Open Journal of Molecular and Integrative Physiology
Vol.3 No.4(2013), Article ID:39569,8 pages DOI:10.4236/ojmip.2013.34024
The acid-base regulation by renal proximal tubule
![]()
Department of Internal Medicine, The University of Tokyo Hospital, Tokyo, Japan
Email: *georgeseki-tky@umin.ac.jp
Copyright © 2013 Shoko Horita et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 21 August 2013; revised 21 September 2013; accepted 28 September 2013
Keywords: PRTA; NHE; NBCe1; V-ATPase
ABSTRACT
The kidney plays quite an important role in the regulation of acid-base homeostasis. The dysfunction of renal acid-base regulation causes diseases such as developmental disorder, bone malformation, calcification of eye and brain, etc. In the kidney, this regulation is performed, to a considerable part, in the proximal tubule of the nephron. In the luminal side the key player is sodium-proton exchanger type 3 (NHE3), whereas sodium-bicarbonate cotransporter (NBCe1) plays the critical role in the basolateral side. In the cytoplasm there is carbonic anhydrase type 2 (CAII) that intermediates the conversion of CO2/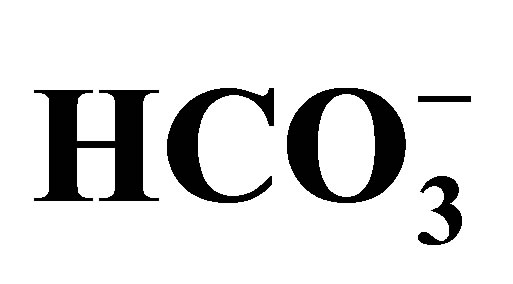 . Interestingly, in human, mutations have been found in NBCe1 and CAII but not in NHE3 so far. Mutations of NBCe1 lead to severe proximal renal tubular acidosis (pRTA) and other systemic manifestations. In animal model studies, however, the relative contribution of NHE3 to proximal tubule functions remains controversial. Recently, V-ATPase with renal specific subunits is suggested to have some roles in the regulation of proximal tubule functions. In this review, we will discuss the regulation of acid-base transport in the proximal tubule and the updates.
. Interestingly, in human, mutations have been found in NBCe1 and CAII but not in NHE3 so far. Mutations of NBCe1 lead to severe proximal renal tubular acidosis (pRTA) and other systemic manifestations. In animal model studies, however, the relative contribution of NHE3 to proximal tubule functions remains controversial. Recently, V-ATPase with renal specific subunits is suggested to have some roles in the regulation of proximal tubule functions. In this review, we will discuss the regulation of acid-base transport in the proximal tubule and the updates.
1. THE LUMINAL SIDE: NHES AND RENAL LUMINAL PROTON TRANSPORT
The Na+/H+ exchangers, NHEs mediate counter-transport, which translocate Na+ and H+ across the cell membrane [1-4]. In human there are ten NHEs, NHE1 to 9 and sperm-specific NHE [3,5].
The existence of NHEs in the kidney was first predicted by Pitts et al. [6,7]. Later Murer [8], Kinsella and Aronson [9] demonstrated the NHE activity by functional studies using brush border membrane vesicles. The first mammalian NHE, NHE1 was cloned by Sardet et al. in 1989 [10].
There are NHE3 and NHE8 in the luminal side of the proximal tubule [2,11-16]. In the mammal NHE3 exists not only in the apical side of renal proximal tubule and thick ascending limb, but also in the gastrointestinal tract, gall bladder, epididymis, and brain [2]. NHE3 consists of 834 amino acids and is thought to have twelve transmembrane domains and long C-terminal intracellular domain [17]. This is similar to other transporters, like Na+-glucose cotransporter and Na+, K+-2Cl− cotransporter.
About two-thirds of filtered NaCl and water are reabsorbed via NHEs in the luminal side of the proximal tubule [2]. NHEs play an important role in the reabsorption of bicarbonate as well, because they provide proton into the lumen, which is titrated with bicarbonate by carbonic anhydrase in the lumen then comes into the proximal tubular cell in the form of CO2. CO2 is again transformed to bicarbonate by intracellular carbonic anhydrase and reabsorbed via the basolateral membrane by Na+-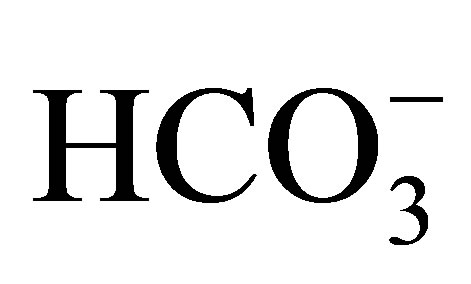 cotransporter (NBCe1).
cotransporter (NBCe1).
2. STUDIES IN NHE3−/− MICE: WHICH IS THE MAJOR PLAYER?
NHE3 and NHE8 exist predominantly in the luminal side of the renal proximal tubule. In neonatal mice and rats NHE8 is dominant. After weaning, NHE3 dominates in the proximal tubule instead of NHE8 [16,18]. It is still controversial about how much NHE3 contributes to apical H+ secretion, as NHE3 deficiency has not been found in human and the studies using NHE3−/− mice have shown the controversial results.
The first NHE3−/− mice was generated by Schultheis et al. [19]. The NHE3 null mice had only a mild metabolic acidosis of 3 to 5 mEq/l less than that of wild type mice. Wang et al. investigated the function of proximal convoluted tubules in NHE3−/− mice by in vivo microperfusion [20]. In wild-type mice the addition of 100 µM 5-(NEthyl-N-isopropyl) amiloride (EIPA), which should inhibit both NHE3 and NHE8 activities [21], reduced 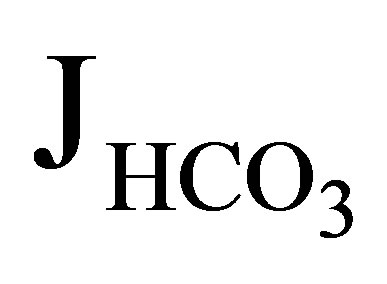 (rate of net
(rate of net 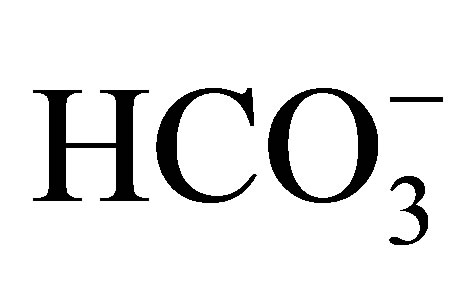 absorption) by approximately 50%. In NHE3 null mice, by contrast, the addition of EIPA did not reduce
absorption) by approximately 50%. In NHE3 null mice, by contrast, the addition of EIPA did not reduce  and Jv (rate of net fluid absorption). On the other hand, the addition of bafilomycin, a V-ATPase inhibitor, reduced
and Jv (rate of net fluid absorption). On the other hand, the addition of bafilomycin, a V-ATPase inhibitor, reduced 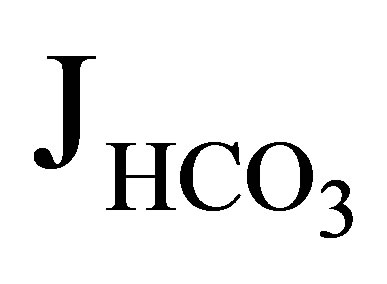 significantly (51.2 to 21.1 pmol/ min/mm) in these mice. However it did not affect Jv in both wild-type and NHE3 null mice. From these results, they concluded that NHE3 is largely, if not completely, responsible for the EIPA-sensitive NHE activity in the luminal side of the proximal tubules. These results further indicated that V-ATPase mediates a significant component of
significantly (51.2 to 21.1 pmol/ min/mm) in these mice. However it did not affect Jv in both wild-type and NHE3 null mice. From these results, they concluded that NHE3 is largely, if not completely, responsible for the EIPA-sensitive NHE activity in the luminal side of the proximal tubules. These results further indicated that V-ATPase mediates a significant component of 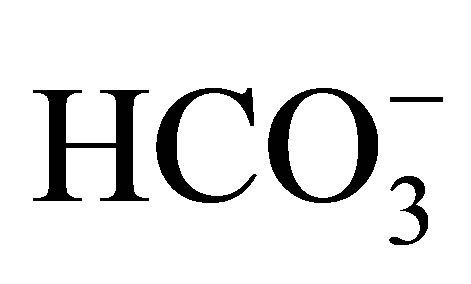 reabsorption in the proximal tubule.
reabsorption in the proximal tubule.
On the other hand, Baum et al. used NHE3−/−/NHE2−/− mice [22,23] to investigate the role of NHE3 in the proximal tubule. In in vitro microperfusion study, the luminal NHE activity in the proximal tubule of the NHE3−/− mice was about 50% of the wild-type, and most of the residual NHE activity was inhibited by 100 µM EIPA. The addition of 50 µM HOE694, which should suppress the NHE2 activity, had no effect, suggesting that NHE2 did not have a significant role in proximal tubule acidification. They speculated that there may be some residual apical EIPA-sensitive NHE activity. Another interpretation would be that EIPA, an inhibitor of NHEs, might inhibit Na+-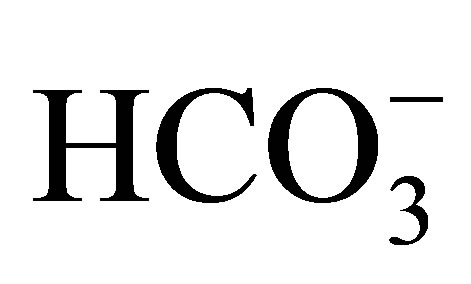 cotransport activity.
cotransport activity.
Recently, Baum et al. investigated whether NHE8 can compensate for the absence of NHE3 in mice [24]. NHE8 is shown to exist in the intestine, mediating the salt reabsorption in the intestine of the neonatal mice [17, 25]. In NHE3−/− mice the expression of NHE8 protein in the renal cortex and cortical brush border membrane was higher than that of wild type mice. The luminal NHE activity in the proximal tubule of NHE3+/+NHE8−/− mice was similar to that in wild-type mice. However, it was about 50% of wild-type in NHE3−/−NHE8+/+ mice, and about 10% of wild-type in NHE3−/−NHE8−/− mice. These results indicate that while NHE3 is the predominant NHE isoform in the luminal side of the proximal tubule, NHE8 can also contribute to the luminal NHE activity, especially in the absence of NHE3.
At present, the reason why Wang et al. failed to detect the EIPA-sensitive component in 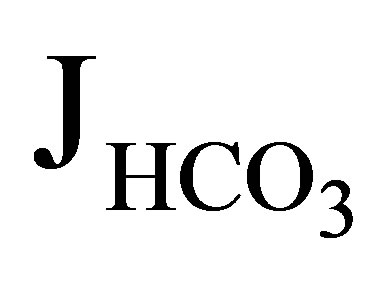 in NHE3−/− mice remains speculative, but the methodological difference may be responsible. Furthermore, the reason why NHE3−/−NHE8−/− mice showed only a mild acidosis similar to that in NHE3−/− mice remains unknown. Probably, future studies are required to definitely determine the relative contributions of NHE3, NHE8, and V-ATPase to the net proximal bicarbonate absorption.
in NHE3−/− mice remains speculative, but the methodological difference may be responsible. Furthermore, the reason why NHE3−/−NHE8−/− mice showed only a mild acidosis similar to that in NHE3−/− mice remains unknown. Probably, future studies are required to definitely determine the relative contributions of NHE3, NHE8, and V-ATPase to the net proximal bicarbonate absorption.
3. THE BASOLATERAL SIDE: NBCe1
There are a number of acid-base transporters in the basolateral side of the proximal tubule, among them NBCe1 plays a major role in the absorption of bicarbonate coupled with sodium [26]. NBCe1 was first cloned from salamander by Romero et al. [27]. Igarashi et al. found two patients who had renal proximal tubular acidosis (pRTA) with different NBCe1 mutations [28]. These patients also had band keratopathy, cataract, glaucoma, cerebral calcification, mental retardation and short stature.
NBCe1 is encoded by the gene SLC4A4 and has five splicing variants [29,30], among them NBCe1A is mainly expressed in the proximal tubule. NBCe1B is expressed widely in many tissues including the pancreas duct and corneal endothelium, while NBCe1C is almost limited in the brain [31-33]. The structures of major NBCe1 variants are shown in Figure 1 [26].
NBCe1A and NBCe1B differ only in the N-terminus, whereas NBCe1C is identical to NBCe1B in the N-terminus but differs in C-terminus. The N-terminus of NBCe1B and NBCe1C has a binding site of IRBIT, inositol 1,4,5-triphosphate (IP3) receptor-binding protein [34].
One of the proposed models of NBCe1 topology is shown in Figure 2 [29,35,36]. This is hypothesized based on the model of anion exchanger 1 [37]. According to
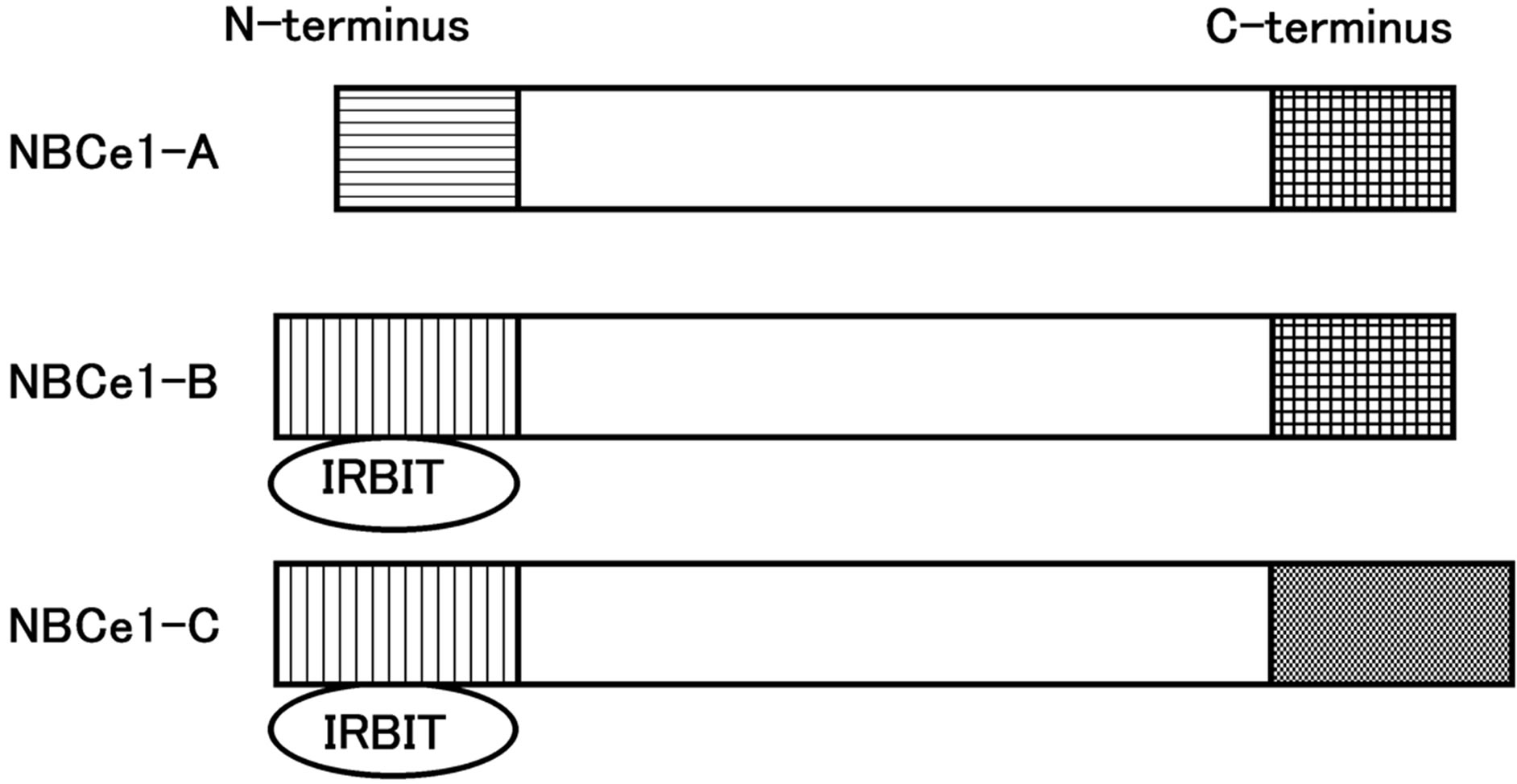
Figure 1. The structure of major NBCe1 variants.
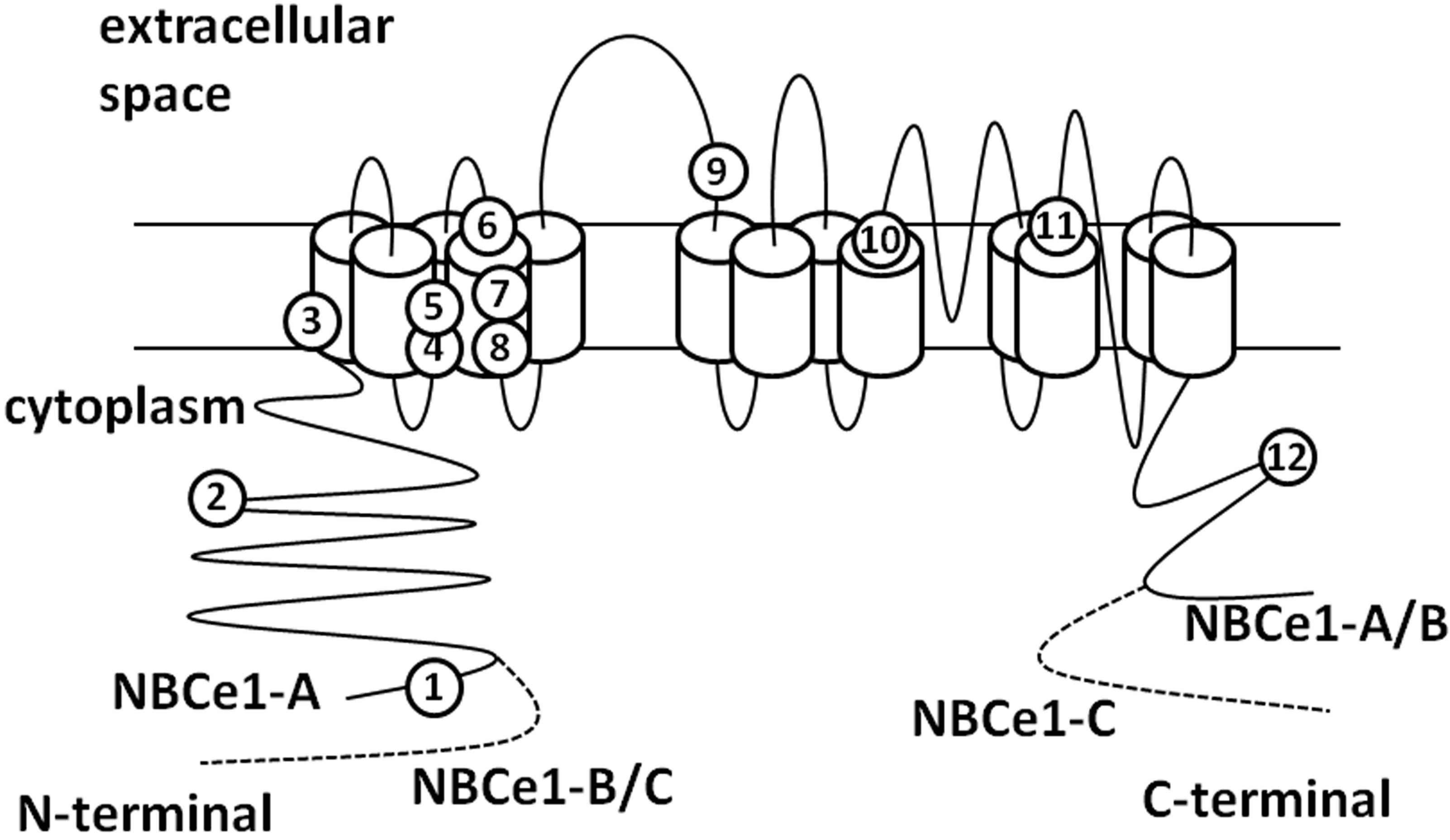
Figure 2. The topology of NBCe1. The numbers show the sites of pRTA-related mutations as follows: 1: Q29X, 2: R298S, 3: S427L, 4: T485S, 5: G486R, 6: R510H, 7: W516X, 8: L522P, 9: N721TxfsX29, 10: A799V, 11: R881C, 12: S982NfsX4.
this model, there are 13 transmembrane domains and a long extraloop with glycosylation sites between TM5 and TM6. Kurtz and colleagues propose another topology model of NBCe1A with 14 TM sites [38].
4. NBCe1 MUTATIONS AND RENAL TUBULAR ACIDOSIS
Igarashi et al. [39] described about a patient with systemic symptoms, including severe proximal renal tubular acidosis (pRTA), short stature, mental retardation, glaucoma, cataracts and band keratopathy. Later the mutations of NBCe1, encoded by SLC4A4, were found to be responsible for these abnormalities [28]. So far 12 symptomatic mutations of SLC4A4 have been reported; eight missense mutations, R298S, S427L, T485S, G486R, R510H, L522P, A799V and R881C [28,40-43], two nonsense mutations, Q29X and W516X [44,45], and two frameshift mutations, N721TxfsX29 and S982NfsX4 [46, 47]. Almost all of these mutations cause severe pRTA, but in S982NfsX4 the acidosis is not so severe (serum 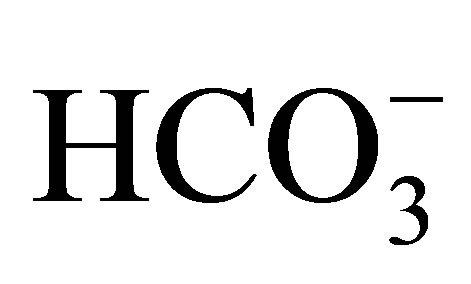 around 15 to 17 mmol/l). In Xenopus oocytes expression study this mutation shows normal function, while in immunohistological study in polarized mammalian cells this shows cytoplasmic retention. The S982NfsX4 mutant is speculated to induce the clinical manifestations via cytoplasmic retention in vivo, but some of the mutant may reach the plasma membrane of proximal tubule, resulting in the relatively mild acidosis. Most of the other symptomatic mutations are found to be in the deep transmembrane sites [48]. The reason why there is no tight relationship between the severity of acidosis and the degree of NBCe1 inactivation remains speculative, but the trafficking defects caused by some NBCe1 mutants may be partly responsible [40,42,47].
around 15 to 17 mmol/l). In Xenopus oocytes expression study this mutation shows normal function, while in immunohistological study in polarized mammalian cells this shows cytoplasmic retention. The S982NfsX4 mutant is speculated to induce the clinical manifestations via cytoplasmic retention in vivo, but some of the mutant may reach the plasma membrane of proximal tubule, resulting in the relatively mild acidosis. Most of the other symptomatic mutations are found to be in the deep transmembrane sites [48]. The reason why there is no tight relationship between the severity of acidosis and the degree of NBCe1 inactivation remains speculative, but the trafficking defects caused by some NBCe1 mutants may be partly responsible [40,42,47].
5. ANALYSIS IN NBCe1 W516X KNOCK-IN MICE
A homozygous W516X mutation of NBCe1 was recently found in Taiwan [45]. The patient was a Chinese girl of 16 years old, presenting severe pRTA (serum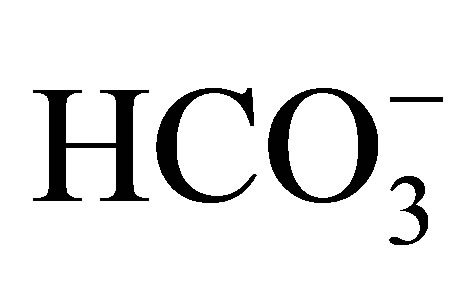 : 10 mmol/l), growth retardation and typical ocular abnormalities such as glaucoma, band keratopathy, and cataract.
: 10 mmol/l), growth retardation and typical ocular abnormalities such as glaucoma, band keratopathy, and cataract.
In order to clarify the pathophysiology of this mutation, we and others created knock-in mice carrying W516X mutation. In NBCe1 W516X/W516X mice a very low amount of the truncated NBCe1 mRNA was detected, suggesting that nonsense-mediated mRNA decay (NMD) is involved in this mechanism. The homozygous mice had severe acidosis (serum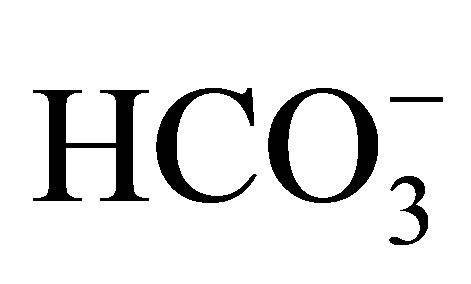 : 3.9 mmol/l) due to pRTA, growth retardation, hyperaldosteronism, anemia, splenomegaly, and early death before weaning. These phenotypes were very similar to those previously reported in NBCe1−/− mice [49]. Interestingly, pRTA has not been reported in humans carrying the heterozygous NBCe1 mutations. However, both NBCe1W516X/+ and NBCe1−/+ mice showed a mild acidosis, suggesting that the compensatory ability of distal tubule acidification might be higher in humans than in mice.
: 3.9 mmol/l) due to pRTA, growth retardation, hyperaldosteronism, anemia, splenomegaly, and early death before weaning. These phenotypes were very similar to those previously reported in NBCe1−/− mice [49]. Interestingly, pRTA has not been reported in humans carrying the heterozygous NBCe1 mutations. However, both NBCe1W516X/+ and NBCe1−/+ mice showed a mild acidosis, suggesting that the compensatory ability of distal tubule acidification might be higher in humans than in mice.
In physiological studies using isolated proximal tubules of NBCe1W516X/W516X mice, the NBCe1 activity was severely reduced to less than twenty percent of that in wild-type mice. Additionally, the rate of bicarbonate absorption from the proximal tubule was reduced to less than twenty percent of that in wild-type mice. These results confirmed the indispensable role of NBCe1 in 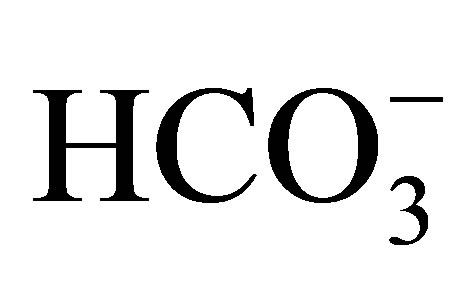 reabsorption in the proximal tubule. In the alkali-treated mice, the symptoms such as bone dysplasia, splenomegaly, and anemia were significantly attenuated. Administration of sodium bicarbonate elongated their life as long as seventy days. This may be a good model to try the effect of PTC124, which is expected to rescue the nonsense mutations [50,51].
reabsorption in the proximal tubule. In the alkali-treated mice, the symptoms such as bone dysplasia, splenomegaly, and anemia were significantly attenuated. Administration of sodium bicarbonate elongated their life as long as seventy days. This may be a good model to try the effect of PTC124, which is expected to rescue the nonsense mutations [50,51].
6. SINGLE NUCLEOTIDE POLYMORPHISMS (SNPS) OF THE NBCe1 AND THEIR CHARACTERS
Recently SNPs of NBCe1 are reported in sequence [http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=8671]. Among them we have analyzed four missense mutations (E122G, S356Y, K558R, and N640I) [35]. In the electrophysiological study in Xenopus oocytes, E122G, S356Y and N640I showed almost normal function, whereas K558R had about half activity of the wildtype. The sodium affinity of K558R was similar as wildtype. In the expression study in polarized MDCK cells, these four mutants (E122G, S356Y, K558R, and N640I) showed the similar basolateral expression as wild-type, suggesting that these mutants have normal trafficking ability. In the functional analysis in HEK cells, the K558R showed significantly reduced pH recovery, while the other SNPs showed normal activity.
This suggests that K558 may play an important role in the transport function of NBCe1. It is predicted that K558 lies in the transmembrane segment 5 [37,38]. As the other SNPs do not lie in the transmembrane site (E122 and S356 in the N-terminal cytoplasmic region, N640 in the extracellular loop between TM5 and TM6), this study also confirms the importance of the transmembrane regions in the normal function of NBCe1. At present, the clinical significance of K558R SNP remains unknown.
7. VACUOLAR H+-ATPASE IN THE KIDNEY: RENAL SPECIFIC SUBTYPES AND STUDIES OF MUTATIONS
The vacuolar H+-ATPase (V-ATPase) is a multisubunit complex which is ubiquitously expressed in various eukaryotic cells [52,53]. V-ATPase is committed to ATP-driven H+ transport across membranes, including plasma membrane, lysosome, the Golgi, secretary vesicle, and endosome. As these intracellular organelles need to keep their pH in optimal range to perform the proper functions, the acidification process by the V-ATPase is crucial.
The structure of the V-ATPase is very complicated and large, constituted of two domains, V0 and V1. V0 domain is membrane-bound, including one a, five c, one d and one H domains. The a subunit has four isoforms, a1, a2, a3 and a4. a1 is expressed ubiquitously, a2 is expressed in lung, kidney and spleen. a3 is in osteoclasts, while a4 is in kidney, epididymis and inner ear [54-58]. Mutations in a4 subunit cause distal renal tubular acidosis (dRTA) with or without hearing loss in human.
V1 domain consists of eight subunits, A to H. Among them the B subunit plays an essential role. There are two subtypes in B subunit, B1 and B2 in human. The expression of B1 is restricted to renal intercalated cells, inner ear, epididymis, and ciliary body, on the other hand B2 is expressed ubiquitously [59-62]. Mutations in B1 subunit in human cause dRTA accompanied with sensorineural hearing loss. Figure 3 shows the simplified scheme of V-ATPase.
8. RENAL SPECIFIC SUBTYPES AND ANALYSIS OF a4 SUBUNIT MUTATIONS
There are four subtypes of “a” subunits of V-ATPase, a1
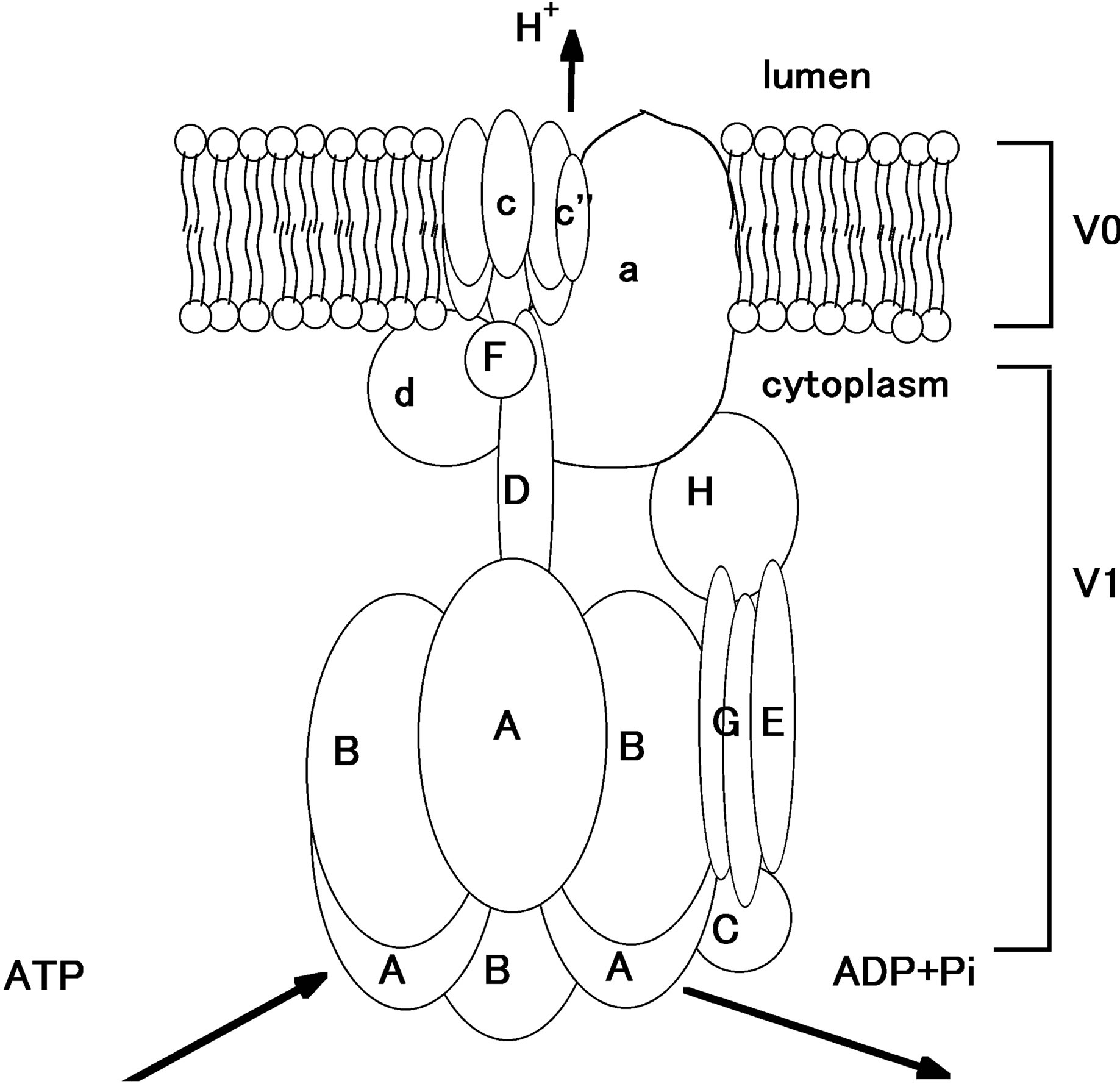
Figure 3. The simplified scheme of V-ATPase [53,63]. In V1, the cytoplasmic part, ATP is hydrolyzed to ADP and Pi, producing energy to drive proton out of the cytoplasm. Proton is exported to luminal side from V0 part.
to a4. Among them a4 subunit is kidney-specific [64]. At first mutations in ATP6V0A4, the gene coding a4 subunit, were shown to cause recessive distal renal tubular acidosis (dRTA) with preserved hearing [65], however later it was found that there are patients carrying homozygous mutations in ATP6V0A4 with both dRTA and hearing loss [66]. The reason why only a subset of a4 deficient patients develops hearing disorder has not been clarified yet.
As previously mentioned, V-ATPase was thought to have some roles in H+ secretion in the proximal tubule [20]. Recently some groups investigated the details of a4 subunit deficiency using targeted gene knockout mice. Norgett et al. [67] made Atp6v0a4 KO mice and clarified that a4 null mice had severe metabolic acidosis, hypokalemia, hyperchloridemia, and early nephrocalcinosis. These mice also lost the sense of smell and had severe hearing impairment, showing elevated threshold of auditory brainstem response and loss of endocochlear potential. a4 null mice died without alkali treatment, and even if they survived the weaning period they had a mild acidemia and alkali urine. Heterozygous mice did not show academia. When they were challenged with acid, however, they became more acidic than the wild-type mice.
On the other hand, a4 knockout mice produced by Hennings et al. [68] had profound deafness and enlarged endolymphatic fluid compartments, like that in pendrin knockout mice [69]. It was also clarified that in the inner ear a4 colocalizes with pendrin. Pendrin is one of the anion exchangers, coded by SLC26A4 [70-72]. Disruption of pendrin causes Pendred syndrome, which shows various phenotypes like sensorineural deafness and thyroid dysfunction [70]. They speculate that pendrin and V-ATPase co-operate in the regulation of endolymph homeostasis.
Interestingly, Hennings et al. also proposed that a4 deficiency causes proximal tubule dysfunction, as these mice showed trafficking disorder of NaPi-IIa (sodiumphosphate transporter), proteinuria, phosphaturia and accumulation of lysosomal material in the proximal tubule. They also reanalyzed the clinical data with recessive dRTA and found that the patients with a4 mutations had severer acidosis, lower serum bicarbonate, and lower blood pH than the patients with B1 mutations. The patients with a4 mutations were diagnosed earlier than the patients with B1 mutations. It remains unknown, however, whether a4 deficiency affects proximal functions also in human.
The reason why these a4 null mice (created by Norgett et al. and Hennings et al.) show the different phenotypes is not entirely clear. Both mice show metabolic acidosis, hypokalemia, and hyperchloridemia. While Norgett’s mice do not show hypercalcemia and hypercalciuria seen in human dRTA, Hennings’ mice show hypercalcemia and hyperphosphateueria (urine calcium not measured). Different background of the mice may be at least partially responsible for these different phenotypes.
9. CONCLUSIONS
Renal proximal tubule plays an important role in the regulation of acid-base homeostasis. Recently there have been some remarkable progresses in the investigation of proximal tubule acid-base transporters such as NHEs and NBCe1. As proximal tubule reabsorbs most of the bicarbonate filtered in the glomeruli, it is quite important to clarify the detailed functions of these transporters and their regulations. Moreover, V-ATPase, a main acid-base regulator of the distal tubule, is now suggested to have some regulatory roles in the proximal tubule.
The animal model studies, especially using transgenic mice, have considerably contributed to the detailed analysis of these transporters. Some models mimic the human phenotypes of these deficient diseases to some extent. In some cases, however, the phenotypes of human and mice are not identical, showing the limitation of these approaches.
Clinically, acid-base balance is quite important in the development of neonatal period, keeping fluid homeostasis, bone metabolism, and even the survival of the individual. We hope that the further understanding of acid/base transporters will lead to the development of more effective treatment of acid/base disorders.
REFERENCES
- Bobulescu, I.A., Di Sole, F. and Moe, O.W. (2005) Na+/ H+ exchangers: Physiology and link to hypertension and organ ischemia. Current Opinion in Nephrology and Hypertension, 14, 485-494. http://dx.doi.org/10.1097/01.mnh.0000174146.52915.5d
- Bobulescu, I.A. and Moe, O.W. (2009) Luminal Na+/H+ exchange in the proximal tubule. Pflügers Archiv, 458, 5-21. http://dx.doi.org/10.1007/s00424-008-0595-1
- Orlowski, J. and Grinstein, S. (2007) Emerging roles of alkali cation/proton exchangers in organellar homeostasis. Current Opinion in Cell Biology, 19, 483-492. http://dx.doi.org/10.1016/j.ceb.2007.06.001
- Slepkov, E.R., Rainey, J.K., Sykes, B.D. and Fliegel, L. (2007) Structural and functional analysis of the Na+/H+ exchanger. Biochemical Journal, 401, 623-633. http://dx.doi.org/10.1042/BJ20061062
- Wang, D., King, S.M., Quill, T.A., Doolittle, L.K. and Garbers, D.L. (2003) A new sperm-specific Na+/H+ exchanger required for sperm motility and fertility. Nature Cell Biology, 5, 1117-1122. http://dx.doi.org/10.1038/ncb1072
- Pitts, R.F. and Lotspeich, W.D. (1946) Bicarbonate and the renal regulation of acid base balance. American Journal of Physiology, 147, 138-154.
- Pitts, R.F., Ayer, J.L., Schiess, W.A. and Miner, P. (1949) The renal regulation of acid-base balance in man. III. The reabsorption and excretion of bicarbonate. Journal of Clinical Investigation, 28, 35-44. http://dx.doi.org/10.1172/JCI102050
- Murer, H., Hopfer, U. and Kinne, R. (1976) Sodium/ proton antiport in brush-border-membrane vesicles isolated from rat small intestine and kidney. Biochemical Journal, 154, 597-604.
- Kinsella, J.L. and Aronson, P.S. (1980) Properties of the Na+-H+ exchanger in renal microvillus membrane vesicles. American Journal of Physiology, 238, F461-F469.
- Sardet, C., Franchi, A. and Pouysségur, J. (1989) Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell, 56, 271-280. http://dx.doi.org/10.1016/0092-8674(89)90901-X
- Biemesderfer, D., Pizzonia, J., Abu-Alfa, A., Exner, M., Reilly, R., Igarashi, P. and Aronson, P.S. (1993) NHE3: A Na+/H+ exchanger isoform of renal brush border. American Journal of Physiology, 265, F736-F742.
- Amemiya, M., Loffing, J., Lötscher, M., Kaissling, B., Alpern, R.J. and Moe, O.W. (1995) Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney International, 48, 1206- 1215. http://dx.doi.org/10.1038/ki.1995.404
- Wu, M.S., Biemesderfer, D., Giebisch, G. and Aronson, P.S. (1996) Role of NHE3 in mediating renal brush border Na+-H+ exchange. Adaptation to metabolic acidosis. Journal of Biological Chemistry, 271, 32749-32752. http://dx.doi.org/10.1074/jbc.271.51.32749
- Goyal, S., Vanden Heuvel, G. and Aronson, P.S. (2003) Renal expression of novel Na+/H+ exchanger isoform NHE8. American Journal of Physiology. Renal Physiology, 284, F467-F473.
- Goyal, S., Mentone, S. and Aronson, P.S. (2005) Immunolocalization of NHE8 in rat kidney. American Journal of Physiology. Renal Physiology, 288, F530-F538. http://dx.doi.org/10.1152/ajprenal.00229.2004
- Becker, A.M., Zhang, J., Goyal, S., Dwarakanath, V., Aronson, P.S., Moe, O.W. and Baum, M. (2007) Ontogeny of NHE8 in the rat proximal tubule. American Journal of Physiology. Renal Physiology, 293, F255-F261. http://dx.doi.org/10.1152/ajprenal.00400.2006
- Yun, C.H., Tse, C.M., Nath, S.K., Levine, S.A., Brant, S.R. and Donowitz, M. (1995) Mammalian Na+/H+ exchanger gene family: Structure and function studies. American Journal of Physiology, 269, G1-G11.
- Twombley, K., Gattineni, J., Bobulescu, I.A., Dwarakanath, V. and Baum, M. (2010) Effect of metabolic acidosis on neonatal proximal tubule acidification. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology, 299, R1360-R1368. http://dx.doi.org/10.1152/ajpregu.00007.2010
- Schultheis, P.J., Clarke, L.L., Meneton, P., Miller, M.L., Soleimani, M., Gawenis, L.R., Riddle, T.M., Duffy, J.J., Doetschman, T., Wang, T., Giebisch, G., Aronson, P.S., Lorenz, J.N. and Shull, G.E. (1998) Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nature Genetics, 19, 282-285. http://dx.doi.org/10.1038/969
- Wang, T., Yang, C.L., Abbiati, T., Schultheis, P.J., Shull, G.E., Giebisch, G. and Aronson, P.S. (1999) Mechanism of proximal tubule bicarbonate absorption in NHE3 null mice. American Journal of Physiology, 277, F298-F302.
- Zhang, J., Bobulescu, I.A., Goyal, S., Aronson, P.S., Baum, M.G. and Moe, O.W. (2007) Characterization of Na+/H+ exchanger NHE8 in cultured renal epithelial cells. American Journal of Physiology. Renal Physiology, 293, F761-F766. http://dx.doi.org/10.1152/ajprenal.00117.2007
- Choi, J.Y., Shah, M., Lee, M.G., Schultheis, P.J., Shull, G.E., Muallem, S. and Baum, M. (2000) Novel amiloride-sensitive sodium-dependent proton secretion in the mouse proximal convoluted tubule. Journal of Clinical Investigation, 105, 1141-1146. http://dx.doi.org/10.1172/JCI9260
- Schultheis, P.J., Clarke, L.L., Meneton, P., Harline, M., Boivin, G.P., Stemmermann, G., Duffy, J.J., Doetschman, T., Miller, M.L. and Shull, G.E. (1998) Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. Journal of Clinical Investigation, 101, 1243-1253. http://dx.doi.org/10.1172/JCI1249
- Baum, M., Twombley, K., Gattineni, J., Joseph, C., Wang, L., Zhang, Q., Dwarakanath, V. and Moe, O.W. (2012) Proximal tubule Na+/H+ exchanger activity in adult NHE8-/-, NHE3-/-, and NHE3-/-/NHE8-/- mice. American Journal of Physiology. Renal Physiology, 303, F1495- F1502. http://dx.doi.org/10.1152/ajprenal.00415.2012
- Zachos, N.C., Tse, M. and Donowitz, M. (2005) Molecular physiology of intestinal Na+/H+ exchange. Annual Review of Physiology, 67, 411-443. http://dx.doi.org/10.1146/annurev.physiol.67.031103.153004
- Seki, G., Yamada, H., Horita, S., Suzuki, M., Yamazaki, O., Van Paesschen, W., Yang, S.S. and Lin, S.H. (2011) Physiological and pathophysiological roles of the electrogenic
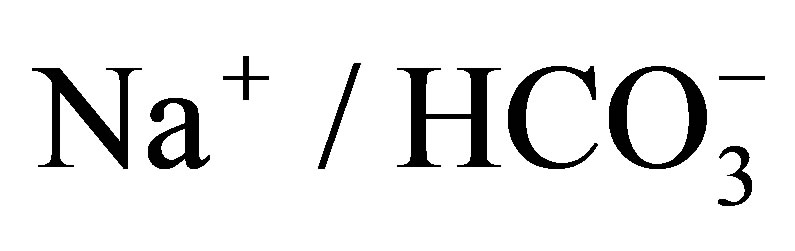 cotransporter NBCe1. Open Journal of Molecular and Integrative Physiology, 1, 9-16. http://dx.doi.org/10.4236/ojmip.2011.12002
cotransporter NBCe1. Open Journal of Molecular and Integrative Physiology, 1, 9-16. http://dx.doi.org/10.4236/ojmip.2011.12002 - Romero, M.F., Hediger, M.A., Boulpaep, E.L. and Boron, W.F. (1997) Expression cloning and characterization of a renal electrogenic
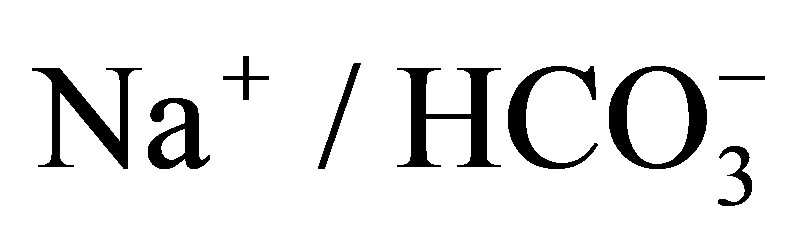 cotransporter. Nature, 387, 409-413. http://dx.doi.org/10.1038/387409a0
cotransporter. Nature, 387, 409-413. http://dx.doi.org/10.1038/387409a0 - Igarashi, T., Inatomi, J., Sekine, T., Cha, S.H., Kanai, Y., Kunimi, M., Tsukamoto, K., Satoh, H., Shimadzu, M., Tozawa, F., Mori, T., Shiobara, M., Seki, G. and Endou, H. (1999) Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nature Genetics, 23, 264-266. http://dx.doi.org/10.1038/15440
- Romero, M.F., Fulton, C.M. and Boron, W.F. (2004) The SLC4 family of
 transporters. Pflügers Archiv, 447, 495-509. http://dx.doi.org/10.1007/s00424-003-1180-2
transporters. Pflügers Archiv, 447, 495-509. http://dx.doi.org/10.1007/s00424-003-1180-2 - Liu, Y., Xu, J.Y., Wang, D.K., Wang, L. and Chen, L.M. (2011) Cloning and identification of two novel NBCe1 splice variants from mouse reproductive tract tissues: A comparative study of NCBT genes. Genomics, 98, 112- 119. http://dx.doi.org/10.1016/j.ygeno.2011.04.010
- Romero, M.F. and Boron, W.F. (1999) Electrogenic
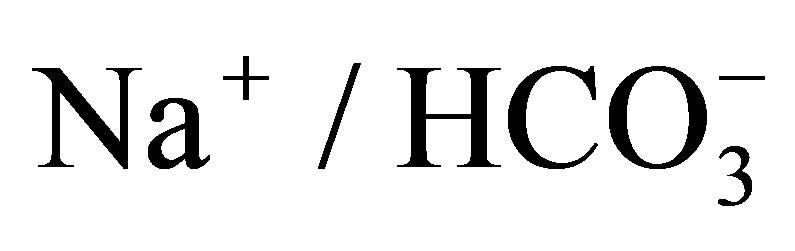 cotransporters: Cloning and physiology. Annual Review of Physiology, 61, 699-723. http://dx.doi.org/10.1146/annurev.physiol.61.1.699
cotransporters: Cloning and physiology. Annual Review of Physiology, 61, 699-723. http://dx.doi.org/10.1146/annurev.physiol.61.1.699 - Abuladze, N., Song, M., Pushkin, A., Newman, D., Lee, I. Nicholas, S. and Kurtz, I. (2000) Structural organization of the human NBC1 gene: kNBC1 is transcribed from an alternative promoter in intron 3. Gene, 251, 109-122. http://dx.doi.org/10.1016/S0378-1119(00)00204-3
- Bevensee, M.O., Schmitt, B.M., Choi, I., Romero, M.F. and Boron, W.F. (2000) An electrogenic Na(+)-HCO(-)(3) cotransporter (NBC) with a novel COOH-terminus, cloned from rat brain. American Journal of Physiology— Cell Physiology, 278, C1200-C1211.
- Shirakabe, K., Priori, G., Yamada, H., Ando, H., Horita, S., Fujita, T., Fujimoto, I., Mizutani, A., Seki, G. and Mikoshiba, K. (2006) IRBIT, an inositol 1,4,5-trisphosphate receptor-binding protein, specifically binds to and activates pancreas-type
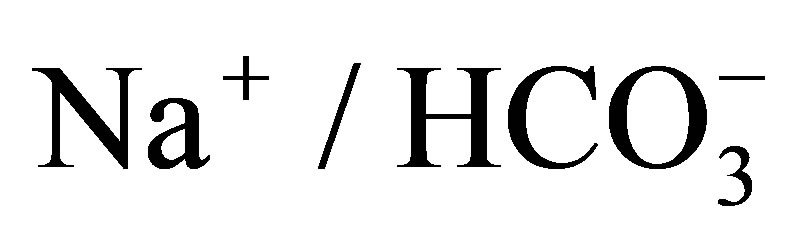 cotransporter 1 (pNBC1). Proceedings of the National Academy of Sciences of the United States of America, 103, 9542-9547. http://dx.doi.org/10.1073/pnas.0602250103
cotransporter 1 (pNBC1). Proceedings of the National Academy of Sciences of the United States of America, 103, 9542-9547. http://dx.doi.org/10.1073/pnas.0602250103 - Yamazaki, O., Yamada, H., Suzuki, M., Horita, S., Shirai, A., Nakamura, M., Seki, G. and Fujita, T. (2011) Functional characterization of nonsynonymous single nucleotide polymorphisms in the electrogenic
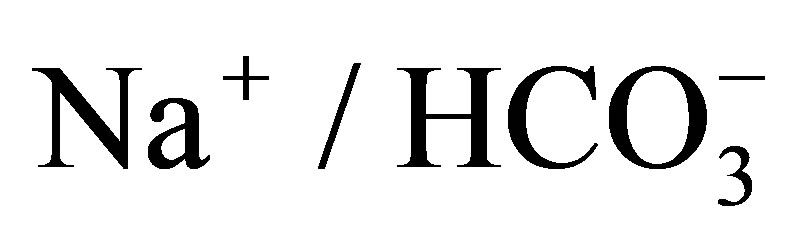 cotransporter NBCe1A. Pflügers Archiv, 461, 249-259. http://dx.doi.org/10.1007/s00424-010-0918-x
cotransporter NBCe1A. Pflügers Archiv, 461, 249-259. http://dx.doi.org/10.1007/s00424-010-0918-x - Zhu, Q., Lee, D.W. and Casey, J.R. (2003) Novel topology in C-terminal region of the human plasma membrane anion exchanger, AE1. Journal of Biological Chemistry, 278, 3112-3120. http://dx.doi.org/10.1074/jbc.M207797200
- Boron, W.F. (2006) Acid-base transport by the renal proximal tubule. Journal of the American Society of Nephrology, 17, 2368-2382. http://dx.doi.org/10.1681/ASN.2006060620
- Zhu, Q., Kao, L., Azimov, R., Abuladze, N., Newman, D., Pushkin, A., Liu, W., Chang, C. and Kurtz, I. (2010) Structural and functional characterization of the C-terminal transmembrane region of NBCe1-A. Journal of Biological Chemistry, 285, 37178-37187. http://dx.doi.org/10.1074/jbc.M110.169201
- Igarashi, T., Ishii, T., Watanabe, K., Hayakawa, H., Horio, K., Sone, Y. and Ohga, K. (1994) Persistent isolated proximal renal tubular acidosis—A systemic disease with a distinct clinical entity. Pediatric Nephrology, 8, 70-71. http://dx.doi.org/10.1007/BF00868266
- Horita, S., Yamada, H., Inatomi, J., Moriyama, N., Sekine, T., Igarashi, T., Endo, Y., Dasouki, M., Ekim, M., Al-Gazali, L., Shimadzu, M., Seki, G. and Fujita, T. (2005) Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. Journal of the American Society of Nephrology, 16, 2270-2278. http://dx.doi.org/10.1681/ASN.2004080667
- Dinour, D., Chang, M.H., Satoh, J., Smith, B.L., Angle, N., Knecht, A., Serban, I., Holtzman, E.J. and Romero, M.F. (2004) A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. Journal of Biological Chemistry, 279, 52238-52246. http://dx.doi.org/10.1074/jbc.M406591200
- Suzuki, M., Vaisbich, M.H., Yamada, H., Horita, S., Li, Y., Sekine, T., Moriyama, N., Igarashi, T., Endo, Y., Cardoso, T.P., de Sá, L.C., Koch, V.H., Seki, G. and Fujita, T. (2008) Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflügers Archiv, 455, 583-593. http://dx.doi.org/10.1007/s00424-007-0319-y
- Demirci, F.Y., Chang, M.H., Mah, T.S., Romero, M.F. and Gorin, M.B. (2006) Proximal renal tubular acidosis and ocular pathology: A novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Molecular Vision, 12, 324-330.
- Igarashi, T., Inatomi, J., Sekine, T., Seki, G., Shimadzu, M., Tozawa, F., Takeshima, Y., Takumi, T., Takahashi, T., Yoshikawa, N., Nakamura, H. and Endou, H. (2001) Novel nonsense mutation in the
 cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. Journal of the American Society of Nephrology, 12, 713-718.
cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. Journal of the American Society of Nephrology, 12, 713-718. - Lo, Y.F., Yang, S.S., Seki, G., Yamada, H., Horita, S., Yamazaki, O., Fujita, T., Usui, T., Tsai, J.D., Yu, I.S., Lin, S.W. and Lin, S.H. (2011) Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kidney International, 79, 730-741. http://dx.doi.org/10.1038/ki.2010.523
- Inatomi, J., Horita, S., Braverman, N., Sekine, T., Yamada, H., Suzuki, Y., Kawahara, K., Moriyama, N., Kudo, A., Kawakami, H., Shimadzu, M., Endou, H., Fujita, T., Seki, G. and Igarashi, T. (2004) Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflügers Archiv, 448, 438-444. http://dx.doi.org/10.1007/s00424-004-1278-1
- Suzuki, M., Van Paesschen, W., Stalmans, I., Horita, S., Yamada, H., Bergmans, B.A., Legius, E., Riant, F., De Jonghe, P., Li, Y., Sekine, T., Igarashi, T., Fujimoto, I., Mikoshiba, K., Shimadzu, M., Shiohara, M., Braverman, N., Al-Gazali, L., Fujita, T. and Seki, G. (2010) Defective membrane expression of the
 cotransporter NBCe1 is associated with familial migraine. Proceedings of the National Academy of Sciences of the United States of America, 107, 15963-15968. http://dx.doi.org/10.1073/pnas.1008705107
cotransporter NBCe1 is associated with familial migraine. Proceedings of the National Academy of Sciences of the United States of America, 107, 15963-15968. http://dx.doi.org/10.1073/pnas.1008705107 - Zhu, Q., Kao, L., Azimov, R., Newman, D., Liu, W., Pushkin, A., Abuladze, N. and Kurtz, I. (2010) Topological location and structural importance of the NBCe1-A residues mutated in proximal renal tubular acidosis. Journal of Biological Chemistry, 285, 13416-13426. http://dx.doi.org/10.1074/jbc.M109.093286
- Gawenis, L.R., Bradford, E.M., Prasad, V., Lorenz, J.N., Simpson, J.E., Clarke, L.L., Woo, A.L., Grisham, C., Sanford, L.P., Doetschman, T., Miller, M.L. and Shull, G.E. (2007) Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1
 cotransporter. Journal of Biological Chemistry, 282, 9042-9052. http://dx.doi.org/10.1074/jbc.M607041200
cotransporter. Journal of Biological Chemistry, 282, 9042-9052. http://dx.doi.org/10.1074/jbc.M607041200 - Welch, E.M., Barton, E.R., Zhuo, J., Tomizawa, Y., Friesen, W.J., Trifillis, P., Paushkin, S., Patel, M., Trotta, C.R., Hwang, S., Wilde, R.G., Karp, G., Takasugi, J., Chen, G., Jones, S., Ren, H., Moon, Y.C., Corson, D., Turpoff, A.A., Campbell, J.A., Conn, M.M., Khan, A., Almstead, N.G., Hedrick, J., Mollin, A., Risher, N., Weetall, M., Yeh, S., Branstrom, A.A., Colacino, J.M., Babiak, J., Ju, W.D., Hirawat, S., Northcutt, V.J., Miller, L.L., Spatrick, P., He, F., Kawana, M., Feng, H., Jacobson, A., Peltz, S.W. and Sweeney, H.L. (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature, 447, 87-91. http://dx.doi.org/10.1038/nature05756
- Du, M., Liu, X., Welch, E.M., Hirawat, S., Peltz, S.W. and Bedwell, D.M. (2008) PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proceedings of the National Academy of Sciences of the United States of America, 105, 2064-2069. http://dx.doi.org/10.1073/pnas.0711795105
- Wagner, C.A., Finberg, K.E., Breton, S., Marshansky, V., Brown, D. and Geibel, J.P. (2004) Renal vacuolar H+- ATPase. Physiological Reviews, 84, 1263-1314. http://dx.doi.org/10.1152/physrev.00045.2003
- Nishi, T. and Forgac, M. (2002) The vacuolar (H+)-ATPases—Nature’s most versatile proton pumps. Nature Reviews Molecular Cell Biology, 3, 94-103. http://dx.doi.org/10.1038/nrm729
- Brody, L.C., Abel, K.J., Castilla, L.H., Couch, F.J., McKinley, D.R., Yin, G., Ho, P.P., Merajver, S., Chandrasekharappa, S.C. and Xu, J. (1995) Construction of a transcription map surrounding the BRCA1 locus of human chromosome 17. Genomics, 25, 238-247. http://dx.doi.org/10.1016/0888-7543(95)80131-5
- Peng, S.B., Crider, B.P., Xie, X.S., Stone, D.K. (1994) Alternative mRNA splicing generates tissue-specific isoforms of 116-kDa polypeptide of vacuolar proton pump. Journal of Biological Chemistry, 269, 17262-17266.
- Peng, S.B., Li, X., Crider, B.P., Zhou, Z., Andersen, P., Tsai, S.J., Xie, X.S. and Stone, D.K. (1999) Identification and reconstitution of an isoform of the 116-kDa subunit of the vacuolar proton translocating ATPase. Journal of Biological Chemistry, 274, 2549-2555. http://dx.doi.org/10.1074/jbc.274.4.2549
- Frattini, A., Orchard, P.J., Sobacchi, C., Giliani, S., Abinun, M., Mattsson, J.P., Keeling, D.J., Andersson, A.K., Wallbrandt, P., Zecca, L., Notarangelo, L.D., Vezzoni, P. and Villa, A. (2000) Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nature Genetics, 25, 343-346. http://dx.doi.org/10.1038/77131
- Smith, A.N., Borthwick, K.J. and Karet, F.E. (2002) Molecular cloning and characterization of novel tissue-specific isoforms of the human vacuolar H(+)-ATPase C, G and d subunits, and their evaluation in autosomal recessive distal renal tubular acidosis. Gene, 297, 169-177. http://dx.doi.org/10.1016/S0378-1119(02)00884-3
- Karet, F.E., Finberg, K.E., Nelson, R.D., Nayir, A., Mocan, H., Sanjad, S.A., Rodriguez-Soriano, J., Santos, F., Cremers, C.W., Di Pietro, A., Hoffbrand, B.I., Winiarski, J., Bakkaloglu, A., Ozen, S., Dusunsel, R., Goodyer, P., Hulton, S.A., Wu, D.K., Skvorak, A.B., Morton, C.C., Cunningham, M.J., Jha, V. and Lifton, R.P. (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nature Genetics, 21, 84-90. http://dx.doi.org/10.1038/5022
- Nelson, R.D., Guo, X.L., Masood, K., Brown, D., Kalkbrenner, M. and Gluck, S. (1992) Selectively amplified expression of an isoform of the vacuolar H(+)-ATPase 56-kilodalton subunit in renal intercalated cells. Proceedings of the National Academy of Sciences of the United States of America, 89, 3541-3545. http://dx.doi.org/10.1073/pnas.89.8.3541
- van Hille, B., Richener, H., Schmid, P., Puettner, I., Green, J.R. and Bilbe, G. (1994) Heterogeneity of vacuolar H(+)-ATPase: differential expression of two human subunit B isoforms. Biochemical Journal, 303, 191- 198.
- Bernasconi, P., Rausch, T., Struve, I., Morgan, L. and Taiz, L. (1990) An mRNA from human brain encodes an isoform of the B subunit of the vacuolar H(+)-ATPase. Journal of Biological Chemistry, 265, 17428-17431.
- Karet, F.E. (2005) Physiological and metabolic implications of V-ATPase isoforms in the kidney. Journal of Bioenergetics and Biomembranes, 37, 425-429. http://dx.doi.org/10.1007/s10863-005-9484-x
- Smith, A.N., Finberg, K.E., Wagner, C.A., Lifton, R.P., Devonald, M.A., Su, Y. and Karet, F.E. (2001) Molecular cloning and characterization of Atp6n1b: A novel fourth murine vacuolar H+-ATPase a-subunit gene. Journal of Biological Chemistry, 276, 42382-42388. http://dx.doi.org/10.1074/jbc.M107267200
- Smith, A.N., Skaug, J., Choate, K.A., Nayir, A., Bakkaloglu, A., Ozen, S., Hulton, S.A., Sanjad, S.A., Al-Sabban, E.A., Lifton, R.P., Scherer, S.W. and Karet, F.E. (2000) Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nature Genetics, 26, 71-75. http://dx.doi.org/10.1038/82492
- Stover, E.H., Borthwick, K.J., Bavalia, C., Eady, N., Fritz, D.M., Rungroj, N., Giersch, A.B., Morton, C.C., Axon, P.R., Akil, I., Al-Sabban, E.A., Baguley, D.M., Bianca, S., Bakkaloglu, A., Bircan, Z., Chauveau, D., Clermont, M.J., Guala, A., Hulton, S.A., Kroes, H., Li Volti, G., Mir, S., Mocan, H., Nayir, A., Ozen, S., Rodriguez Soriano, J., Sanjad, S.A., Tasic, V., Taylor, C.M., Topaloglu, R., Smith, A.N. and Karet, F.E. (2002) Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. Journal of Medical Genetics, 39, 796-803. http://dx.doi.org/10.1136/jmg.39.11.796
- Norgett, E.E., Golder, Z.J., Lorente-Cánovas, B., Ingham, N., Steel, K.P. and Frankl, F.E. (2012) Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proceedings of the National Academy of Sciences of the United States of America, 109, 13775-13780. http://dx.doi.org/10.1073/pnas.1204257109
- Hennings, J.C., Picard, N., Huebner, A.K., Stauber, T., Maier, H., Brown, D., Jentsch, T.J, Vargas-Poussou, R., Eladari, D. and Hübner, C.A. (2012) A mouse model for distal renal tubular acidosis reveals a previously unrecognized role of the V-ATPase a4 subunit in the proximal tubule. EMBO Molecular Medicine, 4, 1057-1071. http://dx.doi.org/10.1002/emmm.201201527
- Everett, L.A., Belyantseva, I.A., Noben-Trauth, K., Cantos, R., Chen, A., Thakkar, S.I., Hoogstraten-Miller, S.L., Kachar, B., Wu, D.K. and Green, E.D. (2001) Targeted disruption of mouse Pds provides insight about the innerear defects encountered in Pendred syndrome. Human Molecular Genetics, 10, 153-161. http://dx.doi.org/10.1093/hmg/10.2.153
- Everett, L.A., Glaser, B., Beck, J.C., Idol, J.R., Buchs, A., Heyman, M., Adawi, F., Hazani, E., Nassir, E., Baxevanis, A.D., Sheffield, V.C. and Green, E.D. (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Genetics, 17, 411-422. http://dx.doi.org/10.1038/ng1297-411
- Wagner, C.A., Mohebbi, N., Capasso, G. and Geibel, J.P. (2011) The anion exchanger pendrin (SLC26A4) and renal acid-base homeostasis. Cellular Physiology and Biochemistry, 28, 497-504. http://dx.doi.org/10.1159/000335111
- Wangemann, P. (2011) The role of pendrin in the development of the murine inner ear. Cellular Physiology and Biochemistry, 28, 527-534. http://dx.doi.org/10.1159/000335113
NOTES
*Corresponding author.