 Vol.3, No.3, 234-248 (2011) Natural Science http://dx.doi.org/10.4236/ns.2011.33030 Copyright © 2011 SciRes. OPEN ACCESS Receptor binding specificity and origin of 2009 H1N1 pandemic influenza virus Wei Hu Department of Computer Science, Houghton College, Houghton, USA; wei.hu@houghton.edu Received 31 December 2010; revised 2 February 2011; accepted 3 February 2011. ABSTRACT Recently, a genetic variant of 2009 H1N1 has become the predominant virus circulating in the southern hemisphere, particularly Australia and New Zealand, and in Singapore during the win- ter of 2010. It was associated with several vac- cine breakthroughs and fatal cases. We ana- lyzed three reported mutations D94N, N125D, and V250A in the HA protein of this genetic variant. It appeared that the reason for D94N and V250A to occur in pairs was to maintain the HA binding to human type receptor, so the virus could replicate in humans efficiently. Guided by this interpretation, we discovered a new muta- tion V30A that could compensate for N125D as V250A did for D94N. We demonstrated that the presence of amino acids 30A and 125N in HA enhanced the binding to human type receptor, while 30V and 125D favored the receptors of avian type and of A/South Carolina/1/18 (H1N1). Furthermore, a combination of 94D, 125D, and 250V made the primary binding preference similar to that of A/South Carolina/1/18 (H1N1) and a combination of 94N, 125D, and 250A re- sulted in the primary binding affinity for avian type receptor, which clearly differed from that of A/California/07/2009 (H1N1), a strain used in the vaccine for 2009 H1N1. We also re-examined the origin of 2009 H1N1 to refine our knowledge of this important issue. Although the NP, PA, PB1, and PB2 of 2009 H1N1 were closest to North American swine H3N2 in sequence identity, their interaction patterns were closest to swine H1N1 in North America. Keywords: 2009 H1N1; Hemagglutinin; Influenza; Informational Spectrum Method; Mutation; Receptor Binding Specificity 1. INTRODUCTION New influenza viruses arise through genetic reassort- ment. The 2009 H1N1 virus is a novel virus with its eight gene segments derived from North American and Eurasian swine lineages [1]. Intensive research on this virus has been conducted, including a series of papers of our own [2-12]. These papers covered the mutations and correlated mutations in HA and NA, the stalk motifs in NA, HA receptor binding specificity, novel host markers, interactions of the proteins of 2009 H1N1. Although the World Health Organization (WHO) declared an end to the 2009 H1N1 influenza pandemic on August 10, 2010, continued surveillance of the evolution of the 2009 H1N1 virus is still warranted. The 2009 H1N1 virus remained genetically stable since it emerged in March 2009. However, a genetic variant of 2009 H1N1 was first discovered in Singapore in early 2010, and then was spread to Australia and New Zealand during the 2010 winter influenza season of the southern hemisphere. This variant became the predomi- nant virus circulating in these three countries and was linked to several vaccine breakthroughs and fatal cases. As such, a vaccine update might be needed sooner than expected [13]. Several mutations were identified in genes HA, NA, PB2, PB1, NP, and NS1 of this variant, including three mutations D94N, N125D, and V250A in the HA protein. To examine the impact of these mutations, a structural homology model of HA from the A/Brisbane/10/2010 (H1N1) virus based on the A/California/04/2009 (H1N1) structure was constructed. Mutation N125D was found to be centrally located in the classical Sa epitope, poten- tially affecting antigenicity, and mutation V250A is lo- cated at an internal beta sheet below the receptor binding pocket facing the Sa epitope [13]. Additionally, two mu- tations D94N and V250A tended to occur in pairs in the HA of this variant exclusively circulating in Australia and New Zealand so far [13]. Given the potential sig- nificance of the mutations observed in this variant, it is imperative to further investigate their roles in HA recep-  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 235235 tor recognition. In general, human influenza viruses bind preferen- tially to α2,6 receptors, typically found in the upper air- way of humans, whereas avian influenza viruses tend to bind to α2,3 receptors, also found in the lower respira- tory tract of humans. It is well documented that muta- tions in the HA protein may alter HA receptor binding selection. For example, mutations E190D, Q226L, or G228S in H1, H2, or H3 could switch binding prefer- ence from avian to human type receptor [14-17]. In [7], the informational spectrum method (ISM) was success- fully applied to quantify the effects of mutations in the HA protein on its binding affinity. Position 222 resides in the receptor binding site of the HA protein of 2009 H1N1 and therefore may play a critical role in HA bind- ing specificity. One of the findings in [7] indicated that mutation D222G in the HA of 2009 H1N1 enhances the selection for avian type receptors, and reduces the selec- tion for human type receptors. This finding was subse- quently verified in an experiment [18] and mutation D222G was further found to be associated with severe clinical outcome [18-20]. Another recent experiment [21] showed that mutation D94N in H5 HA of avian origin increased the binding of HA to human type receptor, while decreased the binding to avian type receptor. The purpose of this study was to elucidate the impact of mutations D94N, N125D, and V250A in the HA pro- tein of this variant on receptor affinity with ISM, a bio- informatics technique developed in [7,22-24]. Also we sought to explore the origin of 2009 H1N1 and its con- nections with the swine lineages to enrich our under- standing of this novel virus. 2. MATERIALS AND METHODS 2.1. Sequence Data Protein and gene sequences of influenza were re- trieved from the Influenza Virus Resource (http://www. ncbi/nlm.nih.giv/geno mes/FLU/FLU.html) of the Na- tional Center for Biotechnology Information (NCBI) and the EpiFlu Database (http://platform.gisaid.org) of GI- SAID. Only the full length and unique sequences were selected. All sequences used in this study were aligned with MAFFT [25]. 2.2. Informational Spectrum Method The informational spectrum method is a bioinformat- ics technique that can be used to analyze protein se- quences. Prior to this analysis, the protein sequences have to be translated into numerical sequences. One such approach is to assign each amino acid to its electron-ion interaction potential (EIIP), which represents the average energy of the valence electrons in the amino acid (Table 1). The application of EIIP to protein function analysis assumes that the strength of the electromagnetic field surrounding the protein is indicative of its biological function. This method was successful in revealing vari- ous protein properties. The numerical sequence 1, 2,xm m of a protein sequence is transformed into the frequency do- main using DFT. The DFT coefficients n are de- fined as 2 1, 2,,2 jnm N nxmen N where N is the length of sequence m The energy density spectrum is defined as 2,1,2,,2SnXnX nXnnN The informational spectrum (IS) of a sequence m comprises the frequencies and the amplitudes of its DFT. Peak frequencies of IS of a protein sequence reflect its biological or biochemical functions. To determine the same biological or biochemical functions of a group of protein sequences, a consensus informational spectrum (CIS) can be used, which is defined as the product of energy density spectrum Sn of each sequence in the group. A measure of similarity for each peak is a signal- to-noise ratio (S/N), which is defined as a ratio of signal density to the mean value of the whole spectrum [22]. The theory of CIS [26] states that: 1) One peak only exits for a group of protein se- quences sharing the same biological function. 2) No signal peak exists for biologically unrelated protein sequences. Table 1. The electron-ion interaction potential (EIIP) of amino acids used to encode amino acids. Amino acid EIIP Amino acid EIIP L 0.0000 Y 0.0516 I 0.0000 W 0.0548 N 0.0036 Q 0.0761 G 0.0050 M 0.0823 E 0.0057 S 0.0829 V 0.0058 C 0.0829 P 0.0198 T 0.0941 H 0.0242 F 0.0946 K 0.0371 R 0.0959 A 0.0373 D 0.1263 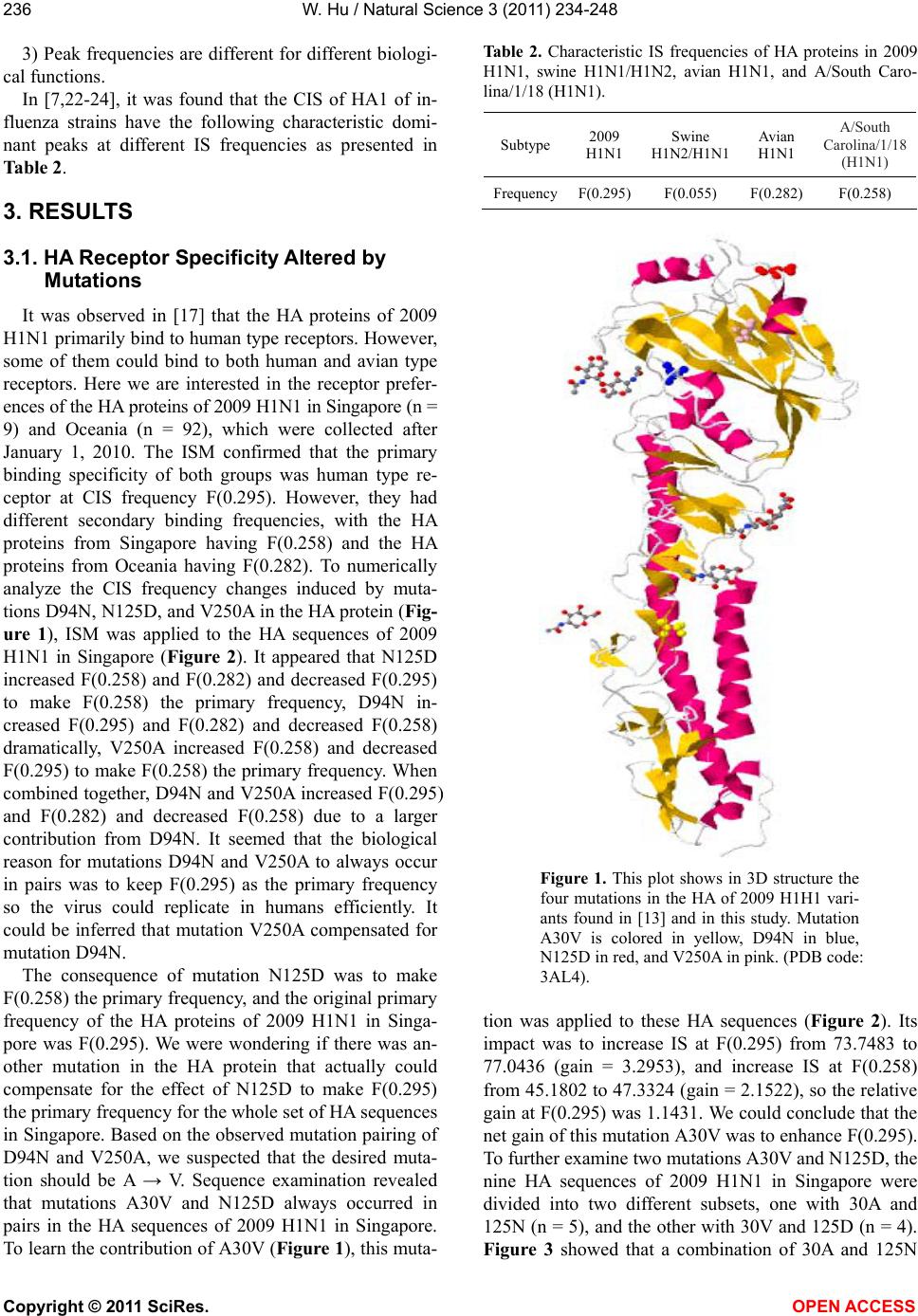 W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 236 3) Peak frequencies are different for different biologi- cal functions. In [7,22-24], it was found that the CIS of HA1 of in- fluenza strains have the following characteristic domi- nant peaks at different IS frequencies as presented in Table 2. 3. RESULTS 3.1. HA Receptor Specificity Altered by Mutations It was observed in [17] that the HA proteins of 2009 H1N1 primarily bind to human type receptors. However, some of them could bind to both human and avian type receptors. Here we are interested in the receptor prefer- ences of the HA proteins of 2009 H1N1 in Singapore (n = 9) and Oceania (n = 92), which were collected after January 1, 2010. The ISM confirmed that the primary binding specificity of both groups was human type re- ceptor at CIS frequency F(0.295). However, they had different secondary binding frequencies, with the HA proteins from Singapore having F(0.258) and the HA proteins from Oceania having F(0.282). To numerically analyze the CIS frequency changes induced by muta- tions D94N, N125D, and V250A in the HA protein (Fig- ure 1), ISM was applied to the HA sequences of 2009 H1N1 in Singapore (Figure 2). It appeared that N125D increased F(0.258) and F(0.282) and decreased F(0.295) to make F(0.258) the primary frequency, D94N in- creased F(0.295) and F(0.282) and decreased F(0.258) dramatically, V250A increased F(0.258) and decreased F(0.295) to make F(0.258) the primary frequency. When combined together, D94N and V250A increased F(0.295) and F(0.282) and decreased F(0.258) due to a larger contribution from D94N. It seemed that the biological reason for mutations D94N and V250A to always occur in pairs was to keep F(0.295) as the primary frequency so the virus could replicate in humans efficiently. It could be inferred that mutation V250A compensated for mutation D94N. The consequence of mutation N125D was to make F(0.258) the primary frequency, and the original primary frequency of the HA proteins of 2009 H1N1 in Singa- pore was F(0.295). We were wondering if there was an- other mutation in the HA protein that actually could compensate for the effect of N125D to make F(0.295) the primary frequency for the whole set of HA sequences in Singapore. Based on the observed mutation pairing of D94N and V250A, we suspected that the desired muta- tion should be A → V. Sequence examination revealed that mutations A30V and N125D always occurred in pairs in the HA sequences of 2009 H1N1 in Singapore. To learn the contribution of A30V (Figure 1), this muta- Table 2. Characteristic IS frequencies of HA proteins in 2009 H1N1, swine H1N1/H1N2, avian H1N1, and A/South Caro- lina/1/18 (H1N1). Subtype 2009 H1N1 Swine H1N2/H1N1 Avian H1N1 A/South Carolina/1/18 (H1N1) FrequencyF(0.295) F(0.055) F(0.282) F(0.258) Figure 1. This plot shows in 3D structure the four mutations in the HA of 2009 H1H1 vari- ants found in [13] and in this study. Mutation A30V is colored in yellow, D94N in blue, N125D in red, and V250A in pink. (PDB code: 3AL4). tion was applied to these HA sequences (Figure 2). Its impact was to increase IS at F(0.295) from 73.7483 to 77.0436 (gain = 3.2953), and increase IS at F(0.258) from 45.1802 to 47.3324 (gain = 2.1522), so the relative gain at F(0.295) was 1.1431. We could conclude that the net gain of this mutation A30V was to enhance F(0.295). To further examine two mutations A30V and N125D, the nine HA sequences of 2009 H1N1 in Singapore were divided into two different subsets, one with 30A and 125N (n = 5), and the other with 30V and 125D (n = 4). Figure 3 showed that a combination of 30A and 125N 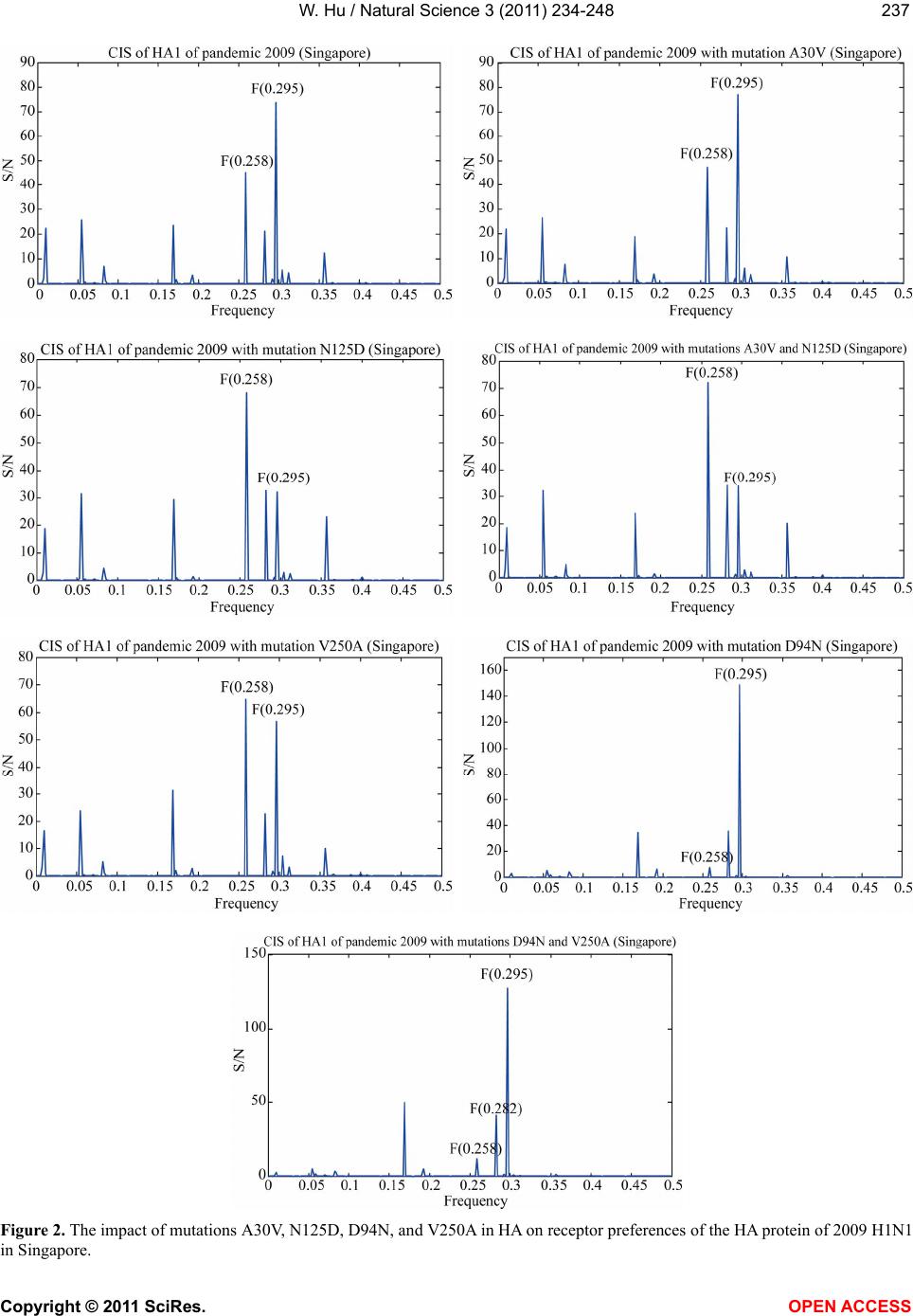 W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 237237 Figure 2. The impact of mutations A30V, N125D, D94N, and V250A in HA on receptor preferences of the HA protein of 2009 H1N1 in Singapore. 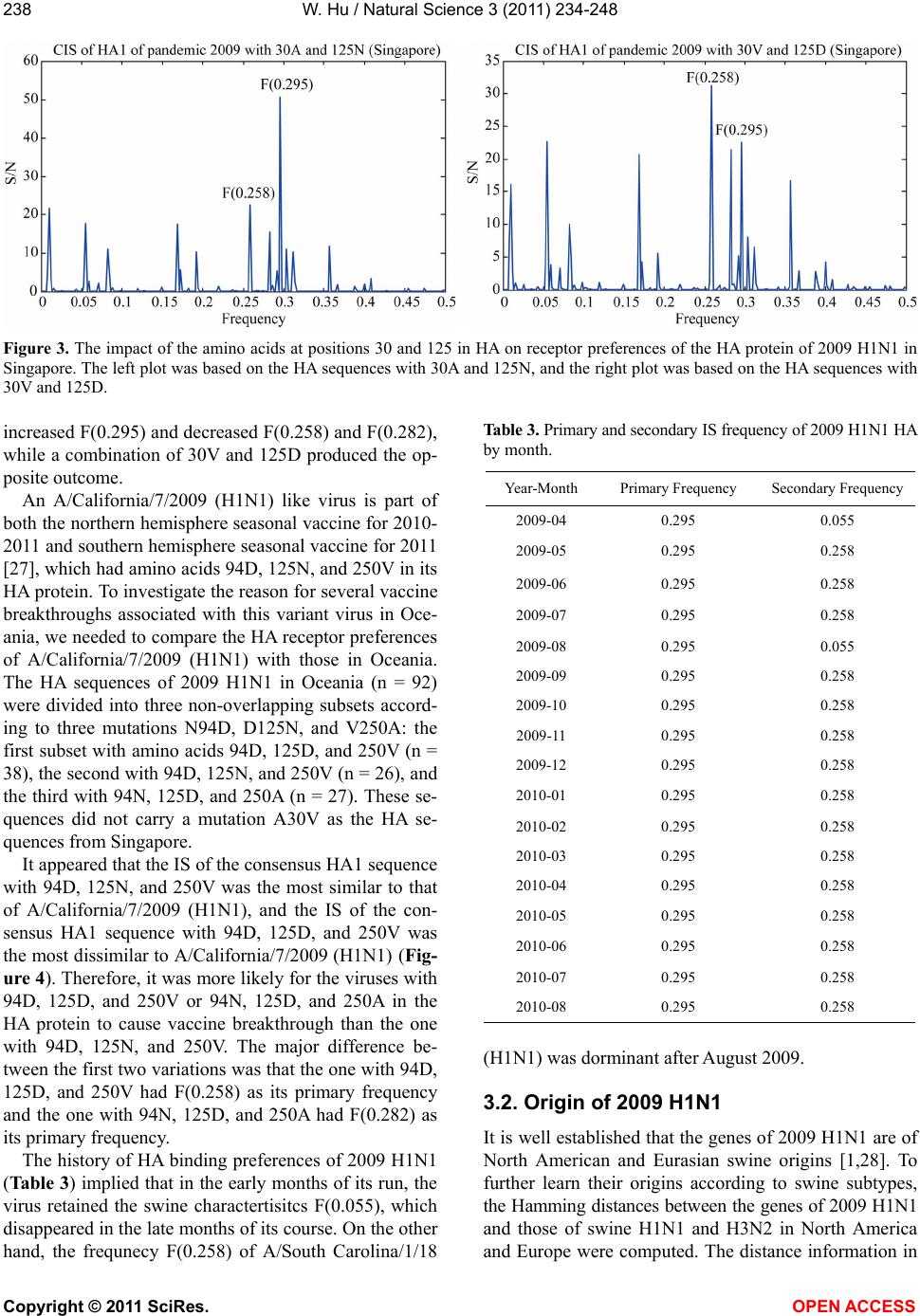 W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 238 Figure 3. The impact of the amino acids at positions 30 and 125 in HA on receptor preferences of the HA protein of 2009 H1N1 in Singapore. The left plot was based on the HA sequences with 30A and 125N, and the right plot was based on the HA sequences with 30V and 125D. increased F(0.295) and decreased F(0.258) and F(0.282), while a combination of 30V and 125D produced the op- posite outcome. An A/California/7/2009 (H1N1) like virus is part of both the northern hemisphere seasonal vaccine for 2010- 2011 and southern hemisphere seasonal vaccine for 2011 [27], which had amino acids 94D, 125N, and 250V in its HA protein. To investigate the reason for several vaccine breakthroughs associated with this variant virus in Oce- ania, we needed to compare the HA receptor preferences of A/California/7/2009 (H1N1) with those in Oceania. The HA sequences of 2009 H1N1 in Oceania (n = 92) were divided into three non-overlapping subsets accord- ing to three mutations N94D, D125N, and V250A: the first subset with amino acids 94D, 125D, and 250V (n = 38), the second with 94D, 125N, and 250V (n = 26), and the third with 94N, 125D, and 250A (n = 27). These se- quences did not carry a mutation A30V as the HA se- quences from Singapore. It appeared that the IS of the consensus HA1 sequence with 94D, 125N, and 250V was the most similar to that of A/California/7/2009 (H1N1), and the IS of the con- sensus HA1 sequence with 94D, 125D, and 250V was the most dissimilar to A/California/7/2009 (H1N1)(Fig- ure 4). Therefore, it was more likely for the viruses with 94D, 125D, and 250V or 94N, 125D, and 250A in the HA protein to cause vaccine breakthrough than the one with 94D, 125N, and 250V. The major difference be- tween the first two variations was that the one with 94D, 125D, and 250V had F(0.258) as its primary frequency and the one with 94N, 125D, and 250A had F(0.282) as its primary frequency. The history of HA binding preferences of 2009 H1N1 (Table 3) implied that in the early months of its run, the virus retained the swine charactertisitcs F(0.055), which disappeared in the late months of its course. On the other hand, the frequnecy F(0.258) of A/South Carolina/1/18 Table 3. Primary and secondary IS frequency of 2009 H1N1 HA by month. Year-Month Primary Frequency Secondary Frequency 2009-04 0.295 0.055 2009-05 0.295 0.258 2009-06 0.295 0.258 2009-07 0.295 0.258 2009-08 0.295 0.055 2009-09 0.295 0.258 2009-10 0.295 0.258 2009-11 0.295 0.258 2009-12 0.295 0.258 2010-01 0.295 0.258 2010-02 0.295 0.258 2010-03 0.295 0.258 2010-04 0.295 0.258 2010-05 0.295 0.258 2010-06 0.295 0.258 2010-07 0.295 0.258 2010-08 0.295 0.258 (H1N1) was dorminant after August 2009. 3.2. Origin of 2009 H1N1 It is well established that the genes of 2009 H1N1 are of North American and Eurasian swine origins [1,28]. To further learn their origins according to swine subtypes, the Hamming distances between the genes of 2009 H1N1 and those of swine H1N1 and H3N2 in North America and Europe were computed. The distance information in  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 239239 Tables 4 and 5 suggested that the HA gene of 2009 H1N1 was derived from swine H1N1 in North America, the NA gene from swine H1N1 in Europe, the M1 and M2 genes from swine H3N2 in Europe, and the NS1, NS2, NP, PA, PB1, and PB2 genes from H3N2 in North America. Hamming distances in Figure 5 gave further evidence that gene segments PA, PB1, and PB2 of 2009 H1N1 Figure 4. The IS of HA1 of A/California/7/2009 (H1N1) and IS of consensus HA1 of pandemic 2009 (Oceania). The three consen- suses were taken from the HA1 sequences with specific amino acids at positions 94, 125, and 250. Table 4. Hamming distances between consensus protein sequences of 2009 H1N1 and those of North American and Eurasian swine viruses were calculated. The minimum distance in each protein is marked with an asterisk. Protein HA NA M1 M2 NS1 NS2 NP PA PB1 PB2 Dist(H1N1 2009, H1N1 N_America) 48* 76 11 10 17 8 12 50 43 46 Dist(H1N1 2009, H3N2 N_America) 13 13 13* 5* 8* 14* 15* 9* Dist(H1N1 2009, H1N1 Europe) 108 24* 5 5* 42 15 32 31 26 25 Dist(H1N1 2009, H3N2 Europe) 3* 5* 42 14 35 32 28 26 Table 5. Hamming distances between consensus gene sequences of 2009 H1N1 and those of North American and Eurasian swine viruses were calculated. The minimum distance in each protein is marked with an asterisk. Gene HA NA M1 M2 NS1 NS2 NP PA PB1 PB2 Dist(H1N1 2009, H1N1 N_America) 149*267 91 21 48 21 92 373 423 358 Dist(H1N1 2009, H3N2 N_America) 97 24 32* 13* 63* 88* 101*82* Dist(H1N1 2009, H1N1 Europe) 431 81* 29 8 126 47 233 281 296 356 Dist(H1N1 2009, H3N2 Europe) 28* 7* 127 51 234 281 301 366  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 240  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 241241 Figure 5. Hamming distances by year between consensus protein sequences of 2009 H1N1 and those of the swine viruses that were closest to 2009 H1N1 as indicated in Table 5. were introduced to swine lineages around 1998, and gene segments NA and M of 2009 H1N1 were intro- duced around 1979 [1]. The findings in [1] also demon- strated that the introduction of HA, NP, and NS of 2009 H1N1 to swine lineages occurred around 1918. However, our analysis suggested that the NP and NS segments were introduced around 1998, and the HA segment around 1957, occurring much later than preciously pre- dicted. Codon usage bias is a unique molecular feature of many organisms including influenza viruses. This bias could influence host adaptation and the virulence of the influenza viruses because their replication relies on host cells. As the 2009 H1N1 virus originated from swine lineages, it is subject to host selection pressure after cross-species transmission, which could be reflected in the codon usage of its genes. To learn the subtle codon usage differences between 2009 H1N1 and its closest swine ancestors, the condon usage patterns of their genes were displayed in Figure 6. It demonstrated that the HAs of 2009 H1N1 and North American swine H1N1 had different codon usage in Asp, His, Gln, and Val, the NAs of 2009 H1N1 and Eurasian swine H1N1 had different codon usage in Leu, Lys, and Tyr, the Matrix genes (M1 + M2) of 2009 H1N1 and Eurasian swine H3N2 had different codon usage in Asn, Cys, Glu, Lys, and Tyr, and the genes NS1, NS2, NP, PA, PB1, and PB2 concatenated together (NS1 + NS2 + NP + PA + PB1 + PB2) of 2009 H1N1 and North American swine H3N2 had different codon usage in His. The ac- tual differences in codon usage were summarized in Ta- ble 6, which showed that 2009 H1N1 made more use of AAC for Asn than swine virus, and less TAC for Tyr than swine virus. Overall, host selection pressure on human influenza viruses does not favor the use of G or C nucleotides and the use of a G nucleotide at the third codon position [29]. 3.3. Comparison of NP, PA, PB1 and PB2 of 2009 H1N1 with Those of Swine Lineages The influenza viral polymerase, composed of proteins PB1, PB2, and PA, is critical in viral RNA synthesis, host adaptation, and virulence by interacting with NP. To  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 242 Figure 6. Codon bias of genes of pandemic 2009 and genes of swine viruses that were closest to those of pandemic 2009.  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 243243 Table 6. Actual codon bias of 2009 H1N1 and swine virus observed in Figure 6. Gene 2009 H1N1 Swine virus HA More GAC for Asp Less GAC for Asp HA Less CAC for His More CAC for His HA More CAG for Gln Less CAG for Gln HA Less CCG for Pro More CCG for Pro HA Less (GTC + GTG) for ValMore (GTC + GTG) for Val NA Less TTC for Phe More TTC for Phe NA More AAC for Asn Less AAC for Asn NA Less CTG for Leu More CTG for Leu NA Less AAG For Lys More AAG For Lys NA Less TAC for Tyr More TAC for Tyr M1 + M2 More AAC for Asn Less AAC for Asn M1 + M2 More TGC for Cys Less TGC for Cys M1 + M2 Less TAC for Tyr More TAC for Tyr M1 + M2 Less GAG for Glu More GAG for Glu M1 + M2 More AAG for Lys Less AAG for Lys NS1 + NS2 + NP + PA + PB1 + PB2 More CAC for His Less CAC for His study the interactions between these four proteins of influenza viruses of avian, human, 2009 H1N1 and swine origins, the correlated residue pairs that had a positive mutual information (MI) value were counted according to their location in the proteins [9]. It uncov- ered that in avian, human, 2009 H1N1, and swine vi- ruses, the inter-protein correlation from (NP, PA), (NP, PB1), (NP, PB2), (PA, PB1), (PA, PB2), (PB1, PB2) was stronger than the intra-protein correlation (NP, NP), (PA, PA), (PB1, PB1) and (PB2, PB2), with (NP, NP) being the weakest. Further, the correlation pattern of 2009 H1N1 was more similar to that of avian and human in- fluenza than to swine, in spite of the swine origin of 2009 H1N1. Using the same approach, we discovered that the interaction pattern of the four proteins of North American swine H1N1 was most similar to that of 2009 H1N1, although the sequence identity of the four pro- teins of North American swine H3N2 was most similar to that of 2009 H1N1 (Figure 7). Our findings rein- forced the concept that sequence identity is only one of the many factors to measure the similarity of two influ- enza viruses. Many of the classical markers for adaptation of avian or swine viruses to human hosts do not exist in 2009 H1N1, implying that other previously unrecognized mo- lecular determinants are accountable for its capability to infect humans. The study in [4,5] discovered novel host markers in the proteins of 2009 H1N1 that were not pre- sent in the traditional host markers. These novel markers were identified by the significant residue positions that could separate 2009 H1N1 from human viruses sub- tracted by the characteristic positions in avian and swine viruses, which were marked as (a) for avian and (s) for swine positions in Tables 7-10. To further examine the important positions in NP, PA, PB1, and PB2 of 2009 H1N1, we compared them to those of swine lineages in North America and Europe, and avian, human, swine (general) viruses (Tables 7-10). There were several important positions in the NP, PA, PB1, and PB2 of avian, human, swine (general), swine H1N1 in North America and in Europe that shared the same amino acid but 2009 H1N1 had a different amino acid at the same positions, reflecting the uniqueness of this novel virus. These positions were 53 and 316 in NP, 186, 204, 213, 275, 336, and 626 in PA, 12, 175, 216, 298, 364, 386, and 728 in PB1, and 54, 684 in PB2. At positions 353, 377, 444, 498 in NP, 362, 388, 407 in PA, 179, 339, 361, 486, 584, 638, 741 in PB1, and 65, 147, 225, 590, 591, 645 in PB2, swine H3N2 in North Amer- ica and 2009 H1N1 shared the same amino acid, but differed from other viruses (Tables 7-10). The amino acid (serine) at position 186 in PA of 2009 H1N1 was found to be necessary for its compatibility with PB2 and PB1 subunits [30], and the amino acids at positions 590 and 591 in PB2 were the SR polymorphism uncovered in [31]. The PB2 of 2009 H1N1 does not carry the human signature 627K, yet this virus replicates in humans and  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 244 Figure 7. Averaged correlated pair counts in each individual protein and between proteins of 2009 H1N1 and swine viruses. Table 7. This table contains the consensus amino acids at the sites in NP that have high importance in separating 2009 H1N1 from human viruses [4,5]. The novel host sites in this protein are the positions without a ‘a’ (for avian) or a ‘s’ (for swine) or both. Position 21 31(a,s) 53 119 189(s) 190 217(a) 289(s) 313(a,s)316 Avian N R E I M V I Y F I Human N K E I M V S Y Y I 2009 H1N1 D R D V I A V H V M Swine D R E V I A I H F I Swine H1N1(North America) D R E V I A I H F I Swine H1N1(Europe) N R E I M V I Y F I Swine H3N2 (North America) D R E V I A I H F I Swine H3N2 (Europe) N R E I M V I Y F I Position 350(s) 353 371 373(a) 377 430(s) 433 444 456(s) 498 Avian T V M T S T T I V N Human T S M A S T T I V N 2009 H1N1 K I V T N S N V L S Swine K V V A S S N I L N Swine H1N1(North America) K V V A S I N I L N Swine H1N1(Europe) T V M T I T T I V N Swine H3N2 (North America) K I V A N S N V L S Swine H3N2 (Europe) T V M T I T T I V N  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 245245 Table 8. This table contains the consensus amino acids at the sites in PA that have high importance in separating 2009 H1N1 from human viruses [4,5]. The novel host sites in this protein are the positions without a ‘a’ (for avian) or a ‘s’ (for swine) or both. Position 28(a,s) 55(a) 85 100(a,s)186 204 213 256 262 275 277 Avian P D T V G R R R K P S Human L N T A G R R R K P S 2009 H1N1 P D I V S K K K R L H Swine P D T V G R R R K P S Swine H1N1(North America) P N N V G R R R K P F Swine H1N1(Europe) P D T V G R R R R P S Swine H3N2 (North America) S D T V G R R Q K P S Swine H3N2 (Europe) P D T V G R R R R P F Position 336 337(a,s) 356(a)362 388 400(a,s)404(a,s) 407 552(a,s) 626 Avian L A K K S S A I T K Human L S R K S L S I S K 2009 H1N1 M A R R G P A V T R Swine L A K K S P A I T K Swine H1N1(North America) L A R K S F A I T K Swine H1N1(Europe) L A K K G M A I T K Swine H3N2 (North America) L A K R G P A V T K Swine H3N2 (Europe) L A K K G M A I T K Table 9. This table contains the consensus amino acids at the sites in PB1 that have high importance in separating 2009 H1N1 from human viruses [4,5]. The novel host sites in this protein are the positions without a ‘a’ (for avian) or a ‘s’ (for swine) or both. Position 12 175 179 216 298 327(a,s)339(s)361(a,s) 364 386(s) Avian V D M S L R I S L R Human V D M S L K I S L R 2009 H1N1 I N I G I R M R I K Swine V D M S L R I N L R Swine H1N1(North America) V D M S L R V N L R Swine H1N1(Europe) V D M S L R I S L R Swine H3N2 (North America) V D I S L R M R L R Swine H3N2 ((Europe) V D M S L R I S L R Position 435 486 517(s)584(a,s)587 618 638(s)728 741(a,s) Avian T R I R A E E I A Human T R I R A E E I A 2009 H1N1 I K V Q V D D V S Swine T R I R A E E I A Swine H1N1(North America) A R I R A K E I A Swine H1N1(Europe) T R V H T E E I A Swine H3N2 (North America) T K I Q A E D I S Swine H3N2 ((Europe) T R V H A E E I A  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 246 Table 10. This table contains the consensus amino acids at the sites in PB2 that have high importance in separating 2009 H1N1 from human viruses [4,5]. The novel host sites in this protein are the positions without a ‘a’ (for avian) or a ‘s’ (for swine) or both. Position 9(a) 54 64(a,s) 65(s) 81(a,s) 105(a,s)147 184(s) 199(a,s) Avian D K M E T T I T A Human N K T E M V I T S 2009 H1N1 D R M D T T T A A Swine D K M E T T I T A Swine H1N1(North America) N K I E T A I M S Swine H1N1(Europe) D K M E T T I T A Swine H3N2 (North America) D K M D T T T T A Swine H3N2 ((Europe) D K M E T T I T A Position 225 292(a,s)315 340(s) 453 475(a) 559 567(a,s)588(a,s) Avian S I M R P L T D A Human S T M R H M T N I 2009 H1N1 G V I K S L I D T Swine S I M R P L T D A Swine H1N1(North America) S T V M K R I S I Swine H1N1(Europe) S T I M K R I P V Swine H3N2 (North America) G A I M K K I P I Swine H3N2 ((Europe) S I I M K R I P V Position 590 591(s) 613(a,s)627(a,s) 645 661(a,s) 667 674(a,s)684(a) Avian G Q V E M A V A A Human G Q T K M T I T A 2009 H1N1 S R V E L A V A S Swine G Q V E M A V A A Swine H1N1(North America) G Q V K M S A N A Swine H1N1(Europe) G Q A E M A A T A Swine H3N2 (North America) S R V E L A A T A Swine H3N2 ((Europe) G Q A E M A A T A are efficiently transmitted in humans. The SR polymor- phism was recently identified in [31] as a mechanism for 2009 H1N1 to partially overcome the lack of K627 by enhancing polymerase activity. However, as early as in 2002, the SR occurred in the PB2 of swine H3N2 in North America (A/swine/Iowa/H02AS8/2002(H3N2)), but none was found in the PB2 of Eurasian swine H1N1 and H3N2. Even though the majority of the PB2 proteins of North American swine H1N1 had GQ, some of them had SR, as early as in 2002 (A/swine/Iowa/ H02NJ56371/2002(H1N1)), and GR in 2008 (A/swine/ Nebraska/02013/2008(H1N1)). Typically, GQ was cou- pled with 627K, though SR and GR were tied with 627E. 4. CONCLUSIONS A genetic variant of 2009 H1N1 recently emerged as a predominant virus in Australia, New Zealand, and Sin- gapore during the winter season of 2010 in the southern hemisphere. Our ISM analysis on the three mutations D94N, N125D, and V250A found in [13,32] suggested that the biological reason for the mutation pairing of D94N, and V250A was to keep the human type receptor as its primary binding preference so the virus could rep- licate in humans efficiently. Mutation V250A compen- sated for D94N. Based on this interpretation, we searched for and uncovered a new mutation A30V that  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 247247 compensated for N125D. We quantitatively investigated how mutations A30V, D94N, N125D, and V250A in the HA protein of this variant may affect its HA receptor binding affinity. In summary, mutation A30V increased IS frequency F(0.295) and decreased F(0.258), while V250A did the opposite. At the same time, mutation D94N increased F(0.295) and decreased F(0.258) and F(0.282), whereas N125D functioned the opposite. When combined together, D94N and V250A increased F(0.295) and F(0.282) and decreased F(0.258), but A30V and N125D produced the opposite. Our ISM re- sults also implied that the recent vaccine breakthroughs were partially caused by the alteration of HA receptor binding specificity resulted from these HA mutations. As the second task of our investigation, we revisited the origin of 2009 H1N1 to refine our understanding of this important issue. Our findings illustrated that the HA gene of 2009 H1N1 came from that of swine H1N1 in North America, the NA gene from Eurasian swine H1N1, the M1 and M2 genes from Eurasian swine H3N2, and the NS1, NS2, NP, PA, PB1, and PB2 genes from swine H3N2 in North America. In addition, our analysis pro- vided the timeline for the occurrence of genes of swine lineages most similar to those of 2009 H1N1. Although the four proteins NP, PA, PB1, and PB2 of 2009 H1N1 were closest to those of North American swine H3N2 in sequence identity, their interaction patterns were closest to those of swine H1N1 in North America. 5. ACKNOWLEDGEMENTS We thank Houghton College for its financial support. REFERENCES [1] Garten, R.J., Davis, C.T., Russell, C.A., Shu, B., Lind- strom, S., Balish, A., et al. (2009) Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Scienc e, 325, 197-201. doi:10.1126/science.1176225 [2] Hu, W. (2009) Analysis of correlated mutations, stalk motifs, and phylogenetic relationship of the 2009 influ- enza A virus neuraminidase sequences. Journal of Bio- medical Science and Engineering, 2, 550-558. doi:10.4236/jbise.2009.27080 [3] Hu, W. (2010) The Interaction between the 2009 H1N1 influenza A hemagglutinin and neuraminidase: Mutations, co-mutations, and the NA stalk motifs. Journal of Bio- medical Science and Engineering, 3, 1-12. [4] Hu, W. (2010) Novel host markers in the 2009 pandemic H1N1 influenza A virus. Journal of Biomedical Science and Engineering, 3, 584-601. doi:10.4236/jbise.2010.36081 [5] Hu, W. (2010) Nucleotide host markers in the influenza A viruses. Journal of Biomedical Science and Engineering, 3, 684-699. doi:10.4236/jbise.2010.37093 [6] Hu, W. (2010) Identification of highly conserved do- mains in hemagglutinin associated with the receptor binding specificity of influenza viruses: 2009 H1N1, Avian H5N1, and Swine H1N2. Journal of Biomedical Science and Engineering, 3, 114-123. doi:10.4236/jbise.2010.32017 [7] Hu, W. (2010) Quantifying the effects of mutations on receptor binding specificity of influenza viruses. Journal of Biomedical Science and Engineering, 3, 227-240. doi:10.4236/jbise.2010.33031 [8] Hu, W. (2010) Subtle differences in receptor binding specificity and gene sequences of the 2009 pandemic H1N1 influenza virus. Advances in Bioscience and Bio- technology, 1, 305-314. doi:10.4236/abb.2010.14040 [9] Hu, W. (2010) Correlated mutations in the four influenza proteins essential for viral RNA synthesis, host adapta- tion, and virulence: NP, PA, PB1, and PB2. Natural Sci- ence, 2, 1138-1147. doi:10.4236/ns.2010.210141 [10] King, D., Miller, Z., Jones, W. and Hu, W. (2010) Char- acteristic sites in the internal proteins of avian and human influenza viruses. Journal of Biomedical Science and Engineering, 3, 943-955. doi:10.4236/jbise.2010.310125 [11] Hu, W. (2010) Highly conserved domains in hemaggluti- nin of influenza viruses characterizing dual receptor binding. Natural Science, 2, 1005-1014. doi:10.4236/ns.2009.29123 [12] Hu, W. (2010) Host markers and correlated mutations in the overlapping genes of influenza viruses: M1, M2; NS1, NS2; and PB1, PB1-F2. Natural Science, 2, 1225-1246. doi:10.4236/ns.2010.211150 [13] Barr, I.G., Cui, L., Komadina, N., Lee, R.T., Lin, R.T., Deng, Y., Caldwell, N., Shaw, R. and Maurer-Stroh, S. (2010) A new pandemic influenza A(H1N1) genetic variant predominated in the winter 2010 influenza season in Australia, New Zealand and Singapore. Euro Surveill, 15, 19692. [14] Stevens, J., Blixt, O., Glaser, L., Taubenberger, J.K., Palese, P., Paulson, J.C. and Wilson, I.A. (2006) Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. Journal of Molecular Biology, 355: 1143- 1155. doi:10.1016/j.jmb.2005.11.002 [15] Stevens, J., Blixt, O., Tumpey, T.M., Taubenberger, J.K., Paulson, J.C. and Wilson, I.A. (2006) Structure and re- ceptor specificity of the hemagglutinin from an H5N1 in- fluenza virus. Science, 312, 404-410. doi:10.1126/science.1124513 [16] Matrosovich, M., Tuzikov, A., Bovin, N., Gambaryan, A., Klimov, A., Castrucci, M.R., Donatelli, I. and Kawaoka, Y. (2000) Early alterations of the receptor-binding prop- erties of H1, H2, and H3 avian influenza virus hemag- glutinins after their introduction into mammals. Journal of Virology, 74, 8502-8512. doi:10.1128/JVI.74.18.8502-8512.2000 [17] Karasin, A.I., West, K., Carman, S. and Olsen, C.W. (2004) Characterization of avian H3N3 and H1N1 influ- enza A viruses isolated from pigs in Canada. Journal of Clinical Microbiology, 42, 4349-4354. doi:10.1128/JCM.42.9.4349-4354.2004 [18] Liu, Y., Childs, R.A., Matrosovich, T., Wharton, S., Palma, A.S., Chai, W., Daniels, R., Gregory, V., et al. (2010). Altered receptor specificity and cell tropism of D222G hemagglutinin mutants isolated from fatal cases  W. Hu / Natural Science 3 (2011) 234-248 Copyright © 2011 SciRes. OPEN ACCESS 248 of pandemic A(H1N1) 2009 influenza virus. Journal of Vir olo gy, 84, 12069-12074. doi:10.1128/JVI.01639-10 [19] Kilander, A., Rykkvin, R., Dudman, S. and Hungnes, O. (2010) Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009-2010. Euro Surveill, 15, 19498. [20] Liu, Y., Childs, R.A., Matrosovich, T., et al. (2010) Al- tered receptor specificity and cell tropism of D222G Hemagglutinin mutants isolated from fatal cases of pan- demic A(H1N1) 2009 influenza virus. Journal of Virol- ogy, 84, 12069-12074. doi:10.1128/JVI.01639-10 [21] Su, Y. Yang, H.Y., Zhang, B.J., Jia, H.L. and Tien, P. (2008) Analysis of a point mutation in H5N1 avian in- fluenza virus haemagglutinin in relation to virus entry into live mammalian cells. Archives of Virology, 153, 2253-2261. doi:10.1007/s00705-008-0255-y [22] Veljkovic, V., Niman, H.L., Glisic, S., Veljkovic, N., Perovic, V. and Muller, C.P. (2009) Identification of he- magglutinin structural domain and polymorphisms which may modulate swine H1N1 interactions with human re- ceptor. BMC Structural Biology, 9, 62. doi:10.1186/1472-6807-9-62 [23] Veljkovic, V., Veljkovic, N., Muller, C.P., Müller, S., Glisic, S., Perovic, V. and Köhler, H. (2009) Characteri- zation of conserved properties of hemagglutinin of H5N1 and human influenza viruses: possible consequences for therapy and infection control. BMC Structural Biology, 7, 9-21. [24] Veljkovic, N., Glisic, S., Prljic, J., Perovic, V., Botta, M. andVeljkovic, V. (2008) Discovery of new therapeutic targets by the informational spectrum method. Current Protein and Peptide Science, 9, 493-506. doi:10.2174/138920308785915245 [25] Katoh, K., Kuma, K., Toh, H. and Miyata, T. (2005) MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Research, 33, 511- 518. doi:10.1093/nar/gki198 [26] Cosic, I. (1997) The resonant recognition model of mac- romolecular bioreactivity, theory and application. Birk- hauser Verlag, Berlin. [27] http://www.cdc.gov/flu/about/qa/1011_vac_selection.htm [28] Solovyov, A., Palacios, G., Briese, T., Lipkin, W.I. and Rabadan, R. (2009) Cluster analysis of the origins of the new influenza A(H1N1) virus. European Surveillance, 14, 19224. [29] Wong, E.H., Smith, D.K., Rabadan, R., Peiris, M. and Poon, L.L. (2010) Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evolutionary Biology, 10, 253. doi:10.1186/1471-2148-10-253 [30] Wanitchang, A., Jengarn, J. and Jongkaewwattana, A. (2011) The N terminus of PA polymerase of swine-origin influenza virus H1N1 determines its compatibility with PB2 and PB1 subunits through a strain-specific amino acid serine 186. Virus Research, 155, 325-333. doi:10.1016/j.virusres.2010.10.032 [31] Mehle, A. and Doudna, J.A. (2009) Adaptive strategies of the influenza virus polymerase for replication in humans. Proceedings of National Academy Science of U.S.A., 106: 21312-21316. doi:10.1073/pnas.0911915106 [32] Maurer-Stroh, S., Lee, R.T., Eisenhaber, F., Cui, L., Phuah, S.P. and Lin, R.T. (2010) A new common muta- tion in the hemagglutinin of the 2009 (H1N1) influenza A virus. PLoS Currency, RRN1162.
|