multivariate distribution. We used the discrete multivariate posterior distribution of the parameters to proceed with two model checking procedures. As the studied candidate genes have different length, we simulated 10,000 times a 500 bp fragment from the posterior parameter distribution to be compared with the set of 500 bp long non overlapping fragments obtained through sliding window procedure on our candidate loci. We first applied a procedure similar to that used by DIYABC, by positioning the mean π, ΘW and Tajima’s D calculated over the 44 observed 500 bp long non overlapping fragment in the corresponding posterior predictive distribution. We then compared the posterior predictive and observed distributions of π, ΘW and Tajima’s D using Kolmogorov-Smirnov [54] test. We finally performed outlier detection by identifying the 500 bp observed fragments with Tajima’s D outside the 95% bilateral confidence interval of the posterior predictive distribution. Such outliers were considered as presenting evidence for selection signature.
3. Results
3.1. Candidate Gene Primary Structure
Each gene consensus sequence was built by alignment of the overlapping fragments. The consensus sequence was then compared with the most similar gene used for the primer design in order to predict the 5’ and 3’ UTR, as well as the exonic and intronic regions. Hundred percent of the E. urophylla CAD2 gene (EuCAD2) was sequenced with a total length of 5408 bp, containing about 2500 bp of promoter region (GQ387647). We found two EuC4H copies and hundred percent of both EuC4H1 and EuC4H2 genes were sequenced. The sequenced genomic DNA length was 5020 bp for EuC4H1 (JX270996) and 2850 bp for EuC4H2 (JX270997). The two sequences were aligned with the three C4Hs of Populus trichocarpa Torr & A. Gray ex Hook and the two C4Hs of Populus tremuloides Michx to identify the localization of the exons and introns which have standard GT/AG splicing sites. A dramatic difference between the two EuC4Hs primary structures was observed: EuC4H1 contains 3 exons (785 pb, 134 pb and 599 pb) and 2 introns (676 pb, 1294 pb), while EuC4H2 had 2 exons (866 pb, 745 pb) and 1 intron (105 pb). Actually, the sequence of the EuC4H2 exon2 was highly similar to the exon 2 and the exon 3 of EuC4H1. At the nucleotide level, the similarity between the whole genes EuC4H1 and EuC4H2 is only 25%. However, when we compare the coding sequences, there is 62% similarity. The length of the sequenced EuCesA1 (JX270998), EuCesA2 (JX70999) and EuCesA3 (JX271000) genes are, respectively, 6723 bp, 5529 bp and 5493 bp. EuCesA1 and EuCesA3 have 13 exons while EuCesA2 has 12 exons. When we compare the mRNA nucleotide sequence for each copy pair, we obtain 62% to 64% of similarity. As for the whole sequence of each gene, we get 41% to 46% of similarity.
3.2. Nucleotide Diversity of Candidate Genes
Each gene was sequenced on 18 individuals (Table 1). Sequences are available from GenBank under accession nos. KF970937- KF971152. On the full sample, the number of sites without missing data and gaps varied from 1027 bp (EuC4H2) to 4615 (EuCAD2), with number of segregating sites S varying from 39 (EuC4H2) to 170 (EuCAD2). Taking all genes together, we had 19,798 sites without any gap or missing data, cumulating 661 segregating sites. The overall level of diversity was 0.00651 for π and 0.00744 for θw. However, at a gene level, the diversity varied on a twofold extends: for π, from 0.00479 (EuCesA1) to 0.00899 (EuC4H2) and for θw, from 0.0051 (EuCesA3) to 0.00916 (EuC4H2). For the six studied genes,  ratio always stayed bellow one at the full length scale, varying on almost a ten fold extend, from 0.0167 for EuCesA3 to 0.1341 for EuCesA2. Considering the sliding window analysis with window length of 100 bp and step of 25 bp, 3 gene regions exhibited
ratio always stayed bellow one at the full length scale, varying on almost a ten fold extend, from 0.0167 for EuCesA3 to 0.1341 for EuCesA2. Considering the sliding window analysis with window length of 100 bp and step of 25 bp, 3 gene regions exhibited 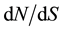 ratio over 1:1.786 between 1360 and 1509 bp in EuCesA2 (beginning of exon 5), 1.029 between 3363 and 3462 bp in EuCesA3 (exon 9) and 2.73 between 5343 and 5442 bp in EuCesA1 (exon12). Tajima’s D values measured at whole gene scale varied from −1.05718 (EuC4H1) to 0.31332 (EuCesA3) and none of them were significantly different from zero according to standard test procedure based on Wright Fisher equilibrium hypothesis. Summary statistics recorded on each gene of the full sample are presented in Table 2. Table 3 presents detailed value of π, θw and Tajima’s D for fragment defined in the 500 bp non overlapping fragment set. Note that three of these fragments have no polymorphism at all. Neither HKA nor MDK test results indicated significant deviation from neutral expectation for the evaluated genes (EuCesa1, EuCesa3 and EuCad2).
ratio over 1:1.786 between 1360 and 1509 bp in EuCesA2 (beginning of exon 5), 1.029 between 3363 and 3462 bp in EuCesA3 (exon 9) and 2.73 between 5343 and 5442 bp in EuCesA1 (exon12). Tajima’s D values measured at whole gene scale varied from −1.05718 (EuC4H1) to 0.31332 (EuCesA3) and none of them were significantly different from zero according to standard test procedure based on Wright Fisher equilibrium hypothesis. Summary statistics recorded on each gene of the full sample are presented in Table 2. Table 3 presents detailed value of π, θw and Tajima’s D for fragment defined in the 500 bp non overlapping fragment set. Note that three of these fragments have no polymorphism at all. Neither HKA nor MDK test results indicated significant deviation from neutral expectation for the evaluated genes (EuCesa1, EuCesa3 and EuCad2).
3.3. Genetic Structure Analysis on SSR Data
Using Structure, we found no cryptic population structure. Increasing group number from 1 to 4 did not increase likelihood model. Average likelihood for K = 1 was −3727, average likelihood for K = 2 was −3764, average likelihood for K = 3 was −3822 and average likelihood for K = 4 was −4122 with low standard error between repetition of K (from 3.28 for K = 1 to 384.05 for K = 1). As an additional proof of no population structure, examination of individual repartition among group for K > 1 shows high admixture level for individual with minor group contribution.
3.4. Demographic History of E. urophylla in Timor Island Based on Microsatellite Data
The reference table was built with a total of 513,800 simulated data points, evenly distributed across scenarios: 102,500 for scenario SNM, 102,361 for scenario BOT, 102,951 for scenario EXP, 102,797 for scenario EXPBOT and 103,191 for scenario BOTEXP. Logistic approach indicated the highest posterior probability for scenario EXP with a mean value of 0.527 and confidence interval [0.502; 0.551]. BOTEXP and EXPBOT had lower probability, respectively 0.311 with confidence interval [0.287, 0.334] for BOTEXP and 0.134 with con-
Table 2. Diversity pattern on candidate genes.
l: length in base pair, excluding gap and missing data, S: number of segregating sites, π: pi, θw: θ defined by Watterson, Dtaj: Tajima’s D, dN/dS: ratio of non synonymous to synonymous polymorphism.
Table 3. Description and summary statistics for the set of 500 bp non overlapping fragment.
“Firstbase” and “lastbase” define the fragment boundaries; π: pi (ref) θw: θ defined by Watterson (ref), Dtaj: Tajima’s D; “Sim P value” indicated significant P values for Tajima’s D calculated using Tajima’s D expectation under sudden expansion demographic history and no selection, *: 0.025 > P and P > 0.975, **: 0.005 > P and P > 0.995.
fidence interval [0.119, 0.149] for EXPBOT. The two remaining scenarios have considerably lower probability, below 1% for scenario BOT and below 5% for scenario SNM. We kept only the scenario EXP for further analysis. Using the new reference table constructed for model checking and using only 2 summary statistics, scenario EXP still appears to be the most likely scenario. Model checking procedure further indicated that scenario EXP is compatible with observed data with a P(simulated < observed) of 0.265 for mean genetic diversity and of 0.403 for mean Garza-Williamson’s M. On the contrary, scenario BOTEXP, the second scenario in terms of posterior probability, was not compatible with observed data as P(simulated < observed) for mean genetic diversity was 0.976. Main characteristics of parameter posterior distributions for scenario EXP are presented in Table 4. For most parameters, the 95% confidence interval appeared to be broad, but examination of the posterior distributions indicated that they generally markedly differed from the priors (Figure S1). The posterior distribution of N2/Ne indicates a marked expansion (5% confidence interval [0.001; 0.110]), with a median value of 0.014. Confidence in parameter estimate is good (Table 4), with a mean bias below 1, except for N2 (mean bias 1.448) and Fact 2 above 0.7 (except again for N2, Fact 2 = 0.634). These values indicate that parameter estimation is reasonably good most parameters except N2. We finally assessed confidence in scenario choice by calculating associated power and alpha risk. The power to identify scenario EXP is good (81.5%) and its alpha risk is moderate (20.35%).
3.5. Determining Selective Status of Candidate Genes Accounting for Demographic History
Following results obtained from ABC analysis conducted on SSR data, we build the expected null distribution of several summary statistics for sequence data conditional to the EXP scenario and used it to assessed neutrality status of the candidate genes. We first evaluated the EXP scenario parameters using candidate gene data. Main characteristics of parameter prior and posterior distributions for scenario EXP are presented in Table 5 and they are graphically displayed in Figure S2. Posterior median value for the ratio of ancestral size to actual size is 0.395, with corresponding 95% being [0.098; 0.778]. Median posterior value for current population size (4μN0) was 0.011 with a narrow confidence interval [0.007; 0.025]. For expansion time expressed in 4N0 generations, median value was 0.273 with broad confidence interval ([0.026; 0.480]).
The parameter posterior distributions obtained for scenario EXP were further used to simulate ten thousand 500 bp long sequence fragments. Tajima’s D, ΘW and π values were calculated on each simulated fragment and used to determine a 95% bilateral confident interval characterizing the expected values of these statistics under a neutral model. The two model checking procedures indicated that observed data are consistent with EXP demographic model and inferred parameters. Positioning Tajima’s D values calculated on the 500 bp non-overlapping observed fragment set allow identifying two fragments presenting outlying values as indicated in Table 3. These two fragments, located in EuCesA2 and EuCesA3 are positioned on Figure 3 along with gene exonic structure, 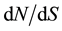 and Tajima’s D sliding window analysis and SNPs associated with wood traits. The first fragment is positioned in EuCesA3, between base 4236 and 5132 and has a positive Tajima’s D (1.395, P value < 0.025). The second outstanding fragment is positioned in EuCesA2, between bases 1 to 922 and has also a positive Tajima’s D value (1.700, P < 0.025) Note that this EuCesA2 fragment contains 2 SNPs found to be associated with Acid Soluble Lignin by Denis et al. [17] .
and Tajima’s D sliding window analysis and SNPs associated with wood traits. The first fragment is positioned in EuCesA3, between base 4236 and 5132 and has a positive Tajima’s D (1.395, P value < 0.025). The second outstanding fragment is positioned in EuCesA2, between bases 1 to 922 and has also a positive Tajima’s D value (1.700, P < 0.025) Note that this EuCesA2 fragment contains 2 SNPs found to be associated with Acid Soluble Lignin by Denis et al. [17] .
Table 4. Parameter estimates for scenario EXP (demographic expansion) using the Approximate Bayesian Computation approach (Cornuet et al. 2008, 2010).
IC 95% indicates 95% confidence interval; MeanBias indicates the average relative bias and Fact2 the average proportion of pseudo observed data set for which the point estimate is at least half and at most twice the true value; N2: ancestral effective population size; Ne: current effective population size; Te: time of expansion in generation number; Mean_μmic: mean mutation rate; Mean_P is average parameter of the Generalized Stepwise model geometric distribution.
Table 5. Prior specifications and posterior estimates for demographic parameter of scenario 3 as inferred from 6 candidate genes using ABC approach (De Mita & Siol 2011).
THETA defines actual population size ´ mutation rate, ANCSIZE is the ratio of ancestral population size to actual population size and DUR indicates the duration of expansion. For each of these three parameters, prior distribution shape (U: uniform) is given with minimum (min) and maximum (max) values. Posterior mean, median and 95% confidence intervals (IC95) are also presented.
4. Discussion
We studied the diversity pattern of six full length candidate genes involved in wood formation in E. urophylla, a tree originating from the Sunda Island and presenting high diversity in various phenotypic traits. This work brings original results to understand impact of natural selection and demography on natural populations for genes coding for crucial traits in trees.
4.1. Genetic Structure, Demographic History of E. urophylla and Consequence on Polymorphism Pattern
E. urophylla is among the rare eucalypts species not originating from Australia. It was formerly found to be not structured at the nuclear level [22] [31] and it is believed to have undergone several cycles of population expansion and bottleneck in relationship with the volcanic nature of Sunda Islands [31] . Our microsatellite data analysis based on a tree sample from seeds originating from a small region of Timor Island is consistent with the absence of genetic structure and with a sudden demographic expansion. If local extinctions due to volcanic eruption may have occurred, it didn’t affect markedly overall diversity pattern in the sampled region where the E. urophylla populations are known to be large and stable. This expansion is confirmed by the ABC analysis we performed on gene sequence data set alone (results not shown). Previous studies on E. urophylla didn’t investigate possible demographic expansion but only tested bottleneck [32] . One of the main consequences of population expansion on polymorphism pattern is an excess of rare variants and thus negatively skewed Tajima’s D values for sequence data. In such a situation, not accounting for population demography may lead to the detection of false positive hits for positive directional selection.
The two data sets we used (microsatellites and genes) are very different in terms of sample size and marker mutation rate and modality. However, it is interesting to compare their respective posterior distribution for ance-
 (a)
(a) (b)
(b)
Figure 3. Sliding window analysis of EuCesA2 (a) and EuCesA3 (b) genes with window length of 100 bp and 25 bp steps. Black thin lines indicate Tajima’s D value and grey thick lines figure dN/dS ratio. Dashed lines materialize the dN/dS threshold value of one. At the bottom of each graph gene structure was schematized with numbered exons represented by grey boxes. 500 bp long regions with outstanding Tajima’s D value are represented by a black horizontal thick line and SNPs found to be associated with wood traits are identified by white triangles (Denis et al. 2013)
stral to current population size ratio in ABC expansion scenario. Indeed it does not depend on mutation rate and is a measure of expansion intensity. Expansion appears much stronger in microsatellites data set (median value of ANCSIZE, the ratio of ancestral population size to actual population size, is 0.014% and 95% confidence interval [0.001; 0.110]) than in gene data set (median value 0.395% and 95% confidence interval [0.098; 0.778]). Two explanations can be proposed for this discrepancy. On the first hand, some uncertainties can be associated with parameter posterior distribution estimation from ABC procedure. For example, in the ABC analysis performed with the microsatellite data set, the mean bias for ancestral size N2 is 1.448, which is beyond the threshold of 1. Poor parameter posterior estimation is often the result of an observed data set insufficiently informative [42] . On the second hand, our gene data set is built only with wood formation genes that are likely impacted by natural selection. For example, balancing selection may generate intermediate frequency variants that may attenuate demographic expansion signal. Finally, the choice of the proper posterior demographic parameters is very important and critical because it strongly affects false discovery rate. In the particular case of this study, we choose to be conservative and to use the posteriors inferred from the candidates themselves to draw neutral expectations, with in mind the fact that they may not exactly correspond to selective neutrality.
4.2. Selection Signature in Wood Related Genes of E. urophylla
We investigated possible natural selection imprint on wood formation candidate genes using several complementary approaches: a test based on site frequency spectrum (Tajima’s D) with ad-hoc correction for neutral expectations, non-synonymous comparison to synonymous polymorphism ( ratios) and two selection tests based on polymorphism to divergence comparison (MDK and HKA tests). As the latter approach required an outgroup sequence, we were not able to implement the two corresponding tests on the full sequence data set.
ratios) and two selection tests based on polymorphism to divergence comparison (MDK and HKA tests). As the latter approach required an outgroup sequence, we were not able to implement the two corresponding tests on the full sequence data set.
None of the studied gene exhibited a clear selection imprint on its full length but we could observe interesting trends in global dN/dS between copies of the same gene family, witnessing possible contrasted constrain level. According to Ohno’s [56] gene family classical model, contrasted constrain level can be expected as at least one member of the family should maintain the original function of the gene, and the other copies possibly undergoing pseudogenization or subfunctionalization. The three members of the CesA gene family we studied are all involved in secondary cell wall formation and possess homologous counterparts across angiosperms [27] . While these three genes seem to be co-expressed in secondary tissues, E. grandis CesA3 copy was found to display the highest expression level in Ranik et al. [27] study. In this work, the three CesA gene copies present very contrasted  ratio, with almost a 10-fold ratio between EuCesA3 (0.0167) and EuCesA2 (0.1341). While these
ratio, with almost a 10-fold ratio between EuCesA3 (0.0167) and EuCesA2 (0.1341). While these  ratios indicate purifying selection in both cases, they suggest more constraints on EuCesA3 than in EuCesA1 and EuCesA2. Similarly, EuC4H2 (
ratios indicate purifying selection in both cases, they suggest more constraints on EuCesA3 than in EuCesA1 and EuCesA2. Similarly, EuC4H2 ( ratio of 0.0217) appears to be more constrained than EuC4H1 (0.1207). The only C4H copy of A. thaliana is expressed in all tissues and responds to light, wounding and fungal infection [28] , indicating that it plays various functions in phenylpropanoid metabolism that open a door for subfunctionalization of gene copies. Indeed citrus sinensis C4H1, ortholog of EuC4H2 is wound inducible unlike C4H2, ortholog of EuC4H1 [30] .
ratio of 0.0217) appears to be more constrained than EuC4H1 (0.1207). The only C4H copy of A. thaliana is expressed in all tissues and responds to light, wounding and fungal infection [28] , indicating that it plays various functions in phenylpropanoid metabolism that open a door for subfunctionalization of gene copies. Indeed citrus sinensis C4H1, ortholog of EuC4H2 is wound inducible unlike C4H2, ortholog of EuC4H1 [30] .
Based on site frequency approaches and by focusing on the 500 bp length non-overlapping fragment set, we found two 500 bp regions that could bear the imprint of natural selection. These two regions both present an excess of sites in intermediate frequencies that can be interpreted as balancing selection. The first region is located between bases 4236 and 5232 of EuCesA3 and covers part of intron 11, exon 12, intron 12 and part of exon 13. This particular region does not itself contain a SNP investigated by Denis et al. [17] but a nearby SNP (EuCesA3-3854) was found to be associated with cellulose and extractive related traits [17] . However, as we do not have proper outgroup sequence for this EuCesA3 region, it is not possible to make any final conclusion about its selective status. The second region is located in EuCesA2 between 1 bp and 922 bp and encompasses the four first exons, the three first introns and part of the fourth exon. It contains two SNPs (EuCesA2-0762 and EuCesA2-853) found to be associated with acid soluble lignin by Denis et al. [17] , among them one being non-synonymous. We also do not have a outgroup sequence for this EuCesA2 particular region but Sexton et al. 2011 found an unexpectedly high concentration of trans-subgeneric conserved SNPs in the nearby promoter interpreted as a signal of balancing selection.
Besides our work, several studies reported evidence of balancing selection in CesA genes in evolutionary distant tree species: in Pinus pinaster (Aiton) [19] , Pinus radiata D. Don [18] , in Eucalyptus genus in general [55] and in E. urophylla [56] in particular. This suggests the wide adaptive importance of CesA genes across trees and raises the question of functional significance of this result. As pinpointed by some authors, wood formation genes may play an important role in plant defence [57] , [58] although the modalities are not clear.
Authors’ Contributions
LCK: data analysis BF, SU DNA extraction and gene study, JP: SSR genotyping, LC-K, BF, SU JMB: manuscript preparation. JMB: funding and overall supervision. All authors read and approved the final manuscript.
References
- Chave, J., Coomes, D., Jansen, S., Lewis, S.L., Swenson, N.G. and Zanne, A.E. (2009) Towards a Worldwide Wood Economics, Spectrum. Ecology Letters, 12, 351-366. http://dx.doi.org/10.1111/j.1461-0248.2009.01285.x
- Weedon, J.T., Cornwell, W.K., Cornelissen, J.H.C., Zanne, A.E., Wirth, C. and Coomes, D.A. (2009) Global Meta- Analysis of Wood Decomposition Rates: A Role for Trait Variation among Tree Species? Ecology Letters, 12, 45-56. http://dx.doi.org/10.1111/j.1461-0248.2008.01259.x
- Gerrienne, P., Gensel, P.G., Strullu-Derrien, C., Lardeux, H., Steemans, P. and Prestianni, C. (2011) A Simple Type of Wood in Two Early Devonian Plants. Science, 333, 837. http://dx.doi.org/10.1126/science.1208882
- Zhang, S.-B., Slik, J.W.F., Zhang, J.-L. and Cao, K.-F. (2011) Spatial Pattern of Wood Traits in China Are Controlled by Phylogeny and the Environment. Global Ecology and Biogeography, 20, 241-250. http://dx.doi.org/10.1111/j.1466-8238.2010.00582.x
- Zobel, B.J. and van Buijtenen, J.P. (1989) Wood Variation. Its Causes and Control. Springer-Verlag, Heidelberg. http://dx.doi.org/10.1007/978-3-642-74069-5
- Allona, I., Quinn, M., Shoop, E., Swope, K., Cyr, S.S., Carlis, J., et al. (1998) Analysis of Xylem Formation in Pine by cDNA Sequencing. Proceedings of the National Academy of Sciences, 95, 9693-9698. http://dx.doi.org/10.1073/pnas.95.16.9693
- Paux, E., Carocha, V., Marques, C., de Sousa, A.M., Borralho, N., Sivadon, P. and Grima-Pettenati, J. (2005) Transcript Profiling of Eucalyptus Xylem Genes during Tension Wood Formation. New Phytologist, 167, 89-100. http://dx.doi.org/10.1111/j.1469-8137.2005.01396.x
- Pavy, N., Paule, C., Parsons, L., Crow, J., Morency, M.-J., Cooke, J., Johnson, J., Noumen, E., Guillet-Claude, C., Butterfield, Y., et al. (2005) Generation, Analysis and Database Integration of 16,500 White Spruce EST Clusters. BMC Genomics, 6, 144. http://dx.doi.org/10.1186/1471-2164-6-144
- Cato, S., McMillan, L., Donaldson, L., Richardson, T., Echt, C. and Gardner, R. (2006) Wood Formation from the Base to the Crown in Pinus radiata, Gradients of Tracheid Wall Thickness, Wood Density, Radial Growth Rate and Gene Expression. Plant Molecular Biology, 60, 565-581. http://dx.doi.org/10.1007/s11103-005-5022-9
- Yuan, Z., Yao, X., Zhang, D.B., Sun, Y. and Huang, H. (2007) Genome-Wide Expression Profiling in Seedlings of the Arabidopsis Mutant uro That Is Defective in the Cell Wall Formation. Journal of Integrative Plant Biology, 49, 1754- 1762. http://dx.doi.org/10.1111/j.1744-7909.2007.00586.x
- Qiu, D., Wilson, I.W., Gan, S., Washusen, R., Moran, G.F. and Southerton, S.G. (2008) Gene Expression in Eucalyptus Branch Wood with Marked Variation in Cellulose Microfibril Orientation and Lacking G-Layers. New Phytologist, 179, 94-103. http://dx.doi.org/10.1111/j.1469-8137.2008.02439.x
- Li, X., Wu, H., Dillon, S. and Southerton, S.G. (2009) Generation and Analysis of Expressed Sequence Tags from Six Developing Xylem Libraries in Pinus radiata D. Don. BMC Genomics, 10, 1-18. http://dx.doi.org/10.1186/1471-2164-10-41
- Thumma, B.R., Nolan, M.R., Evans, R. and Moran, G.F. (2005) Polymorphisms in Cinnamoyl CoA Reductase (CCR) Are Associated with Variation in Microfibril Angle in Eucalyptus spp. Genetics, 171, 1257-1265. http://dx.doi.org/10.1534/genetics.105.042028
- Thumma, B.R., Matheson, B.A., Zhang, D.Q., Meeske, C., Meder, R., Downes, G.M. and Southerton, S.G. (2009) Identification of a Cis-Acting Regulatory Polymorphism in a Eucalypt COBRA-Like Gene Affecting Cellulose Content. Genetics, 183, 1153-1164. http://dx.doi.org/10.1534/genetics.109.106591
- Dillon, S.K., Brawner, J.T., Meder, R., Lee, D.J. and Southerton, S.G. (2012) Association Genetics in Corymbia citriodora subsp. Variegate Identifies Single Nucleotide Polymorphisms Affecting Wood Growth and Cellulosic Pulp Yield. New Phytologist, 195, 596-608. http://dx.doi.org/10.1111/j.1469-8137.2012.04200.x
- Mandrou, E., Hein, P.R.G., Villar, E., Vigneron, P., Plomion, C. and Gion, J.-M. (2012) A Candidate Gene for Lignin Composition in Eucalyptus: Cinnamoyl-CoA Reductase (CCR). Tree Genetics Genomes, 8, 353-364. http://dx.doi.org/10.1007/s11295-011-0446-7
- Denis, M., Favreau, B., Ueno, S., Camus-Kulandaivelu, L., Chaix, G., Gion, J.M., Nourrisier-Montou, S., Polidori, J. and Bouvet, J.M. (2013) Genetic Variation of Wood Chemical Traits and Association with Underlying Genes in Eucalyptus urophylla. Tree Genetics and Genomes, 9, 927-942. http://dx.doi.org/10.1007/s11295-013-0606-z
- Pot, D., McMillan, L., Echt, C., Le Provost, G., Garnier-Géré, P., Cato, S. and Plomion, C. (2005) Nucleotide Variation in Genes Involved in Wood Formation in Two Pine Species. New Phytologist, 167, 101-112. http://dx.doi.org/10.1111/j.1469-8137.2005.01417.x
- Gonzalez-Martinez, S.C., Erzoz, E., Brown, G.R., Wheeler, N.C. and Neale, D.B. (2006) DNA Sequence Variation and Selection of Tag Single-Nucleotide Polymorphisms at Candidate Genes for Drought-Stress Response in Pinus taeda L. Genetics, 172, 1915-1926. http://dx.doi.org/10.1534/genetics.105.047126
- Martin, B. and Cossalter, C. (1975-1976) Les Eucalyptus des Iles de la Sonde. 1re partie. Bois et Forêt des Tropiques 163, 164, 165, 166, 167, 168.
- Eldridge, K., Davidson, J., Harwood, C. and Van Wyk, G. (1993) Eucalypt Domestication and Breeding. Oxford University Press, Oxford.
- Tripiana, V., Bourgeois, M., Verhargen, D., Vigneron, P. and Bouvet, J.-M. (2007) Combining Microsatellites, Growth, and Adaptive Traits for Managing in Situ Genetic Resources of Eucalyptus urophylla. Canadian Journal for Forest Research, 37, 773-785. http://dx.doi.org/10.1139/X06-260
- Mueller, S.C. and Brown Jr., R.M. (1980) Evidence for an Intramembranous Component Associated with a Cellulose Microfibril Synthesizing Complex in Higher Plants. Journal of Cell Biology, 84, 315-326. http://dx.doi.org/10.1083/jcb.84.2.315
- Kimura, S., Laosinchai, W., Itoh, T., Cuib, X., Linderc, C.R. and Brown Jr., R.M. (1999) Immunogold Labeling of Rosette Terminal Cellulose-Synthesizing Complexes in the Vascular Plant Vigna angularis. Plant Cell, 11, 2075-2085. http://dx.doi.org/10.1105/tpc.11.11.2075
- Herth, W. (1983) Arrays of Plasma-Membrane “Rosettes” Involved in Cellulose Microfibril Formation of Spirogyra. Planta, 159, 347-356. http://dx.doi.org/10.1007/BF00393174
- Burton, R.A., Shirley, N.J., King, B.J., Harvey, A.J. and Fincher, G.B. (2004) The CesA Gene Family of Barley. Quantitative Analysis of Transcripts Reveals Two Groups of Co-Expressed Genes. Plant Physiology, 134, 224-236. http://dx.doi.org/10.1104/pp.103.032904
- Ranik, M. and Myburg, A.A. (2006) Six New Cellulose Synthase Genes from Eucalyptus Are Associated with Primary and Secondary Cell Wall Biosynthesis. Tree Physiology, 26, 545-556. http://dx.doi.org/10.1093/treephys/26.5.545
- Raes, J., Rohde, A., Christensen, J.H., van de Peer, Y. and Boerjan, W. (2003) Genome-Wide Characterization of the Lignification Toolbox in Arabidopsis. Plant Physiology, 133, 1051-1071. http://dx.doi.org/10.1104/pp.103.026484
- Nedelkina, S., Jupe, S.C., Blee, K.A., Schalk, M., Werck-Reichert, D. and Bolwell, G.P. (1999) Novel Characteristics and Regulation of a Divergent Cinnamate 4-Hydroxylase (CYP73A15) from French Bean: Engineering Expression in Yeast. Plant Molecular Biology, 39, 1079-1090. http://dx.doi.org/10.1023/A:1006156216654
- Betz, C., McCollum, T.G. and Mayer, R.T. (2001) Differential Expression of Two Cinnamate 4-Hydroxylase Genes in “Valencia” Orange (Citrus sinensis Osbeck). Plant Molecular Biology, 46, 741-748. http://dx.doi.org/10.1023/A:1011625619713
- Payn, K.G., Dvorak, W.S., Janse, B.H. and Myburg, A.A. (2008) Microsatellite Diversity and Genetic Structure of the Commercially Important Tropical Tree Species Eucalyptus urophylla, Endemic to Seven Islands in Eastern Indonesia. Tree Genetics & Genomes, 4, 519-530. http://dx.doi.org/10.1007/s11295-007-0128-7
- Payn, K.G., Dvorak, W.S. and Myburg, A.A. (2007) Chloroplast DNA Phylogeography Reveals the Island Colonisation Route of Eucalyptus urophylla (Myrtaceae). Australian Journal of Botany, 55, 673-683. http://dx.doi.org/10.1071/BT07056
- Beaumont, M.A., Zhang, W.Y. and Balding, D.J. (2002) Approximate Bayesian Computation in Population Genetics. Genetics, 162, 2025-2035.
- Gawel, N. and Jarret, R.L. (1991) Cytoplasmic Genetic Diversity in Bananas and Plantains. Euphytica, 52, 19-23.
- Saghai-Maroof, M.A., Soliman, K.M., Jorgensen, R.A. and Allard, R.W. (1984) Ribosomal DNA Spacer-Length Polymorphisms in Barley, Mendelian Inheritance, Chromosomal Location, and Population Dynamics. Proceedings of the National Academy of Sciences of the United States of America, 81, 8014-8018. http://dx.doi.org/10.1073/pnas.81.24.8014
- Brondani, R.P.V., Brondani, C., Tarchini, R. and Grattapaglia, D. (1998) Development, Characterization and Mapping of Microsatellite Markers in Eucalyptus grandis and Eucalyptus urophylla. Theoretical and Applied Genetics, 97, 816- 827. http://dx.doi.org/10.1007/s001220050961
- Brondani, R.P.V., Brondani, C. and Grattapaglia, D. (2002) Towards a Genus-Wide Reference Linkage Map for Eucalyptus Based Exclusively on Highly Informative Microsatellite Markers. Molecular Genetics and Genomics, 267, 338- 347. http://dx.doi.org/10.1007/s00438-002-0665-6
- van der Nest, M.A., Steenkamp, E.T., Wingfield, B.D. and Wingfield, M.J. (2000) Development of Simple Sequence Repeat (SSR) Markers in Eucalyptus from Amplified Inter-Simple Sequence Repeats (ISSR). Plant Breeding, 119, 433-436. http://dx.doi.org/10.1046/j.1439-0523.2000.00515.x
- Dmitriev, D.A. and Rakitov, R.A. (2008) Decoding of Superimposed Traces Produced by Direct Sequencing of Heterozygous Indels. PLoS Computational Biology, 4, e1000113. http://dx.doi.org/10.1371/journal.pcbi.1000113
- Pritchard, J.K., Stephens, M. and Donnelly, P. (2000) Inference of Population Structure Using Multilocus Genotype data. Genetics, 155, 945-959.
- Hubisz, M.J., Falush, D., Stephens, M. and Pritchard, J.K. (2009) Inferring Weak Population Structure with the Assistance of Sample Group Information. Molecular Ecology Resources, 9, 1322-1332. http://dx.doi.org/10.1111/j.1755-0998.2009.02591.x
- Cornuet, J.M., Santos, F., Beaumont, M.A., Robert, C.P., Marin, J.-M., Balding, D.J., Guillemaud, T. and Estoup, A. (2008) Inferring Population History with DIYABC, a User Friendly Approach to Approximate Bayesian Computation. Bioinformatics, 24, 2713-2719. http://dx.doi.org/10.1093/bioinformatics/btn514
- Cornuet, J.M., Ravigné, V. and Estoup, A. (2010) Inference on Population History and Model Checking Using DNA Sequence and Microsatellite Data with the Software DIYABC (v1.0). Bioinformatics, 11, 401.
- Nei, M. (1987) Molecular Evolutionary Genetics. Columbia University Press, New York.
- Garza, J.C. and Williamson, E.G. (2001) Detection of Reduction in Population Size Using Data from Microsatellite Loci. Molecular Ecology, 10, 305-318. http://dx.doi.org/10.1046/j.1365-294x.2001.01190.x
- Rozas, J., Sanchez-DelBarrio, J.C., Messeguer, X. and Rozas, R. (2003) DnaSP, DNA Polymorphism Analyses by the Coalescent and Other Methods. Bioinformatics, 19, 2496-2497. http://dx.doi.org/10.1093/bioinformatics/btg359
- Watterson, G.A. (1975) Number of Segragating Sites in Genetic Models without Recombination. Theoretical Population Biology, 7, 256-276. http://dx.doi.org/10.1016/0040-5809(75)90020-9
- Tajima, F. (1989) Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics, 123, 585-595.
- Roth, C. and Liberles, D. (2006) A Systematic Search for Positive Selection in Higher Plants (Embryophytes). BMC Plant Biology, 6, 12. http://dx.doi.org/10.1186/1471-2229-6-12
- Hudson, R.R., Kreitman, M. and Aguade, M. (1987) A Test of Neutral Molecular Evolution Based on Nucleotide Data. Genetics, 116, 153-159.
- McDonald, J.H. and Kreitman, M. (1991) Adaptive Protein Evolution at the Adh Locus in Drosophila. Nature, 351, 652-654. http://dx.doi.org/10.1038/351652a0
- De Mita, S. and Siol, M. (2012) EggLib: Processing, Analysis and Simulation Tools for Population Genetics and Genomics. BMC Genetics, 13, 27. http://dx.doi.org/10.1186/1471-2156-13-27
- Przeworski, M. (2003) Estimating the Time since Fixation of a Beneficial Allele. Genetics, 164, 1667-1676.
- Corder, G.W. and Foreman, D.I. (2009) Nonparametric Statistics for Non-Statisticians: A Step-by-Step Approach. John Wiley & Sons, Inc., Hoboken. http://dx.doi.org/10.1002/9781118165881
- Sexton, T.R. (2011) Candidate Gene Discovery, Genotyping and Association with Wood Quality Traits in Eucalyptus pillularis (Blackbutt). Ph.D. Thesis, Southern Cross University, Lismore.
- Ohno, S. (1970) Evolution by Gene Duplication. Springer, New York, USA. http://dx.doi.org/10.1007/978-3-642-86659-3
- Quang, T.H., Hallingbäck, H., Gyllenstrand, N., von Arnold, S. and Claphan, D. (2012) Expression of Genes of Cellulose and Lignin Synthesis in Eucalyptus urophylla and Its Relation to Some Economic traits. Trees, 26, 893-901. http://dx.doi.org/10.1007/s00468-011-0664-5
- Hawkins, S. and Boudet, A. (2003) “Defence Lignin” and Hydroxycinnamyl Alcohol Dehydrogenase Activities in Wounded Eucalyptus gunnii. Forest Pathology, 33, 91-104. http://dx.doi.org/10.1046/j.1439-0329.2003.00308.x
Appendix
Table S1. Primers used for gene amplification.
Table S2. Prior setting for demographic scenario of E. urophylla in Timor using the Approximate Bayesian Computation approach (Cornuet et al. 2008, 2010) and based on 17 SSR loci.SNM Standard Neutral Model (constant population size), BOT: bottleneck, EXP: demographic expansion, EXPBOT: recent expansion and past bottleneck and BOTEXP: recent bottleneck and past expansion. N, N1, N3, N4, Nb, Ne, Nbe, Nr, Nex,






Figure S1. Density function for parameter prior (grey) and posterior (black) distributions for scenario 3. Ne and N2 define population sizes and Te expansion time expressed in generation number. Μ_micA is the average mutation rate and Pmean is the average parameter of the Generalized Stepwise model geometric distribution. Note that the effective prior of N2 and Ne are not uniforms because of the constraint Ne > N2 we apply on these parameters. We also represented Ne/N2 which represent the inverse of population expansion.








Figure S2. Density fonction for parameter prior (grey) and posterior (black) distributions for scenario 4 N3, Nr and N3 define population sizes. T1 defines bottleneck time and T2 defines expansion time expressed in generation number. Μ_micA is the average mutation rate and Pmean is the average parameter of the Generalized Stepwise model geometric distribution. Note that the effective prior of Nr, Nbe, N3, T1 and T2 are not uniforms because of the constraints (Nbe > Nr, Nr < N3 and T1 < T2) we apply on these parameters.
NOTES

*Corresponding author.