American Journal of Molecular Biology
Vol.4 No.3(2014), Article
ID:47873,16
pages
DOI:10.4236/ajmb.2014.43015
Endometrial Carcinogenesis and Molecular Signaling Pathways
Xianyong Ma1*, Charles X. Ma2, Jianghui Wang1
1The Department of Pathology, Yale University School of Medicine, New Haven, USA
2University of Connecticut School of Medicine, Farmington, USA
Email: *Xian-yong.ma@yale.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 April 2014; revised 2 May 2014; accepted 1 June 2014
ABSTRACT
The Endometrial Cancer (EC) is the most common gynecologic malignancy that starts in the endometrium of women. Carcinogenesis of EC is associated with several critical regulatory molecules, which involve in different signaling pathways. A number of signaling pathways have been identified to be involved in the multiple-step development of EC, including PI3K/AKT/mTOR signaling pathway, WNT/β-catenin signal transduction cascades (including APC/β-catenin pathway), MAPK/ ERK pathway, VEGF/VEGFR ligand receptor signaling pathway, ErbB signaling pathway, P53/P21 and P16INK4a/pRB signaling pathways. This review mainly focuses on the molecular signaling pathways relevant to human endometrial cancer and discusses those critical capabilities of transforming endometrial cells, including evading apoptosis; enhancing cell proliferation; blocking differentiation; and inducing angiogenesis.
Keywords:Signaling Pathway, Endometrial Cancer (EC), Carcinogenesis

1. Introduction
Abnormal endometrial proliferation associates with the most common gynecological diseases, including the endometrial cancer, endometriosis and adenomyosis. The Endometrial Cancer (EC) is a type of malignancy that starts in the endometrium. Carcinogenesis of endometrial tissue can originate from abnormal hormone estrogen level/bleeding, chemical insult, radiation and viral infection, as well as genetic predispositions. Tumors developed in the epithelial layer of endometrium are classified as endometrial carcinomas (98% of endometrial tumors), while tumors developed in the muscle layer or stromal tissue are called sarcomas (2%). Given the high prevalence of the endometrial carcinoma, this review mainly focuses on the molecular signaling pathways relevant to this type of caner.
The human endometrium is a dynamically remodeling tissue undergoing around 300 cycles of regeneration, differentiation, and shedding during the reproductive life of women. These cycles are controlled by a number of signaling pathways including estrogen and progesterone hormone signals. Mutations and abnormal expression of the genes related to these signaling molecules have been linked with the multistep development of endometrial carcinogenesis, and the proteins encoded by these genes are the key mediators of several signaling pathways, such as PI3K/AKT/mTOR signaling pathway, WNT/b-catenin signal transduction cascades (including APC/bcatenin signaling), MAPK/ERK pathway, VEGF/VEGFR ligand receptor signaling pathway, ErbB signaling pathway and P53-P16INK4a signaling pathways. Based on the fundamental biological function of these signaling pathways and pathological features of EC, we propose that four capabilities of transforming endometrial cells are critical to the primary carcinogenesis and metastasis: 1) evading apoptosis, 2) enhancing cell proliferation, 3) blocking differentiation, 4) inducing angiogenesis; other functions such as cell cycle arrest, microsatellite instability (even other pathological functions) can be integrated into these capabilities. In this paper we will discuss on the pathological roles of these pathways during the endometrial carcinogenesis.
2. Signaling Pathways and Carcinogenesis of Endometrial Cancer
2.1. PI3K/AKT/mTOR and EC
This signaling pathway integrates both intracellular and extracellular signals and serves as a central regulator of basic biological processes of cells such as metabolism, growth, proliferation, survival and angiogenesis (see Figure 1). mTOR nucleates at least two distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [1] . mTOR pathway was identified to be activated during tumor formation, and is deregulated in human diseases including cancer and type 2 diabetes [2] . High-frequency mutation of PTEN within this pathway was detected from type I endometrial cancer, and accompanied the PI3K/AKT/mTOR activation [3] [4] . Molecular alterations in AKT are rare, but both type I and type II endometrial cancers showed a
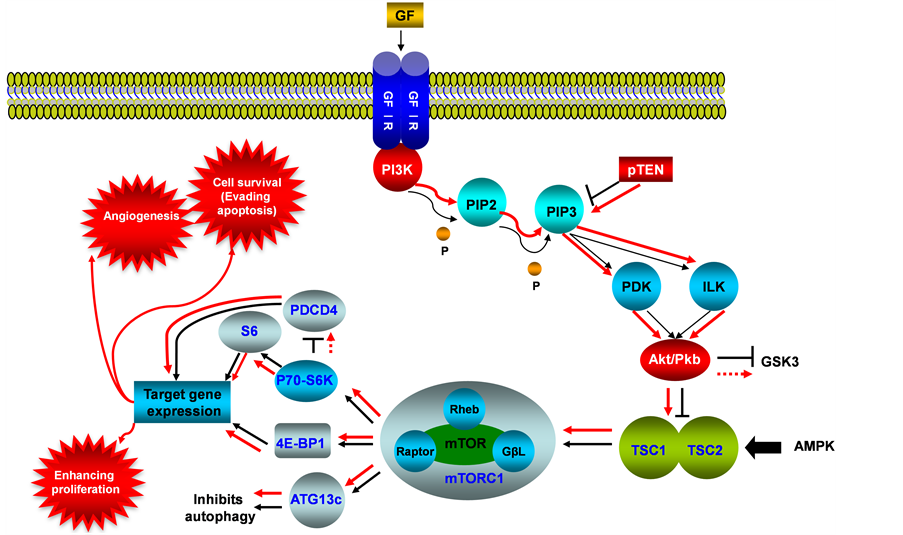
Figure 1. PI3K/AKT/mTOR signaling pathway in endometrial cancer. This illustration encompasses mediators involved in PI3K/AKT/mTOR pathway, alternated genes or proteins (PI3K, PTEN and AKT/PKB, labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for this signaling pathway results in causing tumor angiogenesis, evading apoptosis and enhancing proliferation.
high rate of mutations in the PIK3CA gene, which encodes the PI3K catalytic subunit alpha [5] PIK3CA mutations were detected in 20% - 30% of endometrial cancers [6] . Amplification of the PIK3CA gene, however, is much more common in type II endometrial cancer, with a prevalence of about 46% [7] [8] . PTEN antagonizes PI3K function and negatively regulates AKT activities, which encompass mainly cell survival and anti-apoptosis. Research also showed that PTEN gene mutations were associated with the histology, early stage, and favorable clinical behavior of endometrial cancers [9] . Loss of PTEN activity in endometrial cancers is associated with increased activity of the PI3K with resultant phosphorylation of its downstream substrate AKT [10] . In turn, AKT upregulates mTOR activity, and hyper activation of mTOR signaling enhances translation of mRNAs encoding growth factors, cell cycle regulators, survival proteins, and angiogenic factors, therefore, dysregulation of the PI3K/AKT/ mTOR pathway as a result of genetic mutations and amplifications has been thought to be a conduit for carcinogenesis [11] [12] . Interestingly, in a study of metastatic endometrial cancers, frequent mutations of KRAS, FGFR2 and PIK3CA were identified [13] [14] . The cellular location of the AKT activity is important in the behavior of the cancer, the nuclear p-AKT level was significantly higher in grade 1 (well-differentiated, G1) than in grade 3 (poorly differentiated, G3) cancers and associated with estrogen receptor (ER-alpha) expression. Hence, the nuclear p-AKT level was significantly correlated with the prognosis for EC of G1, and possibly resulting in inhibition of apoptosis. Activation of AKT frequently occurs in endometrial cancer, but is independent of PTEN or PIK3CA activation status. Expression of p-AKT was basically detected in the cytoplasm but some samples had both cytoplasmic and nuclear staining [15] , also activation of AKT pathway by mutated PIK3CA or PTEN is possibly associated with favorable prognosis of EC [16] . PTEN also plays a role in cellular signaling by inhibiting the MAPK kinase pathway. MAPK pathway is another major intracellular signaling cascade, which transmits signals through phosphorylation by tyrosine kinase-type receptors on cell membranes, and will be reviewed in the next section of this paper.
2.2. MARK/ERK Pathway and EC
The mitogen-activated protein kinase (MAPK) pathway is also known as RAS-RAF-MEK-ERK signaling pathway. It regulates cell proliferation and differentiation via RAF-MEK-ERK signaling cascade (see Figure 2). MAPK is activated by the upstream growth factors and their receptors. An autonomous and constitutive activation of MAPK signaling is often induced by the overexpression of growth factors and their receptors in cancer [17] . K-RAS gene encodes a protein that is a member of the small GTPase superfamily and is involved in signal transduction pathways between cell surface receptors and the nucleus, K-RAS mutations have been identified in the 10% - 30% of endometrioid endometrial carcinomas [18] [19] . While some investigators have reported an almost complete absence of K-RAS mutations in serous and clear cell carcinomas of endometrium, other researchers found a higher frequency of K-RAS mutations in Microsatellite Instability (MSI)-positive carcinomas than in MSI-negative tumors [20] [21] . Moreover, K-RAS mutations were detected in endometrial hyperplasias at a similar rate compared to that observed in endometrioid endometrial carcinomas, suggesting that K-RAS mutations could be early events during endometrial carcinogensis [22] . No association has been identified between K-RAS mutations and tumor stage, histologic grade, depth of myometrial invasion, age, or clinical outcome in endometrioid endometrial carcinomas [23] .
2.3. WNT Signaling Pathway and EC
The WNT signaling pathway is one of the most evolutionary-conserved signal transduction pathways. The WNT signaling pathways include b-catenin dependent WNT signaling pathway (i.e. canonical WNT/b-catenin) and b-catenin independent WNT signaling pathway (i.e. Non-canonical, such as WNT/JNK pathway, WNT/calcium pathway) (see Figure 3). Among these WNT signaling pathways, the b-catenin dependent pathway has been associated with human endometrial cancer [24] . Four different intracellular (one canonical while 3 others noncanonical) signaling pathways of WNT are mediated by different downstream mediators and cause different biological activities as illustrated in Figure 3. For canonical WNT signaling pathways, the major effector is the transcription factor β-catenin.
2.3.1. WNT/(-Catenin Signaling Pathway
In the presence of WNT ligand, WNT binds to frizzled family proteins and several co-repressors such as lipoprotein receptor-related proteins (LRP-5/6, RYK or ROR2) [25] , as a result, the APC/AXIN/CK1/GSK3β de-
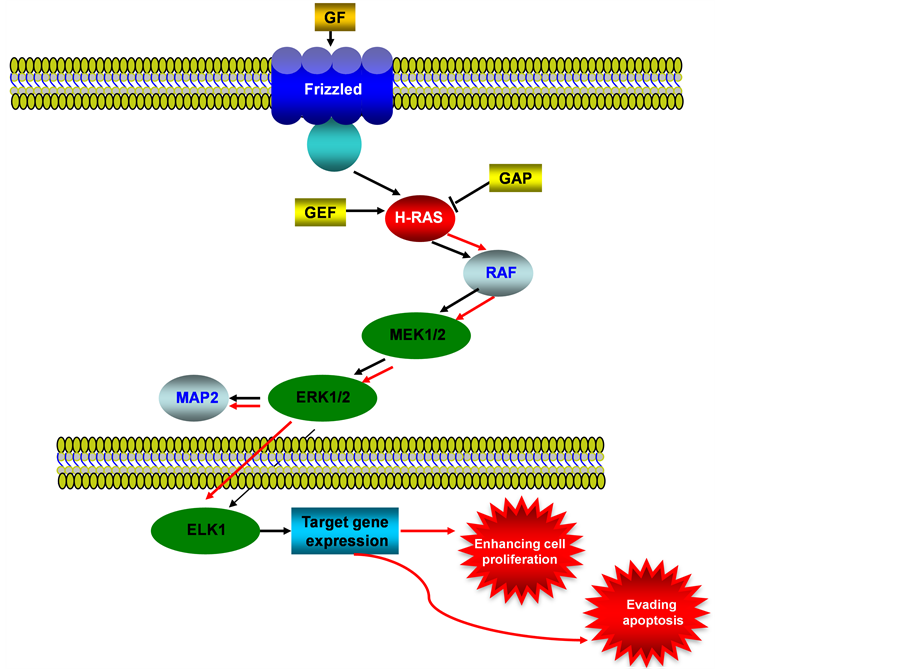
Figure 2. MAPK/ERK signaling pathway in endometrial cancer. This illustration encompasses mediators involved in MARK/ERK pathway, the alternated genes or proteins (RAS, labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for this signaling pathway results in evading apoptosis and enhancing proliferation.
struction complex is inhibited, leading to the stabilization of β-catenin and thus translocation to the nucleus where it interacts with TCF/LEF family transcription factors and then ensues the gene transcription activation. In the absence of WNT ligand, phosphorylated β-catenin is prone to be ubiquitinated and destroyed by the proteasome, thus the expression for TCF/LEF family transcription factors is not activated. The non-canonical WNT signaling pathways are less understood, and they work in a β-catenin independent manner, such as WNT/Calcium pathway, in which the WNT binds to Frizzled receptors, resulting in the release of calcium and activation of calmodulin and other calcium-related enzymes [26] . Moreover, the WNT/JNK/JUN pathway utilizes frizzled receptors, JNK and Rho family GTPases in the absence of β-catenin [27] , Progesterone inhibits WNT signaling by induction of DKK1 and FOXO1 expression, the inhibition of WNT signaling by progesterone was partly circumvented in WNT activated Ishikawa cells [28] and immunohistochemical (IHC) analysis of the WNT target gene CD44 showed that progesterone acted as an inhibitor of WNT signaling in hyperplasia and in well-differentiated endometrial cancer. IHC and loss of heterozygosity (LOH) analysis of tissue-specific inducible mouse model indicated the loss of the APC function in the endometrium leads to cytoplasmic and nuclear β-catenin abnormal accumulation and association with uterine hyperplasia and squamous cell metaplasia [29] . During the menstrual cycle, estradiol can enhance WNT/β-catenin signaling, and constitutive activation of WNT/β-catenin signaling will cause endometrial hyperplasia, which may develop further into endometrial cancer [30] .
2.3.2. APC/(-Catenin Signaling Pathway
The APC/b-catenin signaling pathway plays important roles in normal and tumor cells [31] . In normal cells, in the absence of an extracellular WNT signal, the free (cytoplasmic) b-catenin level is low, since this protein is
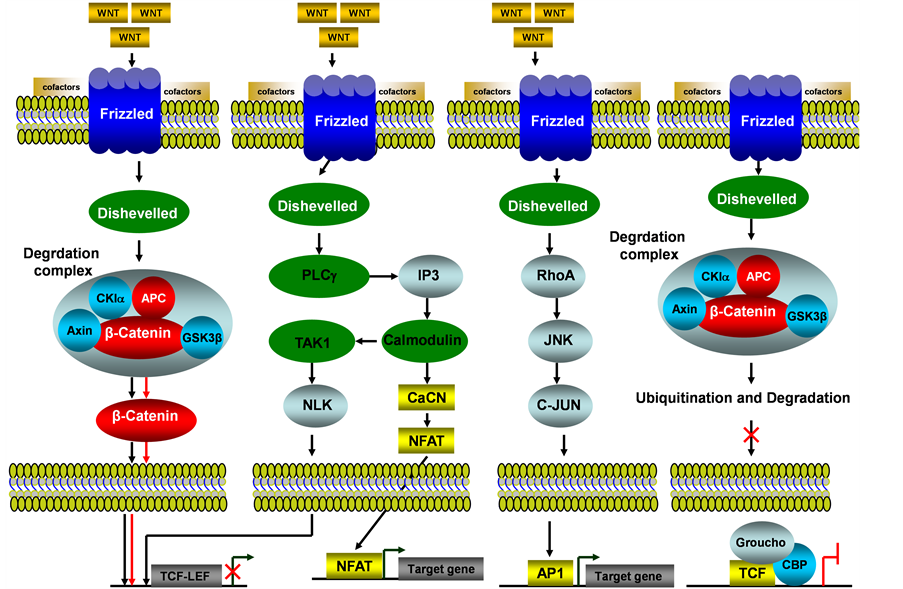
Figure 3. WNT/b-catenin signaling pathway in endometrial cancer. This illustration encompasses b-catenin dependent WNT signaling pathway (i.e. canonical WNT/b-catenin) and b-catenin independent WNT signaling pathway (i.e. Non-canonical, such as WNT/JNK, WNT/calcium and WNT/TAK1 pathways) and even mediators involved in these pathways, the alternated genes or proteins (APC and b-catenin, labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for these signaling pathways result in enhancing proliferation during EC carcinogenesis.
targeted for destruction in the ubiquitin-proteasome system after phosphorylation by glycogen synthase kinase- 3b (GSK-3b) [32] . The subcellular distribution and percentage for b-catenin varies in different stage of endometrial cancer, and nuclear accumulations for b-catenin or g-catenin are associated with activation of TCF-LEF expression in APC/Catenin signaling transduction. An analysis for 40 endometrial carcinomas [33] showed that nuclear catenin expression was found in 13 carcinomas (10 had nuclear b-catenin and 3 g-catenin), and the catenin expression always companied by low-grade endometrial carcinoma, and also frequently associated with a second carcinoma. Another research work [34] showed in 128 endometrial cancer cases, nuclear b-catenin expression was identified in 31.2% in endometrioid endometrial carcinoma (EECs) (total EEC cases employed were 95), and 3% in non endometrioid endometrial carcinoma (NEECs) (the total NEEC cases employed 33), and around 25% EC have b-catenin accumulation in nuclear without mutations for this gene. For g-catenin, the nuclear expression is similar to b-catenin [34] , the wild type and mutated g-catenin induced transforming activity depends on TCF/LEF activation was also identified [35] .
2.4. VEGF/VEGFR Signaling Pathway and EC
VEGFs (Vascular Endothelial Growth Factor) are secreted by tumor cells that stimulate a speed and reversible increase in permeability of microvascular without mast cell degranulation or endothelia cell damage [36] , VEGFs ligands play angiogenic role by binding to specific VEGF receptors, resulted in dimerization of receptor VEGFR and activation of VEGF/VEGFR signaling pathway (Figure 4) [37] [38] . The VEGF receptors are presence on the membrane of endothelial cells, expression of VEGF receptors (VEGFR-1, VEGFR-2 and VEGFR-3) in endothelial cells varies among these 3 receptors, VEGFR-2 is expressed in most of endothelial cells, but VEGFR-1 and VEGFR-3 are expressed only in distinct vascular cells. The function of corepressors
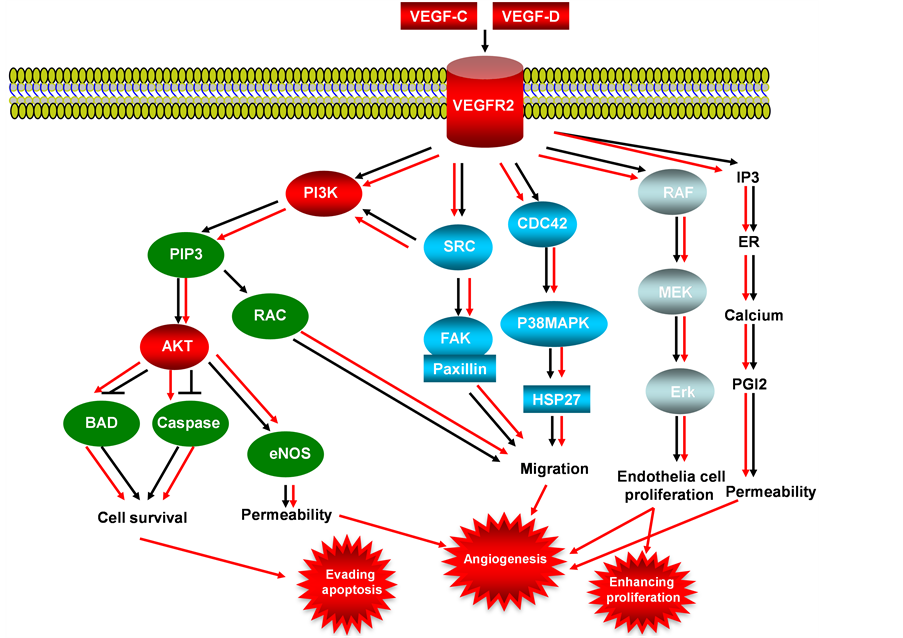
Figure 4. VEGF/VEGFR signaling pathway in endometrial cancer. This illustration encompasses mediators involved in VEGF/VEGFR signaling pathway, the alternated genes or proteins (VEFG-C, VEGF-D, VEGFR-2, PI3K and AKT, labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for these signaling pathways result in enhancing proliferation, evading apoptosis and causing tumor angiogenesis during EC carcinogenesis.
neuropilin-1 (NP-1) and neuropilin-2 (NP-2) are associated with increase of the binding affinity of VEGF ligands to these primary receptors, up regulation of VEGFR in tumor cells suggests the critical role in tumorigenesis of endothelial cells [39] -[41] . VEGFR-3 promotes lymphangiogenesis in lymphatic endothelial cells in adults but not by adult vascular endothelium, and this receptor plays a key role in maintaining vascular integrity by modulating VEGFR-3 activity [42] , related research work showed VEGFR-3 is activated in some solid tumors such as melanoma, in these solid tumors the increased VEGF-C and VEGF-D (VEGFR ligands) are linked to lymph node metastases [43] , and there is also a strong association between lymphovascular density and VEGF-C expression in human malignant mesothelioma [44] . Since angiogenesis is a critical event in the growth and spread of tumors during carcinogenesis [45] , and VEGFs and the angiopoietins are key factors in angiogenesis [46] [47] . The VEGF-C and VEGF-D have both been correlated with poor prognosis in endometrial cancer and atypical complex hyperplasia (ACH), in low grade endometrial tumors, expression of VEGF-B is increased, and experiment showed VEGF-A and VEGF-B, are differentially expressed in benign postmenopausal endometrium and endometrioid endometrial cancer [48] . High expression of VEGF-D in carcinoma cells and stroma cells, are closely related with high level of VEGFR-3 in carcinoma cells and endothelial cells, suggested that VEGF/ VEGFR signaling pathway mediated by VEGF-D/VEGFR-3 plays a significant role to myometrial invasion and lymph node metastasis [49] .
2.5. ErbB Signaling Pathway and EC
The ErbB (Erythroblastoma Virus B) receptor tyrosine kinase family consists of four related receptors: ErbB-1/EGFR/HER1, ErbB-2/HER2, ErbB-3/HER3 and ErbB-4/HER4. ErbB receptors are biochemically typical cell membrane receptor with tyrosine kinase activity that is activated following ligand binding and receptor dimerization. The ErbB receptors are mediators of cell proliferation, migration, differentiation, apoptosis, and cell motility by signaling transduction. Dimerization of ErbB receptors leads to autophosphorylation [50] [51] , thus the phosphorylated tyrosine residues function as docking sites for binding of cytoplasmic proteins containing SRC homology 2 (SH2) and phosphotyrosine binding (PTB) domains [52] [53] , following these modifications, recruitment of proteins then initiates intracellular signaling via following several pathways (Figure 5).
2.5.1. ERB/MARK/ERK Signaling Pathway
The MARK/ERK pathway regulates cell proliferation and survival as described above, for ERB mediating MARK/ERK signaling pathway, the RAS mutation was identified to be involved in early events of endometrial carcinogenesis [18] [19] , and [22] .
2.5.2. ERB/PI3K/AKT/mTOR Signaling Pathway
In this signaling pathway, PI3K is a heterodimer enzyme that contains regulatory subunit P85 and catalytic subunit p110, P85 offers an anchorage site for ErbB (Her) binding, then P110 generates phosphatidylinositol 3, 4, 5-triphosphate as the substrate of protein kinase AKT, which is a critical mediator of PI3K/AKT/mTOR [54] , the AKT causes several down stream transcriptional factors activation including NF-KB, BAD, P27 and GSK3
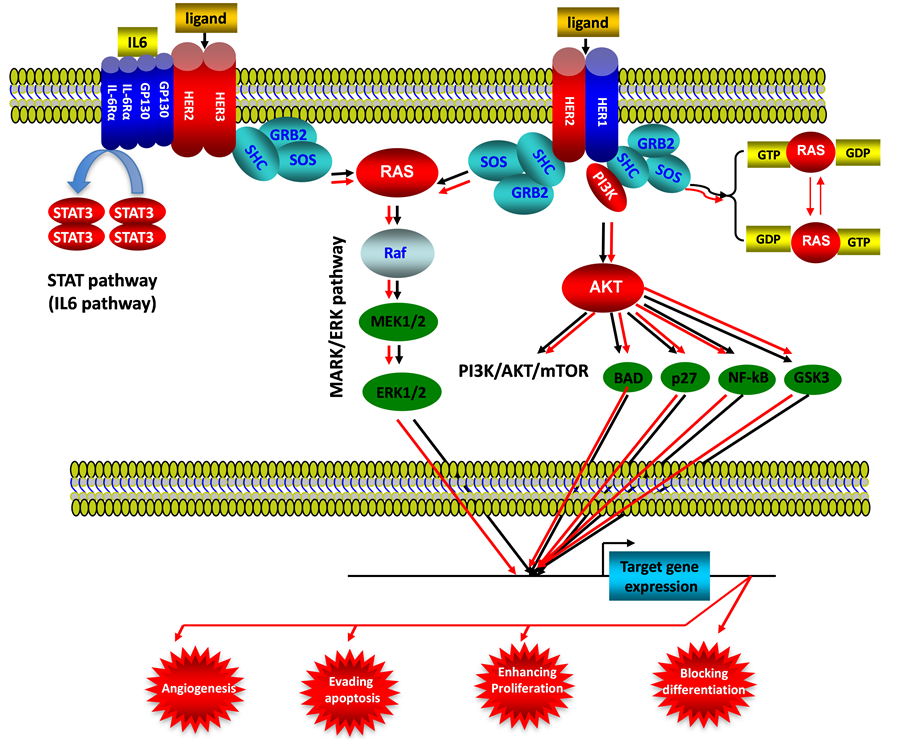
Figure 5. ErbB signaling pathway in endometrial cancer. This illustration encompasses mediators involved in ErbB signaling pathway, the alternated genes or proteins (HER2, HER3, RAS, STAT3 and AKT labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for these signaling pathways result in enhancing proliferation, evading apoptosis, blocking differentiation and causing tumor angiogenesis during EC carcinogenesis.
(See Figure 5), as mentioned in this paper, activation of AKT is closely associated with endometrial carcinogenesis and prognosis by controlling specific gene expression, thereafter occurs new hallmarks for cell transformation such as angiogenesis, evading apoptosis, enhancing proliferation and blocking differentiation.
2.5.3. ERB/STAT Signaling Pathway
Signal transducers and activators of transcription (STAT) pathway are well known to regulate carcinogenesis and tumor progression the dimerized STAT proteins interact with phosphotyrosine residues via their SRC domain (SH2), then translocate into nucleus and active the expression of target gene promoters, increased activity of EGFR and ErbB-2 promote persistent STAT activation and subsequently induce carcinogenesis and tumor progression. Constitutive activation of STAT proteins (especially STAT-3 and STAT-5) is presence in many primary cancers including endometrial cancers [56] -[58] . In EC, STAT3 activates the key oncogene gene such as survivin [57] , Cycline D1 [58] expression to promote EC carcinogenesis.
2.5.4. ERB/SRC Kinase Signaling Pathway
The SRC kinase pathway is critical to regulate cell proliferation, migration, adhesion, angiogenesis, and immune function. SRC is a member of a 10 genes family (SRC, FYN, YES, BLK, FRK, FGR, HCK, LCK, LYN, SRMS). This pathway synergically interacts with other signaling pathways, such as PI3K and STAT pathways to regulate the cell function and possible carcinogenesis [59] [60] . It has been determined that SRC protein (for example, SRC-3) overexpressed in various cancers including EC [61] [62] , and the SRC-3 expression is correlated with the clinical stage, depth of myometrial invasion and differentiation of EC.
2.5.5. ERB/Cγ/PKC Signaling Pathway
Phospholipase Cγ interacts with activated EGFR and ErbB-2 and hydrolyses phosphatidylinositol 4, 5 diphosphate to inositol 1, 3, 5 triphosphate (IP3) and 1, 2 diacylglycerol (DAG), DAG is a cofactor of protein kinase C (PKC) activation, PKC activates MAPK and c-Jun NH2-terminal kinase (JNK) thus regulates the MARK/ERK signaling pathway during EC carcinogenesis [63] [64] . As critical molecule in ErbB signaling pathway, the ErbB-2 protein is localized baso-laterally in the glands and surface epithelial cells [65] [66] , in unselected patients with EC, ErbB-2 amplification/overexpression represents a rare event, in patients with type I EC, it has been reported ΕrbB-2 receptor overexpression in 8% of cases and ErbB-2 gene amplification in 1.4% - 3% of cases [67] [68] . ΕrbB-2 amplification/overexpression is more common in patients with type II EC, for example, Konecny [68] reported that HER2 (ErbB-2) gene amplification with 17% frequency (18/105) in evaluable uterine serous papillary EC specimens. Interestingly, the frequency and regulation patterns of HER2 gene changes dependent on the special populations for same type of tumor, Santin [69] reported that ErbB-2 was overexpression with 70% frequency for black patients, but 24% for white patients. ErbB-2 overexpression is an indicator for type II EC and its treatment prognosis. For ErbB-3, ErbB-4, in endometrium, these two proteins are localized to surface of epithelial cells [70] -[74] , and overexpression of these two genes were detected in endometrial adenocarcinoma (vs. normal controls) [72] , the clinical and pathological implication for ErbB-3 is associated with better survival of cancer cells under treatment conditions [75] [76] . ErbB4 protein was upregulated in type I endometrial carcinoma indicates an oncogenic function in endometrial carcinogenesis [77] .
2.6. P53/P21 and P16INK4/pRB Signaling Pathways and EC
2.6.1. P53/P21 Signaling Pathway
As a multiple function protein, P53 plays a crucial role on cell cycle, and acts as a typical tumor suppressor to prevent carcinogenesis (Figure 6). A numbers of clinic researches have show P53 mutations are closely associated with the endometrial carcinogenesis, Janiec-Jankowska [78] reported in 81 endometrial cases, there were 40 cases (49.4%) had P53 mutations. Tashiro et al. reported in uterine serous carcinoma (USC) cases, there were 90% (19/21) cases contain the P53 mutations, and in endometrial intraepithelial carcinoma (EIC), there were 78% (7/9) cases contain P53 mutations [79] , mutated P53 as a nonfunctional protein accumulates in the cells (especially in nucleus) acts as a dominate negative inhibitor of wild-type P53, leading to the function loss of G1 arrest, resulting in evading apoptosis of cancer cells. Overexpression of P53 was also frequently detected from clinical samples of EC, especially in early stage of endometrial cancer [80] . P53 has been suggested to be an indicator for identifying cases of aggressive carcinoma (especially in the early stage), also as a prognostic indicator for low-stage endometrial cancer [81] .
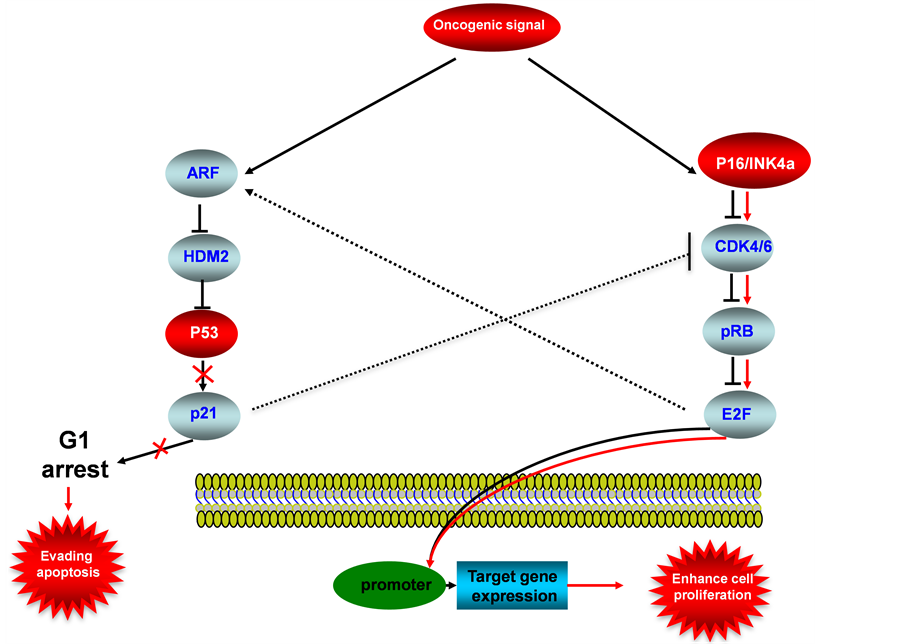
Figure 6. P53-P16INK4a/pRB signaling pathway in endometrial cancer. This illustration encompasses P53 signaling pathway, P16INK4a/pRB signaling pathway and mediators involved in these two pathways, the alternated genes or proteins (P53 and P16INK4a, labeled with red color) in EC carcinogenesis. The flow pattern of the wild type signaling pathway is labeled with black arrow or black line; and the flow pattern of the alternated type signaling pathway is labeled with red arrow or red line. Changes for these signaling pathways result in enhancing proliferation and evading apoptosis during EC carcinogenesis.
2.6.2. P16INK4/pRB Signaling Pathway
P16INK4a (known as P16) is a newly identified tumor suppressor, wild-type P16 acts as a inhibitor of CDK4/6, therefore pRB activation is inhibited, resulted in deactivation of E2F response promoters, thus the specific target genes will not be expressed (Figure 6), the downregulations of P16 was frequently detected in EC, for examples: Nakashima et al detected reduced expression of P16 in 5 of 19 (26%) cervical tumor and 4 of 25 (16%) endometrial tumors [82] . The alterations and abnormal epigenetic modifications for P16INK4a are also play a important role during EC carcinogenesis, Semczuk et al showed 50% P16INK4a gene alterations in sporadic endometrial cancer [83] , high methylation of P16INK4a promoter was been observed up to 75% of sporadic endometrial cancer [84] . Cross talk between P16INK4a/pRB pathway with P53 pathway is linked through the inhibition of wild-type CDK4/6 by P21.
Besides the above genes that we summarized in the different signaling pathways, some other important biomarker for human EC were also identified, such as tumor suppressor gene BCL2, E-cadherin, Baf250, oncogene FGFR2 [85] . The microsatellite instability (MI) was associated with endometrial cancer, five marker of NR-27, NR-21, NR-24, BAT-25 and BAT-26 showed instability in 16, 21, 11, 15 and 17 out of the 80 endometrial tumor samples [86] . All of these changed genes identified in endometrial tumors are listed in Table1
3. Summary
Although morphological basis and several molecular indicators of clinic diagnosis are widely used for endometrial cancer, but we are still in a stage of “blind people touch elephant” to endometrial carcinogenesis even other cancers, all of the molecular evidence of signaling pathways identified to be involved in the carcinogenesis

Table 1 . Genetic/Epigenetic alterations in endometrial cancer.
Caution: Hypermethytlation indicates the modification is on the Promoter region.
of EC are dispersion and disorder, in this paper we try to establish a clear and concise model to show the bird version for carcinogenesis of endometrial tissue based on the discovered proteins and pathways, and we successfully connected the pathological functions from different signaling pathways to four hallmarks of cancer (Figure 7); this model enable us to easily understand the molecular mechanisms for carcinogenesis of EC, and
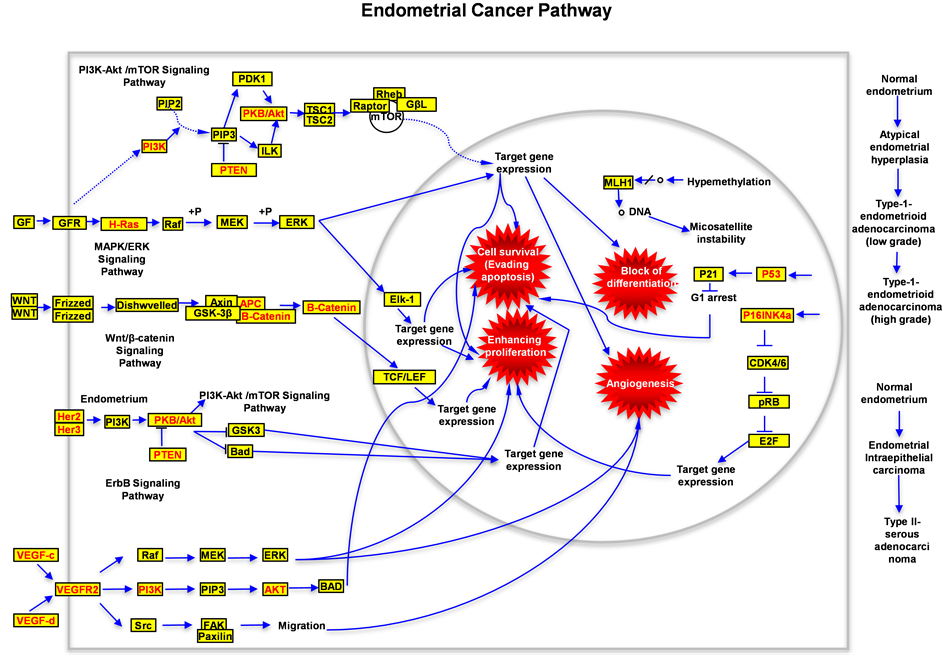
Figure 7. Schematic drawing presents the signaling pathways of human endometrial cancer. All alternated genes or proteins are labeled with red color, the wild type genes or proteins are labeled with black color. In this figure the possible signal transduction flow (including the wild type and mutated type) is indicated. Four critical hallmarkers for carcinogenesis of human endometrial tumor are labeled with red explosions.
will be helpful to design treatment targets and may give some directions to search new treatment strategies for this type of cancer in the future, and this model of EC carcinogenesis may be similar with that of other cancers.
Acknowledgements
We thank Dr. Xiaobing Gao, for reading manuscript and his constructive suggestions.
References
- Laplante, M. and Sabatini, D.M. (2009) mTOR Signaling at a Glance. Journal of Cell Science, 122, 3589-3594. http://dx.doi.org/10.1242/jcs.051011
- Hagiwara, A., Cornu, M., Cybulski, N., Polak, P., Betz, C., Trapani, F., Terracciano, L., Heim, M.H., Rüegg, M.A. and Hallemail, M.N. (2012) Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metabolism, 15, 725-738. http://dx.doi.org/10.1016/j.cmet.2012.03.015
- Risinger, J.I., Hayes, K., Maxwell, G.L., Carney, M.E., Dodge, R.K., Barrett, J.C. and Berchuck, A. (1998) PTEN Mutation in Endometrial Cancers Is Associated with Favorable Clinical and Pathologic Characteristics. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 4, 3005-3010.
- Cancer Genome Atlas Research Networks (2013) Integrated Genomic Characterization of Endometrial Carcinoma. Nature, 497, 67-73. http://dx.doi.org/10.1038/nature12113
- Cheung, L.W.T., Hennessy, B.T., Jie, L., Yu, S.X., Myers, A.P., Djordjevic, B., Lu, Y.L., Stemke-Hale, K., Dyer, M.D., Zhang, F., Ju, Z.L., Cantley, L.C., Scherer, S.E., Liang, H., Lu, K.H., Broaddus, R.R. and Mills, G.B. (2011) High Frequency of PIK3R1 and PIK3R2 Mutations in Endometrial Cancer Elucidates a Novel Mechanism for Regulation of PTEN Protein Stability. Cancer Discovery, 1, 170-185. http://dx.doi.org/10.1158/2159-8290.CD-11-0039
- Oda, K. (2011) Targeting Ras-PI3K/mTOR Pathway and the Predictive Biomarkers in Endometrial Cancer. Gan to Kagakuryoho. Cancer & Chemotherapy, 38, 1084-1087.
- Salvesen, H.B., Carter, S.L., Mannelqvist, M., Duttd, A., Getz, G., Stefansson, I.M., Raeder, M.B., Sos, M.L., Engelsen, I.B., Trovik, J., Wik, E., Greulich, H., Bø, T.H., Jonassen, I., Thomas, R.K., Zanderh, T., Garraway, L.A., Øyan, A.M., Sellers, W.R., Kalland, K.H., Meyerson, M., Akslen, L.A. and Beroukhim, R. (2009) Integrated Genomic Profiling of Endometrial Carcinoma Associates Aggressive Tumors with Indicators of PI3 Kinase Activation. Proceedings of the National Academy of Sciences of the United States of America, 106, 4834-4839. http://dx.doi.org/10.1073/pnas.0806514106
- Dedes, K.J., Wetterskog, D., Ashworth, A., Kaye, S.B. and Jorge, S. (2011) Emerging Therapeutic Targets in Endometrial Cancer. Nature Reviews. Clinical Oncology, 8, 261-271. http://dx.doi.org/10.1038/nrclinonc.2010.216
- Uegaki, K., Kanamori, Y., Kigawa, J., Kawaguchi, W., Kaneko, R., Naniwa, J., Takahashi, M., Shimada, M., Oishi, T., Itamochi, H. and Terakawa, N. (2005) PTEN-Positive and Phosphorylated-Akt-Negative Expression Is a Predictor of Survival for Patients with Advanced Endometrial Carcinoma. Oncology Reports, 14, 389-392.
- Franke, T.F., Hornik C.P., Segev, L., Shostak, G.A. and Sugimoto, C. (2003) PI3K/Akt and Apoptosis: Size Matters. Oncogene, 22, 8983-8998. http://dx.doi.org/10.1038/sj.onc.1207115
- Hennessy, B.T., Smith, D.L., Ram, P.T., Lu, Y.L. and Mill, G.B. (2005) Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nature Reviews. Drug Discovery, 4, 988-1004. http://dx.doi.org/10.1038/nrd1902
- Yap, T.A., Garrett, M.D., Walton, M.I., Raynaud, F., de Bono, J.S. and Workman, P. (2008) Targeting the PI3K-AKT-mTOR Pathway: Progress, Pitfalls, and Promises. Current Opinion in Pharmacology, 8, 393-412. http://dx.doi.org/10.1016/j.coph.2008.08.004
- Lacey Jr., J.V., Yang, H., Gaudet, M.M., Dunning, A., Lissowska, J., Sherman, M.E., Peplonska, B., Brinton, L.A., Healey, C.S., Ahmed, S., Pharoah, P., Easton, D., Chanock, S. and Garcia-Closas, M. (2011) Endometrial Cancer and Genetic Variation in PTEN, PIK3CA, AKT1, MLH1, and MSH2 within a Population-Based Case-Control Study. Gynecologic Oncology, 120, 167-173. http://dx.doi.org/10.1016/j.ygyno.2010.10.016
- Krakstad, C., Birkeland, E., Seidel, D., Kusonmano, K., Petersen, K., Mjøs, S., Hoivik, E.A., Wik, E., Halle, M.K., Øyan, A.M., Kalland, K.-H., Werner, H.M.J., Trovik, J. and Salvesen, H. (2012) High-Throughput Mutation Profiling of Primary and Metastatic Endometrial Cancers Identifies KRAS, FGFR2 and PIK3CA to Be Frequently Mutated. PloS One, 7, Article ID: e52795. http://dx.doi.org/10.1371/journal.pone.0052795
- Abe, N., Watanabe, J., Tsunoda, S., Kuramoto, H. and Okayasu, I. (2011) Significance of Nuclear p-Akt in Endometrial Carcinogenesis: Rapid Translocation of p-Akt into the Nucleus by Estrogen, Possibly Resulting in Inhibition of Apoptosis. International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society, 21, 194-202. http://dx.doi.org/10.1097/IGC.0b013e318207964c
- Mori, N., Kyo, S., Sakaguchi, J., Mizumoto, Y., Ohno, S., Maida, Y., Hashimoto, M., Takakura, M. and Inoue, M. (2007) Concomitant Activation of AKT with Extracellular-Regulated Kinase 1/2 Occurs Independently of PTEN or PIK3CA Mutations in Endometrial Cancer and May Be Associated with Favorable Prognosiss. Cancer Science, 98, 1881-1888. http://dx.doi.org/10.1111/j.1349-7006.2007.00630.x
- Hoshino, R., Chatani, Y., Yamori, T., Tsuruo, T., Oka, H., Yoshida, O., Shimada, Y., Ari-i, S., Wada, H., Fujimoto, J. and Kohno, M. (1999) Constitutive Activation of the 41-/43-kDa Mitogen-Activated Protein Kinase Signaling Pathway in Human Tumors. Oncogene, 18, 813-822. http://dx.doi.org/10.1038/sj.onc.1202367
- Alexander-Sefre, F., Salvesen, H.B., Ryan, A., Singh, N., Akslen, L.A., MacDonald, N., Wilbanks, G. and Jacobs, I.J. (2003) Molecular Assessment of Depth of Myometrial Invasion in Stage I Endometrial Cancer: A Model Based on K-ras Mutation Analysis. Gynecologic Oncology, 91, 218-225. http://dx.doi.org/10.1016/S0090-8258(03)00505-5
- Mizuuchi, H., Nasim, S., Kudo, R., Silverberg, S.G., Greenhouse, S. and Garrett, C.T. (1992) Clinical Implications of K-ras Mutations in Malignant Epithelial Tumors of the Endometrium. Cancer Research, 52, 2777-2781.
- Matias-Guiu, X., Catasus, L., Bussaglia, E., Lagarda, H., Garcia, A., Pons, C., Muñoz, J., Argüelles, R., Machin, P. and Prat, J. (2001) Molecular Pathology of Endometrial Hyperplasia and Carcinoma. Human Pathology, 32, 569-577. http://dx.doi.org/10.1053/hupa.2001.25929
- Prat, J., Gallardo, A., Cuatrecasas, M. and Catasús, L. (2007) Endometrial Carcinoma: Pathology and Genetics. Pathology, 39, 72-87. http://dx.doi.org/10.1080/00313020601136153
- Dobrzycka, B., Terlikowski, S.J., et al. (2009) Mutations of the KRAS Oncogene in Endometrial Hyperplasia and Carcinoma. Folia Histochemica et Cytobiologica/Polish Academy of Sciences, Polish Histochemical and Cytochemical Society, 47, 65-68. http://dx.doi.org/10.2478/v10042-009-0014-2
- Ito, K., Watanabe, K., Nasim, S., Sasano, H., Sato, S., Yajima, A., Silverberg, S.G. and Garrett, C.T. (1996) K-RAS Point Mutations in Endometrial Carcinoma: Effect on Outcome Is Dependent on Age of Patient. Gynecologic Oncology, 63, 238-246. http://dx.doi.org/10.1006/gyno.1996.0313
- Kestler, H.A. and Kuhl, M. (2008) From Individual Wnt Pathways towards a Wnt Signalling Network. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences, 363, 1333-1347. http://dx.doi.org/10.1098/rstb.2007.2251
- Logan, C.Y. and Nusse, R. (2004) The Wnt Signaling Pathway in Development and Disease. Annual Review of Cell and Developmental Biology, 20, 781-810. http://dx.doi.org/10.1186/2045-3701-2-14
- Kuhl, M., Sheldahl, L.C., Malbon, C.C. and Moon, R.T. (2000) Ca2+/Calmodulin-Dependent Protein Kinase II Is Stimulated by Wnt and Frizzled Homologs and Promotes Ventral Cell Fates in Xenopus. Journal of Biological Chemistry, 275, 12701-12711. http://dx.doi.org/10.1074/jbc.275.17.12701
- Veeman, M.T., Axelrod, J.D. and Moon, R.T. (2003) A Second Canon. Functions and Mechanisms of β-Catenin-Independent Wnt Signaling. Developmental Cell, 5, 367-377. http://dx.doi.org/10.1016/S1534-5807(03)00266-1
- Wang, Y., Hanifi-Moghaddam, P., Hanekamp, E.E., Kloosterboer, H.J., Franken, P., Veldscholte, J., van Doorn, H.C., Ewing, P.C., Kim, J.J., Grootegoed, J.A., Burger, C.W., Fodde, R. and Blok, L.J. (2009) Progesterone Inhibition of Wnt/β-Catenin Signaling in Normal Endometrium and Endometrial Cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 15, 5784-5793. http://dx.doi.org/10.1158/1078-0432.CCR-09-0814
- Martens, B., Amman, F., Manoharadas, S., Zeichen, L., Orell, A., Albers, S.V., Hofacker, I. and Blãsi, U. (2013) Alterations of the Transcriptome of Sulfolobus acidocaldarius by Exoribonuclease aCPSF2. PLoS ONE, 8, Article ID: e76569. http://dx.doi.org/10.1371/journal.pone.0076569
- Wang, Y., van der Zee, M., Fodde, R. and Blok, L.J. (2010) Wnt/β-Catenin and Sex Hormone Signaling in Endometrial Homeostasis and Cancer. Oncotarget, 1, 674-684.
- Sharpe, C., Lawrence, N. and Arias, A.M. (2001) Wnt Signalling: A Theme with Nuclear Variations. BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 23, 311-318. http://dx.doi.org/10.1002/bies.1045
- Aberle, H., Bauer, A., Stappert, J., Kispert, A. and Kemler, R. (1997) β-Catenin Is a Target for the Ubiquitin-Proteasome Pathway. The EMBO Journal, 16, 3797-3804. http://dx.doi.org/10.1093/emboj/16.13.3797
- Palacios, J., Catasus, L., Moreno-Bueno, G., Matias-Guiu, X., Prat, J. and Gamallo, C. (2001) Betaand GammaCatenin Expression in Endometrial Carcinoma. Relationship with Clinicopathological Features and Microsatellite Instability. Virchows Archiv: An International Journal of Pathology, 438, 464-469. http://dx.doi.org/10.1007/s004280000371
- Moreno-Bueno, G., Hardisson, D., Sánchez, C., Sarrió, D., Cassia, R., García-Rostán, G., Prat, J., Guo, M., Herman, J.G., Matías-Guiu, X., Esteller, M. and Palacios, J. (2002) Abnormalities of the APC/β-Catenin Pathway in Endometrial Cancer. Oncogene, 21, 7981-7990. http://dx.doi.org/10.1038/sj.onc.1205924
- Kolligs, F.T., Kolligs, B., Hajra, K.M., Hu, G., Tani, M., Cho, K.R. and Fearon, E.R. (2000) Gamma-Catenin Is Regulated by the APC Tumor Suppressor and Its Oncogenic Activity Is Distinct from that of β-Catenin. Genes & Development, 14, 1319-1331.
- Senger, D.R., Galli, S.J., Dvorak, A.M., Perruzzi, C.A., Harvey, V.S. and Dvorak, H.F. (1983) Tumor Cells Secrete a Vascular Permeability Factor that Promotes Accumulation of Ascites Fluid. Science, 219, 983-985. http://dx.doi.org/10.1126/science.6823562
- Ferrara, N. (2002) VEGF and the Quest for Tumour Angiogenesis Factors. Nature Reviews Cancer, 2, 795-803. http://dx.doi.org/10.1038/nrc909
- Hicklin, D.J. and Ellis, L.M. (2005) Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 23, 1011-1027. http://dx.doi.org/10.1200/JCO.2005.06.081
- Kawasaki, T., Kitsukawa, T., Bekku, Y., Matsuda, Y., Sanbo, M., Yagi, T. and Fujisawa, H. (1999) A Requirement for Neuropilin-1 in Embryonic Vessel Formation. Development, 126, 4895-4902.
- Ferrer, F.A., Miller, L.J., Lindquist, R., Kowalczyk, P., Laudone, V.P., Albertsen, P.C. and Kreutzer, D.L. (1999) Expression of Vascular Endothelial Growth Factor Receptors in Human Prostate Cancer. Urology, 54, 567-572. http://dx.doi.org/10.1016/S0090-4295(99)00156-9
- Decaussin, M., Sartelet, H., Robert, C., Moro, D., Claraz, C., Brambilla, C. and Brambilla, E. (1999) Expression of Vascular Endothelial Growth Factor (VEGF) and Its Two Receptors (VEGF-R1-Flt1 and VEGF-R2-Flk1/KDR) in Non-Small Cell Lung Carcinomas (NSCLCs): Correlation with Angiogenesis and Survival. The Journal of Pathology, 188, 369-377. http://dx.doi.org/10.1002/(SICI)1096-9896(199908)188:4<369::AID-PATH381>3.0.CO;2-X
- Dumont, D.J., Jussila, L., Taipale, J., Lymboussaki, A., Mustonen, T., Pajusola, K., Breitman, M. and Alitalo, K. (1998) Cardiovascular Failure in Mouse Embryos Deficient in VEGF Receptor-3. Science, 282, 946-949. http://dx.doi.org/10.1126/science.282.5390.946
- Achen, M.G., Williams, R.A., Minekus, M.P., Thornton, G.E., Stenvers, K., Rogers, P.A.W., Lederman, F., Roufail, S. and Stacker, S.A. (2001) Localization of Vascular Endothelial Growth Factor-D in Malignant Melanoma Suggests a Role in Tumour Angiogenesis. The Journal of Pathology, 193, 147-154. http://dx.doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH757>3.0.CO;2-G
- Ohta, Y., Shridhar, V., Bright, R.K., Kalemkerian, G.P., Du, W., Carbone, M., Watanabe, Y. and Pass, H.I. (1999) VEGF and VEGF Type C Play an Important Role in Angiogenesis and Lymphangiogenesis in Human Malignant Mesothelioma Tumours. British Journal of Cancer, 81, 54-61.
- Folkman, J. (1990) What Is the Evidence that Tumors Are Angiogenesis Dependent? Journal of the National Cancer Institute, 82, 4-6. http://dx.doi.org/10.1093/jnci/82.1.4
- Fong, G.H., Rossant, J., Gertsenstein, M. and Breitman, M.L. (1995) Role of the Flt-1 Receptor Tyrosine Kinase in Regulating the Assembly of Vascular Endothelium. Nature, 376, 66-70. http://dx.doi.org/10.1038/376066a0
- Thurston, G., Suri, C., Smith, K., McClain, J., Sato, T.N., Yancopoulos, G.D. and McDonald, D.M. (1999) Leakage-Resistant Blood Vessels in Mice Transgenically Overexpressing Angiopoietin-1. Science, 286, 2511-2514. http://dx.doi.org/10.1126/science.286.5449.2511
- Holland, C.M., Day, K., Evans, A. and Smith, S.K. (2003) Expression of the VEGF and Angiopoietin Genes in Endometrial Atypical Hyperplasia and Endometrial Cancer. British Journal of Cancer, 89, 891-898. http://dx.doi.org/10.1038/sj.bjc.6601194
- Yokoyama, Y., Charnock-Jones, D.S., Licence, D., Yanaihara, A., Hastings, J.M., Holland, C.M., Emoto, M., Sakamoto, A., Sakamoto, T., Maruyama, H., Sato, S., Mizunuma, H. and Smith, S.K. (2003) Expression of Vascular Endothelial Growth Factor (VEGF)-D and Its Receptor, VEGF Receptor 3, as a Prognostic Factor in Endometrial Carcinoma. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 9, 1361-1369.
- Burgess, A.W., Cho, H.S., Eigenbrot, C., Ferguson, K.M., Garrett, T.P.J., Leahy, D.J., et al. (2003) An Open-and-Shut Case? Recent Insights into the Activation of EGF/ErbB Receptors. Molecular Cell, 12, 541-552. http://dx.doi.org/10.1016/S1097-2765(03)00350-2
- Zhang, H.T., Berezov, A., Wang, Q., Zhang, G., Drebin, J., Murali, R. and Greene, M.I. (2007) ErbB Receptors: From Oncogenes to Targeted Cancer Therapies. The Journal of Clinical Investigation, 117, 2051-2058. http://dx.doi.org/10.1172/JCI32278
- Zhou, S.Y., Margolis, B., Chaudhuri, M., Shoelson, S.E. and Cantley, L.C. (1995) The Phosphotyrosine Interaction Domain of SHC Recognizes Tyrosine-Phosphorylated NPXY Motif. The Journal of Biological Chemistry, 270, 14863-14866. http://dx.doi.org/10.1074/jbc.270.25.14863
- Yarden, Y. and Sliwkowski, M.X. (2001) Untangling the ErbB Signalling Network. Nature Reviews Molecular Cell Biology, 2, 127-137. http://dx.doi.org/10.1038/35052073
- Cuevas, B.D., Lu, Y.L., Mao, M.L., Zhang, J.Y., LaPushin, R., Siminovitch, K. and Mills, G.B. (2001) Tyrosine Phosphorylation of p85 Relieves Its Inhibitory Activity on Phosphatidylinositol 3-Kinase. The Journal of Biological Chemistry, 276, 27455-27461. http://dx.doi.org/10.1074/jbc.M100556200
- Jiang, X.N., Chen, S., Asara, J.M. and Balk, S.P. (2010) Phosphoinositide 3-Kinase Pathway Activation in Phosphate and Tensin Homolog (PTEN)-Deficient Prostate Cancer Cells Is Independent of Receptor Tyrosine Kinases and Mediated by the p110β and p110δ Catalytic Subunits. The Journal of Biological Chemistry, 285, 14980-14989. http://dx.doi.org/10.1074/jbc.M109.085696
- Haura, E.B., Turkson, J. and Jove, R. (2005) Mechanisms of Disease: Insights into the Emerging Role of Signal Transducers and Activators of Transcription in Cancer. Nature Clinical Practice Oncology, 2, 315-324. http://dx.doi.org/10.1038/ncponc0195
- Pallares, J., Martinez-Guitarte, J.L., et al. (2005) Survivin Expression in Endometrial Carcinoma: A Tissue Microarray Study with Correlation with PTEN and STAT-3. International Journal of Gynecological Pathology: Official Journal of the International Society of Gynecological Pathologists, 24, 247-253.
- Catalano, S., Giordano, C., et al. (2009) Evidence That Leptin through STAT and CREB Signaling Enhances Cyclin D1 Expression and Promotes Human Endometrial Cancer Proliferation. Journal of Cellular Physiology, 218, 490-500. http://dx.doi.org/10.1002/jcp.21622
- Irby, R.B., McCarthy, S.M. and Yeatman, T.J. (2004) Osteopontin Regulates Multiple Functions Contributing to Human Colon cancer Development and Progression. Clinical & Experimental Metastasis, 21, 515-523. http://dx.doi.org/10.1007/s10585-004-2873-4
- Summy, J.M. and Gallick, G.E. (2006) Treatment for Advanced Tumors: SRC Reclaims Center Stage. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 12, 1398-1401. http://dx.doi.org/10.1158/1078-0432.CCR-05-2692
- Sakaguchi, H., Fujimoto, J., et al. (2007) Clinical Implications of Steroid Receptor Coactivator (SRC)-3 in Uterine Endometrial Cancers. The Journal of Steroid Biochemistry and Molecular Biology, 104, 237-240. http://dx.doi.org/10.1016/j.jsbmb.2007.03.00762
- Balmer, N.N., Richer, J.K., et al. (2006) Steroid Receptor Coactivator AIB1 in Endometrial Carcinoma, Hyperplasia and Normal Endometrium: Correlation with Clinicopathologic Parameters and Biomarkers. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, 19, 1593-1605. http://dx.doi.org/10.1038/modpathol.3800696
- Schonwasser, D.C., Marais, R.M., Marshall, C.J. and Parker, P.J. (1998) Activation of the Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Pathway by Conventional, Novel, and Atypical Protein Kinase C Isotypes. Molecular and Cellular Biology, 18, 790-798.
- McClellan, M., Kievit, P., Auersperg, N. and Rodland, K. (1999) Regulation of Proliferation and Apoptosis by Epidermal Growth Factor and Protein Kinase C in Human Ovarian Surface Epithelial Cells. Experimental Cell Research, 246, 471-479. http://dx.doi.org/10.1006/excr.1998.4328
- Miturski, R., Semczuk, A. and Jakowicki, J.A. (1998) C-erbB-2 Expression in Human Proliferative and Hyperplastic Endometrium. International Journal of Gynaecology and Obstetrics: The Official Organ of the International Federation of Gynaecology and Obstetrics, 61, 73-74. http://dx.doi.org/10.1016/S0020-7292(98)00008-3
- Ejskjaer, K., Sorensen, B.S., Poulsen, S.S., Forman, A., Nexø, E. and Mogensen, O. (2007) Expression of the Epidermal Growth Factor System in Endometrioid Endometrial Cancer. Gynecologic Oncology, 104, 158-167. http://dx.doi.org/10.1016/j.ygyno.2006.07.015
- Huang, T.H. and Morrison, S.L. (2006) A Trimeric Anti-HER2/neu ScFv and Tumor Necrosis Factor-α Fusion Protein Induces HER2/neu Signaling and Facilitates Repair of Injured Epithelia. The Journal of Pharmacology and Experimental Therapeutics, 316, 983-991. http://dx.doi.org/10.1124/jpet.105.095513
- Konecny, G.E., Santos, L., Winterhoff, B., Hatmal, M., Keeney, G.L., Mariani, A., et al. (2009) HER2 Gene Amplification and EGFR Expression in a Large Cohort of Surgically Staged Patients with Nonendometrioid (Type II) Endometrial Cancer. British Journal of Cancer, 100, 89-95. http://dx.doi.org/10.1038/sj.bjc.6604814
- Santin, A.D., Bellone, S., Siegel, E.R., Palmieri, M., Thomas, M., Cannon, M.J., Kay, H.H., Roman, J.J., Burnett, A. and Pecorelli, S. (2005) Racial Differences in the Overexpression of Epidermal Growth Factor Type II Receptor (HER2/neu): A Major Prognostic Indicator in Uterine Serous Papillary Cancer. American Journal of Obstetrics and Gynecology, 192, 813-818. http://dx.doi.org/10.1016/j.ajog.2004.10.605
- Konecny, G.E., Santos, L., Winterhoff, B., Hatmal, M., Keeney, G.L., Mariani, A., et al. (2009) HER2 Gene Amplification and EGFR Expression in a Large Cohort of Surgically Staged Patients with Nonendometrioid (Type II) Endometrial Cancer. British Journal of Cancer, 100, 89-95. http://dx.doi.org/10.1038/sj.bjc.6604814
- Lemoine, N.R., Barnes, D.M., Hollywood, D.P., Hughes, C.M., Smith, P., Dublin, E., Prigent, S.A., Gullick, W.J. and Hurst, H.C. (1992) Expression of the ERBB3 Gene Product in Breast Cancer. British Journal of Cancer, 66, 1116-1121. http://dx.doi.org/10.1038/bjc.1992.420
- Srinivasan, R., Benton, E., McCormick, F., Thomas, H. and Gullick, W.J. (1999) Expression of the c-erbB-3/HER-3 and c-erbB-4/HER-4 Growth Factor Receptors and Their Ligands, Neuregulin-1 Alpha, Neuregulin-1 Beta, and Betacellulin, in Normal Endometrium and Endometrial Cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 5, 2877-2883.
- Ejskjaer, K., Sorensen, B.S., Poulsen, S.S., Forman, A., Nexø, E. and Mogensen, O. (2007) Expression of the Epidermal Growth Factor System in Endometrioid Endometrial Cancer. Gynecologic Oncology, 104, 158-167. http://dx.doi.org/10.1016/j.ygyno.2006.07.015
- Androutsopoulos, G., Adonakis, G., Liava, A., Ravazoula, P. and Decavalas, G. (2013) Expression and Potential Role of ErbB Receptors in Type II Endometrial Cancer. European Journal of Obstetrics, Gynecology and Reproductive Biology, 168, 204-208. http://dx.doi.org/10.1016/j.ejogrb.2013.01.007
- Contessa, J.N., Abell, A., Mikkelsen, R.B., Valerie, K. and Schmidt-Ullrich, R.K. (2006) Compensatory ErbB3/c-SRC Signaling Enhances Carcinoma Cell Survival to Ionizing Radiation. Breast Cancer Research and Treatment, 95, 17-27. http://dx.doi.org/10.1007/s10549-005-9023-9
- Chobotova, K., Karpovich, N., Carver, J., Manek, S., Gullick, W.J., Barlow, D.H. and Mardon, H.J. (2005) Heparin-Binding Epidermal Growth Factor and Its Receptors Mediate Decidualization and Potentiate Survival of Human Endometrial Stromal Cells. The Journal of Clinical Endocrinology and Metabolism, 90, 913-919. http://dx.doi.org/10.1210/jc.2004-0476
- Saghir, F.S.A., Rose, I.M., et al. (2010) Gene Expression Profiling and Cancer-Related Pathways in Type I Endometrial Carcinoma. International Journal of Gynecological Cancer, 20, 724-731. http://dx.doi.org/10.1111/IGC.0b013e3181e1c14c
- Janiec-Jankowska, A., Konopka, B., Goluda, C. and Najmola, U. (2010) Tp53 Mutations in Endometrial Cancers: Relation to PTEN Gene Defects. International Journal of Gynecological Cancer, 20, 196-202. http://dx.doi.org/10.1111/IGC.0b013e3181c83675
- Tashiro, H., Isacson, C., Levine, R., Kurman, R.J., Cho, K.R. and Hedrick, L. (1997) P53 Gene Mutations Are Common in Uterine Serous Carcinoma and Occur Early in Their Pathogenesis. The American Journal of Pathology, 150, 177-185.
- Kohlberger, P., Gitsch, G., Loesch, A., Tempfer, C., Kaider, A., Reinthaller, A., Kainz, C. and Breitenecker, G. (1996) P53 Protein Overexpression in Early Stage Endometrial Cancer. Gynecologic Oncology, 62, 213-217. http://dx.doi.org/10.1006/gyno.1996.0218
- Ito, K., Watanabe, K., Nasim, S., Sasano, H., Sato, S., Yajima, A., Silverberg, S.G. and Garrett, C.T. (1994) Prognostic Significance of P53 Overexpression in Endometrial Cancer. Cancer Research, 54, 4667-4670.
- Nakashima, R., Fujita, M., Enomoto, T., Haba, T., Yoshino, K., Wada, H., Kurachi, H., Sasaki, M., Wakasa, K., Inoue, M., Buzard, G. and Murata, Y. (1999) Alteration of p16 and p15 Genes in Human Uterine Tumours. British Journal of Cancer, 80, 458-467. http://dx.doi.org/10.1038/sj.bjc.6690379
- Semczuk, A., Boltze, C., Marzec, B., Szczygielska, A., Roessner, A. and Schneider-Stock, R. (2003) p16INK4A Alterations Are Accompanied by Aberrant Protein Immunostaining in Endometrial Carcinomas. Journal of Cancer Research and Clinical Oncology, 129, 589-596. http://dx.doi.org/10.1007/s00432-003-0482-2
- Guida, M., Sanguedolce, F., Bufo, P., Di Spiezio Sardo, A., Bifulco, G., Nappi, C. and Pannone, G. (2009) Aberrant DNA Hypermethylation of hMLH-1 and CDKN2A/p16 Genes in Benign, Premalignant and Malignant Endometrial Lesions. European Journal of Gynaecological Oncology, 30, 267-270.
- O’Hara, A.J. and Bell, D.W. (2012) The Genomics and Genetics of Endometrial Cancer. Advances in Genomics and Genetics, 2012, 33-47. http://dx.doi.org/10.2147/AGG.S28953
- Wong, Y.F., Cheung, T.H., Kit Lo, K.W., Yim, S.F., Yin Chan, L.K., Buhard, O., Duval, A., Hung Chung, T.K. and Hamelin, R. (2006) Detection of Microsatellite Instability in Endometrial Cancer: Advantages of a Panel of Five Mononucleotide Repeats over the National Cancer Institute Panel of Markers. Carcinogenesis, 27, 951-955. http://dx.doi.org/10.1093/carcin/bgi333
NOTES

*Corresponding author.