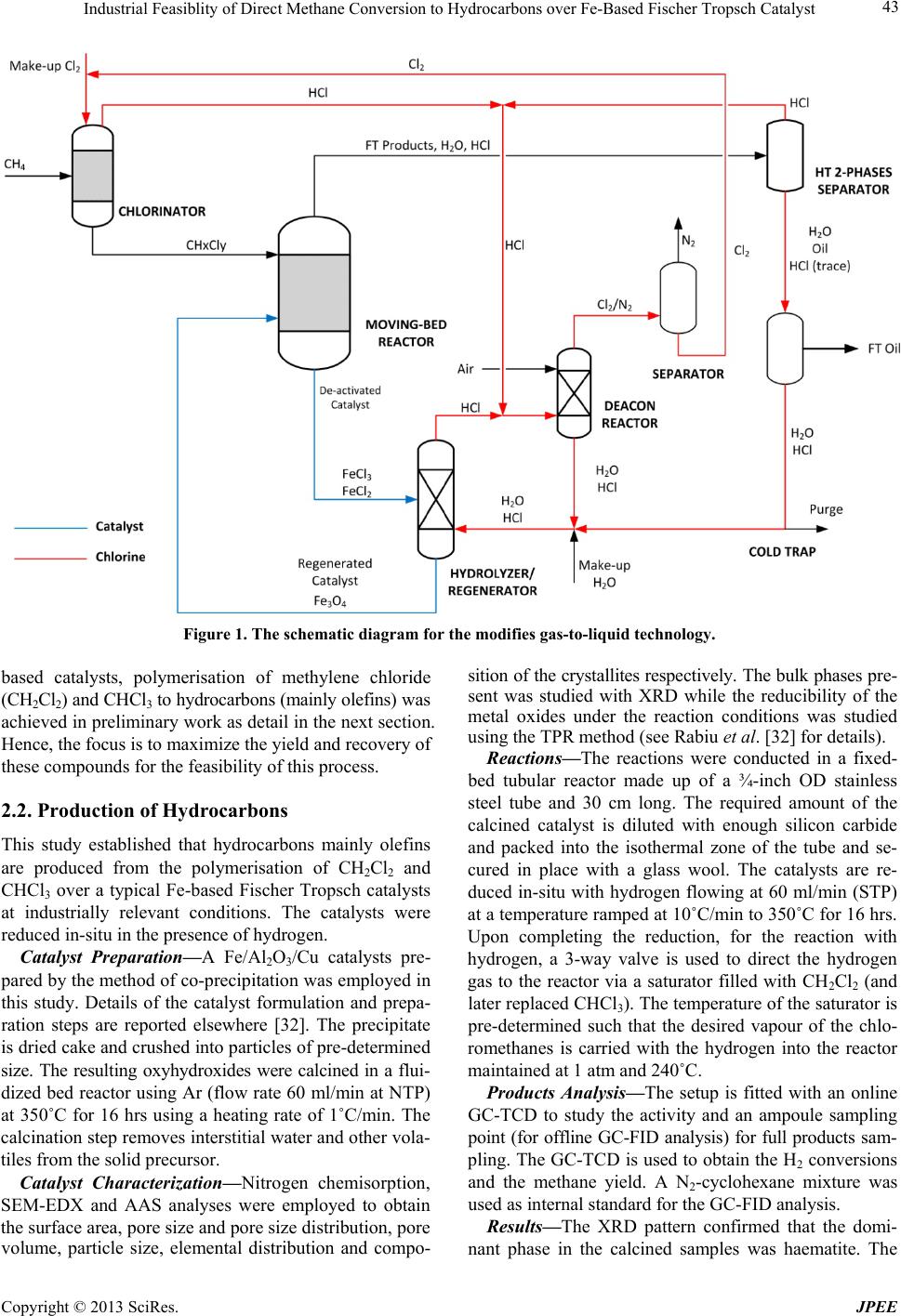
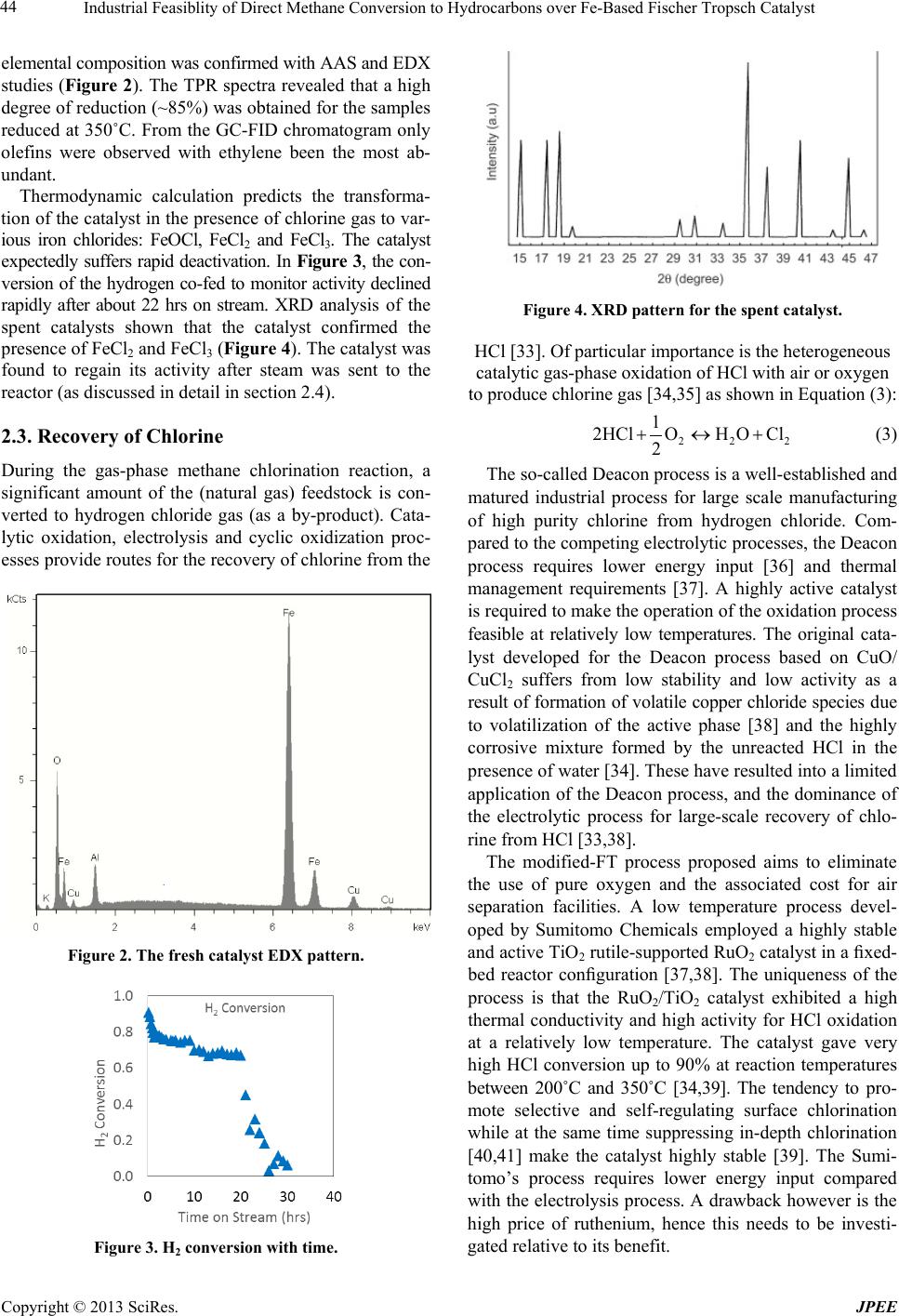
Industrial Feasiblity of Direct Methane Conversion to Hydrocarbons over Fe-Based Fischer Tropsch Catalyst
Copyright © 2013 SciRes. JPEE
er-Tropsch Synthesis,” Applied Catalysis A, V ol . 186, No.
1-2, 1999, pp. 3-12.
[19] I. T. Horváth, “Encyclopedia of Catalysis,” John Wiley &
Sons, New Jersey, 2003.
http://dx.doi.org/10.1016/S0926-860X(99)00160-X
[20] Y. Ogawa, K. Hasenaka and Y. Kawasaki, “Research on
Marketability of GTL: Liquid Fuel from Natural Gas,”
IEEJ, 2000. eneken.ieej.or.jp/en/data/old/pdf/gtl.pdf
[21] J. M. G. Carballo, J. Yang, A. Holmen, et al., “Catalyti c
Effects of Ruthenium Particle Size on the Fischer-
Tropsch Synthesis,” Journal of Catalysis, Vol. 284, No. 1,
2011, pp. 102-108.
[22] A. Tavasoli, A. Khodadadi, Y. Mortazavi, et al., “Lower-
ing Methane and Raising Di s tillates Yields in Fischer-
Tropsch Synthesis by Using Promoted and Unpromoted
Cobalt Catalyst s in a Dual Bed Reactor,” Fuel Processing
Technology, Vol. 87, 2006, pp. 641-647.
http://dx.doi.org/10.1016/j.jcat.2011.09.008
http://dx.doi.org/10.1016/j.fuproc.2006.01.010
[23] P. K. Bakkerud, “Update on Synthesis Gas Production for
GTL,” Catalysis Today, Vol. 106, No. 1-4, 2005, pp. 30-
33. http://dx.doi.org/10.1016/j.cattod.2005.07.147
[24] B. Ghaemmaghami, “GTL: Progress and Prospects—
Study Yields Generic, Coastal-Based GTL Plant,” Oil &
Gas Journal, Vol. 99, No. 11, 2001, pp. 23-31.
[25] M. E. Dry, “The Fischer-Tropsch Process: 1950-2000,”
Catalysis Today, Vol. 71, No. 3-4, 2002, pp. 227-241.
http://dx.doi.org/10.1016/S0920-5861(01)00453-9
[26] D. J. O’Rear and C. L. Kibby, “Control of CO2 Emissions
from a Fischer-Tropsch Facility by Use of Multiple Re-
actors,” United States Patent No. US6992113 B2 2005,
International Application No. PCT/US2004/035610.
[27] V. N. Rozanov and Y. A. Treger, “Kinetics of the Gas-
Phase Thermal Chlorination of Methane,” Kinetics and
Catalysis, Vol. 51, No. 5, 2010, pp. 635-643.
http://dx.doi.org/10.1134/S0023158410050034
[28] Y. A. Treger, V. N. Rozanov, S. V. Sokolova, et al., “Pro-
ducing Ethylene and Propylene from Natural Gas via the
Intermediate Synthesis of Methyl Chloride and Its Sub-
sequent Catalytic Pyrolysis,” Catalysis in Industry, Vol. 4,
No. 4, 2012, pp. 231-235.
http://dx.doi.org/10.1134/S2070050412040186
[29] M. C. Alvarez-Galvan, N. Mota, M. Ojeda, et al., “Dire ct
Methane Conversion Routes to Chemical s and Fuels,”
Catalysis Today, Vol. 171, No. 1, 2011, pp. 15-23.
http://dx.doi.org/10.1016/j.cattod.2011.02.028
[30] M. R. Rahimpour, M. Jafari and D. Iranshahi, “Progress
in Catalytic Naphtha Reforming Process: A Review,” Ap-
plied Energy, Vol. 109, 2013, pp. 79-93.
http://dx.doi.org/10.1016/j.apenergy.2013.03.080
[31] J. He, T. Xu, Z. Wang, et al., “Transformation of Me-
thane to Propylene: A Two-Step Reaction Route Cata-
lyzed by Modified CeO2 Nanocrystals and Zeolites,” An-
gewandte Chemie, Vol. 124, No. 10, 2012, pp. 2488-
2492.
[32] A. M. Rabiu, E. van Steen and M. Claeys, “Further Inves-
tigation into the Formation of Alcohol during Fischer
Tropsch Synthesis on Fe-Based Catalysts,” APCBEE Pro-
cedia, Vol. 3, 2012, pp. 110-115.
http://dx.doi.org/10.1002/ange.201104071
http://dx.doi.org/10.1016/j.apcbee.2012.06.055
[33] K. Iwanaga, K. Seki, T. Hibi, et al., “The Development of
Improved Hydrogen Chloride Oxidation Process,” Vol.
2004, Sumitomo Kagaku, Japan, 2004.
[34] M. A. G. Hevia, A. P. Amrute, T. Schmidt, et al., “Tran-
sient Mechanistic Study of the Gas-Phase HCl Oxidation
to Cl2 on Bulk and Supported RuO2 Catalysts,” Journal of
Catalysis, Vol. 276, No. 1, 2010, pp. 141-151.
http://dx.doi.org/10.1016/j.jcat.2010.09.009
[35] A. P. Amrute, C. Mondelli, M. Moser, et al., “Perfor-
mance, Structure, and Mechanism of CeO2 in HCl Oxida-
tion to Cl2,” Journal of Catalysis, Vol. 286, 2012, pp.
287-297.
[36] J. Perez-Ramirez, C. Mondelli, T. Schmidt, et al., “Sus-
tainable Chlorine R ecycling via Catalysed HCl Oxidation:
From Fundamentals to Implementation,” Energy & Envi-
ronmental Science, Vol. 4, No. 12, 2011, pp. 4786-4799.
http://dx.doi.org/10.1016/j.jcat.2011.11.016
[37] N. López, J. Gómez-Segura, R. P. Marín, et al., “Mecha-
nism of HCl Oxidation (Deacon Process) over RuO2,”
Journal of Catalysis, Vol. 255, No. 1, 2008, pp. 29-39.
http://dx.doi.org/10.1039/c1ee02190g
http://dx.doi.org/10.1016/j.jcat.2008.01.020
[38] M. Hammes, M. Valtchev, M. B. Roth, et al., “A Search
for Alternative Deacon Catalysts,” Applied Catalysis B:
Environmental, Vol. 132-133, 2013, pp. 389-400.
[39] H. Over and R. Schomäcker, “What Makes a Good Cata-
lyst for the Deacon Process?” ACS Catalysis, Vol. 3, No.
5, 2013, pp. 1034-1046.
http://dx.doi.org/10.1016/j.apcatb.2012.11.034
[40] J. P. Hofmann, S. Zweidinger, A. P. Seitsonen , et al., “D y-
namic Response of Chlorine Atoms on a RuO2(110) Mo-
del C atalyst Surface,” Physical Chemistry Chemical Phy-
sics, Vol. 12, No. 47, 2010, pp. 15358-15366.
http://dx.doi.org/10.1039/c0cp01126f
[41] S. Zweidinger, J. P. Hofmann, O. Balmes, et al., “In-Situ
Studies of the Oxidation of HCl over RuO2 Model Cata-
lysts: Stability and Reactivity,” Journal of Catalysis, Vol.
272, No. 1, 2010, pp. 169-175.
http://dx.doi.org/10.1016/j.jcat.2010.02.030
[42] M. Moser, C. Mondelli, T. Schmidt, et al., “Supported
CeO2 Catalyst s in Technical Form for Sustainable Chlo-
rine Production,” Applied Catalysis B: Environmental, Vol.
132-133, 2013, pp. 123-131.
http://dx.doi.org/10.1016/j.apcatb.2012.11.024