Health
Vol.4 No.12A(2012), Article ID:26069,8 pages DOI:10.4236/health.2012.412A203
Lipoprotein lipase and obesity
![]()
1Department of Internal Medicine, Medical Clinic, Aichi Medical University, Nagoya, Japan; *Corresponding Author: info@tonyo.jp
2Okinaka Memorial Institute for Medical Research, Tokyo, Japan
3Department of Biomedical Information Engineering, Graduate School of Medical Science, Yamagata University, Yamagata, Japan
Received 11 October 2012; revised 11 November 2012; accepted 24 November 2012
Keywords: Lipoprotein lipase; Lipoprotein lipase activator; Insulin resistance; Lipid metabolism
ABSTRACT
Obesity is one of the fast-growing major diseases in developed and developing countries. As has been persuasively argued, long-term imbalance between intake and expenditure of fat is a central factor in the etiology of obesity. Obesity aggravates insulin resistance and promotes cardiovascular diseases and atherosclerosis. We hypothesized that elevating lipoprotein lipase (LPL) activity in skeletal muscle would cause an improvement of obesity. To test this hypothesis, we studied the effects of the LPL activator NO-1886 in obese animals. NO- 1886 elevated LPL activity in skeletal muscle, and improved obesity as well as insulin resistance in obese rats. Furthermore, NO-1886 mitigated body weight gain induced by pioglitazone without suppressive effect on the adiponectinincreasing action of pioglitazone. LPL activators hold a lot of promise of curing several diseases shown above in clinical scene.
1. INTRODUCTION
Obesity in adulthood is characterized by adipocyte hypertrophy. Adipose tissue participates in the regulation of energy homeostasis. High-fat diet-induced insulin resistance associated with obesity is a major risk factor for diabetes and cardiovascular disease [1]. Adipose tissue itself serves as the site of triglyceride (TG) storage and free fatty acid release in response to changing energy demands [1].
Lipoprotein lipase (LPL) plays a pivotal role in lipids and the metabolism of lipoprotein [2]. Major functions of LPL include the hydrolysis of TG-rich lipoproteins and release of non-esterified fatty acid (NEFA), which are taken up and used for metabolic energy in peripheral tissue such as muscle, or are re-esterified into TG and stored in adipose tissue. The balance between these competing effects could determine whether increased LPL activity will lead to a reduced rate of weight gain or to increased adiposity through increased rates of adipose tissue storage of TG. An imbalance of LPL activity may alter the partitions of plasma TG between muscle and adipose tissue, and thus influence insulin resistance and obesity.
Institute of Otsuka Pharmaceutical Factory, Inc. synthesized the LPL activator NO-1886 ([4-(4-bromo-2- cyano-phenylcarbamoyl)-benzyl]-phosphonic acid diethyl ester, CAS no.: 133208-93-2, generic name: ibrolipim).
Hara et al. reported that LPL activator NO-1886 treatment in high-fructose diet induced insulin resistance rats decreases the respiratory quotient (RQ) and plasma TG [3]. These results may indicate the elevation of LPL activity ameliorate obesity.
Therefore, we hypothesized that elevating LPL activity would cause an improvement of obesity. To test this hypothesis, we studied the effects of the LPL activator NO-1886 in obese animals.
2. BACKGROUND OF LIPOPROTEIN LIPASE
LPL is a glycoprotein located on the luminal surface of capillary endothelial cells. The active enzyme is a nonconvalent homodimer [4]. The enzyme has an apparent monometric molecular mass of 60,000 daltons on SDSPAGE. The human LPL gene is approximately 30 kb in length [5].
LPL mRNA has been found in human adipose tissue, and also in muscle, adrenal, kidneys, intestine and neonatal, but not adult liver. The mRNA for LPL in humans is highly homologus with that of mice, rats and cows [6, 7].
LPL binds to heparin sulfate on the surface of endothelial cells via the heparin-binding site, which allows the enzyme to be extended into the plasma [8,9]. Following intravenous administration of heparin, LPL can be displaced from the endothelial surface into plasma (post-heparin plasma), where enzyme activity can be measured. The active enzyme bound to heparin sulfate on the capillary endothelium is predominantly in the dimeric form.
TG and monoglyceride are preferred substrates for LPL, which preferentially hydrolyzes 1- and 3-ester bounds in TG, generating 2-monoglyceride, which are converted to 1-monoglyceride by isomerization for further hydrolysis [10].
A small portion of the core TG from chylomicron and very-low-density lipoprotein (VLDL) can be transferred to high-density lipoprotein (HDL). More important contributors to HDL are the surface remnants of the TG-rich lipoproteins that occur as a result of hydrolysis of core TG. Nikkila et al. noted a relationship between LPL activity and HDL cholesterol (HDL-C), especially HDL2 cholesterol (HDL2-C), in many clinical situations [11], and recently, we demonstrated that activation of LPL causes production of HDL2-C by catabolism of TG-rich lipoproteins and enlarges HDL2 particle size in rats [12]. Tsutsumi et al. reported that plasma TG levels were inversely correlated with postheparin plasma LPL activity, while HDL-C levels were positively correlated with the activity of the enzyme in rats [13].
Insulin increases LPL activity, rates of LPL synthesis and LPL mRNA levels in adipocytes [14]. Since insulin does not stimulate LPL gene transcription [4], the increases in steady-state LPL mRNA must be due to changes in mRNA stability (post-transcriptional mechanism).
Insulin-deficient diabetes results in a reduced degradation of VLDL by the reduction of functional (endothelium-bound) LPL activity in myocardium and adipose tissue. And short-term administration of insulin in vivo restores the effects of LPL activity in adipose tissue, but not in myocardium [15].
Whether LPL directly or indirectly promotes or protects against atherosclerosis remains controversial. Misenbock et al. reported that LPL +/− humans have atherogenic lipoproteins, especially in the postprandial state [16]. Katzel et al. found that older, normocholesterolemic, nondiabetic athletic individuals with silent myocardial ischemia have increased insulin resistance, increased postheparin plasma hepatic triglyceride lipase (HTGL) activity, and reduced postprandial response of abdominal adipose tissue LPL activity to feeding [17]. These conditions are associated with low HDL2-C levels and increased postprandial lipemia. The abnormalities in plasma HDL-C and postprandial TG metabolism may increase the risk for coronary artery disease in these subjects.
Reymer et al. studied human LPL mutations. They showed that in approximately 1 in 20 males with proven atherosclerosis, an Asn291Ser mutation in the human LPL gene is associated with significantly reduced plasma HDL-C concentrations and results in a significant decrease in LPL catalytic activity [18]. They showed the relationship between LPL activity and plasma HDL-C concentrations, and suggested that a specific LPL mutation may be a factor in the development of atherosclerosis.
Higher levels of postheparin plasma LPL activity are associated with decrease plasma TG and increased HDL-C [13]. People who are heterozygous for LPL deficiency have increased plasma TG and decreased plasma HDL-C concentrations, a profile associated with increased atherogenic risk [19]. These reports suggest that increased postheparin plasma LPL activity is associated with protection against atherosclerosis in humans.
Fan et al. generated transgenic rabbits expressing human LPL to elucidate the physiological roles of LPL in lipid and lipoprotein metabolism. When the transgenic rabbits were fed a cholesterol-rich diet, the development of hypercholesterolemia and aortic atherosclerosis was dramatically suppressed [20]. Using another model, Shimada et al. established an overexpressed human LPL gene in the heart, skeletal muscle, and adipose tissue of mice. These transgenic mice had 5- and 1.7-fold higher LPL activity in adipose tissue and postheparin plasma, respectively. Also, VLDL triglycerides were greatly reduced and HDL2 was increased 1.4-fold [21]. These results demonstrated that the lipid profile in these LPL transgenic mice is antiatherogenic.
Shimada et al. also created low density lipoprotein (LDL) receptor knockout mice (LDLRKO) that over expressd LPL (LPL/LDLRKO) by mating LPL transgenic mice to LDLRKO mice, and compared their plasma lipoprotein profiles and atherosclerosis with those in non expressing LDLRKO mice. LPL/LDLRKO mice showed marked suppression of mean plasma TG concentrations and a modest decrease in cholesterol concentrations compared to LDLRKO mice. Additionally, larger lipoprotein particles of intermediate density lipoprotein/LDL, which is considered remnant lipoproteins, were selectively reduced in LPL/LDLRKO mice. Therefore, it was showed that the altered lipoprotein profile, in particular the reduced level of remnant lipoproteins, plays a role for protection by LPL against atherosclerosis [22].
Regarding the LPL activator NO-1886, NO-1886 were significantly dose-dependent increases in postheparin plasma LPL activity in normal rats. On the other hand, NO-1886 did not affect postheparin plasma HTGL. NO- 1886 also significantly and dose-dependently increased tissue LPL activity in normal rats. NO-1886 enhanced expression of LPL mRNA in adipose tissue and myocardium, and increased LPL protein mass and LPL activity in postheparin plasma [13].
NO-1886 dose-dependently decreases plasma TG levels, with concomitant increase in plasma HDL-C in rats, hamsters, and rabbits [23]. Endothelial function is closely related to the development of atherosclerosis and is impaired before the development of initial lesions in hypercholesterolemic animals [24]. Aging is associated with a progressive development of dyslipidemia, insulin resistance, and obesity, all of which are risk factors for cardiovascular disease and atherosclerosis [25]. It is known that endothelium-dependent relaxation decreases with age [26]. In our previous works, NO-1886 ameliorated the aging-related deterioration of endotheliumdependent relaxation in thoracic aorta in 10-month-old male rats [27], and also prevented the development of impaired endothelium-dependent relaxation of rat thoracic aorta in 2-year-old male rats [28]. We speculated that NO-1886 might have improved the endotheliumdependent relaxation by normalizing the lipid disorder, in particular by elevating plasma HDL-C, which possesses antioxidant effects [29] and is very important in old rats due to elevated plasma lipid peroxide levels caused by exercise [28].
Long-term administration of NO-1886 to rats with experimental atherosclerosis caused by a high-cholesterol diet significantly inhibited the development of atherosclerotic lesions in the coronary arteries [13]. The results of multiple regression analysis in the studies suggest that plasma HDL-C is a strong protective factor against atherosclerosis in coronary arteries.
Chiba et al. administered NO-1886 to cholesterol-fed New Zealand White rabbits for 20 weeks. NO-1886 increased postheparin plasma LPL activity by 30% - 40% compared with the control group. Plasma HDL-C concentration was 2-fold greater in the NO-1886 group compared to that in the controls, and plasma TG was reduced to the level of normal controls. Post-heparin plasma LPL activity was positively correlated with plasma HDL-C and inversely correlated with plasma TG. The relative atheromatous area in the aorta was reduced to 11% - 14% in the NO-1886 group compared to 51% in the control group. Multiple regression analysis of postheparin plasma LPL activity, plasma HDL-C and TG indicated that plasma HDL-C was the most powerful protector against aortic cholesterol accumulation. A decrease in plasma TG also protected against atherosclerosis, though not as strongly as plasma HDL-C. They concluded that NO-1886 prevented the development of atherosclerosis by increasing LPL activity, resulting in an increase in plasma HDL-C and a decrease in plasma TG, without a significant influence o plasma total cholesterol concentrations [30].
Yin et al. created a diabetic rabbit model with atherosclerosis in the aorta by feeding a high-fat/high-sucrose diet. They administered NO-1886 to these rabbits to determine whether the LPL activator had an antiatherogenic effect. NO-1886 decreased plasma cholesterol and TG, and increased plasma HDL-C. Interestingly, NO-1886 provided protection against the development of atherosclerosis in the aorta. These results suggest that NO-1886 not only ameliorates the lipid disorder, but also lower plasma glucose levels and suppresses atherosclerosis in the aorta of diabetic rabbits [31].
These published papers show that activation of LPL protects against the development of atherosclerosis.
3. OBESITY AND LIPOPROTEIN LIPASE
Adipose tissue participates in the regulation of energy homeostasis as an important endocrine organ that secretes a number of biologically active adipocytokines such as adipsin [32], leptin [33], plasminogen activator inhibitor-1 [34], resistin [35], TNF-α [36], and adiponectin [37]. LPL is one such adipocytokine.
Some reports have suggested that LPL activity in adipose tissue is high in obesity [38,39]. An imbalance of LPL activity may alter the partitions of plasma TG between muscle and adipose tissue, and thus influence insulin resistance and obesity.
Shimada et al. have reported that none of the mice in which human LPL gene expression was induced obese, and that storage and decomposition of fat were balanced in mice as a result of increased activity of hormone-sensitive lipase in adipose tissue [40]. In other words, because of homeostasis, body weight may not be increased in normal animals even by elevation of LPL activity.
Jensen et al. have reported that overexpression of human LPL in skeletal muscle prevents diet-induced obesity in transgenic mice [41]. Accordingly to Ferraro et al., the RQ is inversely correlated with LPL activity in skeletal muscle in Pima Indians, and Pima Indians have a high RQ, which is a risk factor for body weight gain [42].
Hara et al. have also reported that long term administration of LPL activator NO-1886 causes a reduction in RQ in high-fructose-induced diabetic rats without fat accumulation in tissues [3] (Figure 1). The RQ is the steady-state ration of carbon dioxide production to oxygen consumption by whole-body tissue metabolism. Therefore, in general, a decrease in RQ means an increase in fat oxidation. Based on this information, we hypothesized that an LPL activator may improve obesity by activating LPL in skeletal muscle.
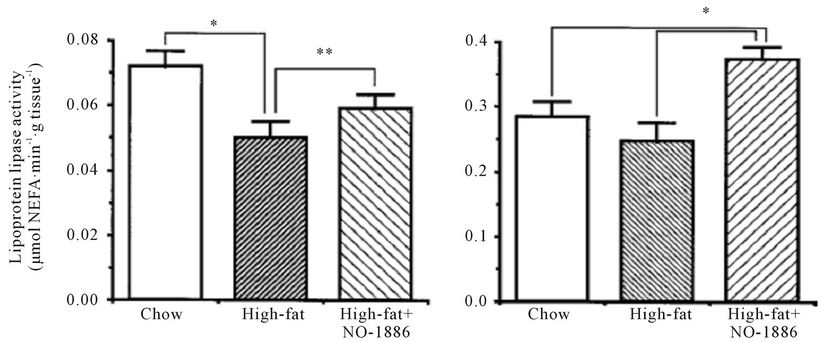
NO-1886 was administered to rats rendered obese with a high-fat diet. NO-1886 suppressed the body weight gain and accumulation of visceral and subcutaneous fat (Figure 2). NO-1886 also increased skeletal muscle LPL activity without affecting adipose tissue LPL activity (Figure 3), and lowered the RQ in obesity rats fed a high-fat diet [43].
It has long been known that uncoupling proteins (UCPs)
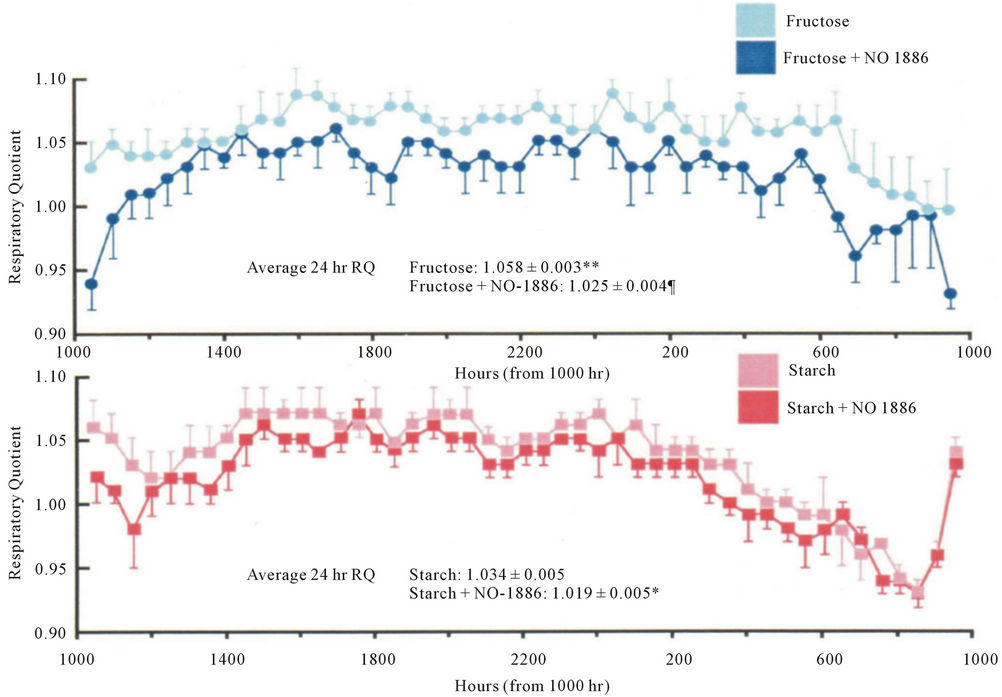
Figure 1. RQ measured over 24 hours in rats fed starch or fructose with or without NO-1886 supplementation. The mean 24-hour RQ was significantly lower in the fructose + NO-1886 group than that in the fructose-only group. Data expressed as mean ± SD. n = 4 measurements of 8 animals in each group. **P < 0.001, *P < 0.01 vs starch fed group. ¶P < 0.01 fructose vs fructose + NO-1886 group.
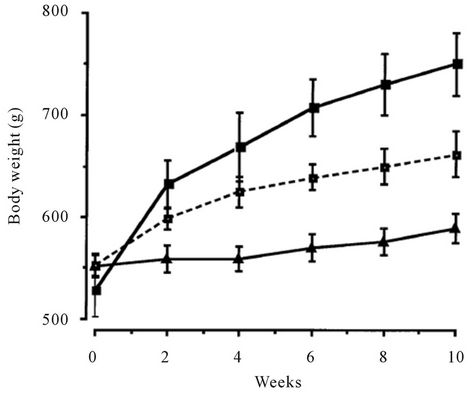
Figure 2. Effects of chow or a high-fat diet with and without NO-1886 (50 mg/kg/day) for 10 weeks on body weight gain in SD rats. Body weight was significantly different between all 3 groups by 6 weeks of feeding/drug treatment and remained so for the duration of treatment. –■–, high-fat; --□--, high-fat + NO-1886; –▲–, chow.
are responsible for facultative thermogenesis in rodents. UCPs play an important role in energy metabolism and obesity [44]. UCP1 expression is restricted to brown adipose tissue (BAT), UCP2 is widely expressed, and UCP3 is found mainly in skeletal muscle [45]. Doi et al. reported that NO-1886 accelerates the expression of fatty acid oxidation-related enzymes, resulting in a reduction of RQ [46]. However, the mechanism for antiobesity effects of NO-1886 remained unclear. To clarify the mechanism, we studied the effects of NO-1886 on the expression of UCP-1, UCP-2, and UCP3 in rats. NO- 1886 did not affect the expression of UCP1 and UCP2 in BAT, mesenteric adipose tissue, and skeletal muscle whereas NO-1886 increased the expression of UCP3 mRNA only in skeletal muscle [47]. Therefore, a possible mechanism for antiobesity effects of NO-1886 in rats may be the enhancement of LPL activity in skeletal muscle and the accompanying increase in UCP3 expression.
The antidiabetic agent pioglitazone is thought to promote the differentiation of adipocytes, convert large-type
 (a)
(a)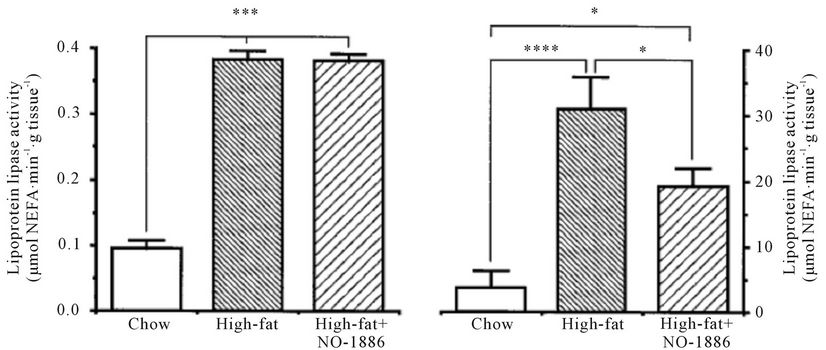 (b)
(b)
Figure 3. Effects of chow or a high-fat diet with and without NO-1886 (50 mg/kg/day) for 10 weeks on soleus skeletal muscle (a), myocardium (b), and edipidymal adipose tissue LPL activity (c) in SD rats. (d) Shows visceral adipose tissue total heparin-releasable LPL activity assuming that the LPL activity in epididymal fat is representative of the visceral fat in general. *P < 0.05, **P < 0.10, ***P < 0.001, ****P < 0.01.
hypertrophic adipocytes into small-type adipocytes, and to increase insulin activity through peroxisome proliferator-activated receptor-γ activation [48]. In addition, thiazolidinediones also improve the serum levels of several adiopocytokines, such as adiponectin and TNF-α, in type 2 diabetic patients [49]. However, as a result of enhanced adipocyte differentiation, pioglitazone treatment was shown to be associated with body weight gain in obese animals and type 2 diabetic patients [49-51]. As the mechanism underlying the body weight gain, Hallakou et al. explained that pioglitazone stimulated the expression of genes involved in lipid metabolism and induced a large increase in glucose utilization in the adipose tissue [52]. Obesity aggravates diabetes and promotes cardiovascular diseases and atherosclerosis, and the body-weight-increasing action of pioglitazone is a disadvantage in diabetic patients. On the other hand, the LPL activator is known to improve both obesity and insulin resistance in obese animals [43,47].
We reported the effect of the simultaneous administration of pioglitazone, which induces a body weight gain, and NO-1886, which has an anti-obesity action, on the body weight, and insulin resistance of obese rats. The concomitant administration of pioglitazone and NO-1886 suppressed the body weight gain in animals fed a highfat diet, confirming that NO-1886 mitigates the body weight-increasing action of pioglitazone. Since an assessment of their effects on insulin resistance showed that both pioglitazone and NO-1886 treated to increase the glucose infusion rate (GIR) in obese animals, these compounds may also improve insulin resistance. The percent increase in the GIR in the pioglitazone + NO- 1886 group was greater than that in either the pioglitazone or the NO-1886 group. Consequently, the improvement of insulin sensitivity may be enhanced by the combined administration of pioglitazone and NO-1886, compared with that observed following the administration of either drug alone. Moreover, NO-1886 might not have an opposing effect on the adiponectin-increasing action of pioglitazone [53]. Thus, the combined administration of pioglitazone and LPL activator may be of great benefit for the treatment of type 2 diabetic patients.
Recently, fatty acid composition has been proposed as one of determinants of insulin resistance. We elucidated that serum saturated as well as unsaturated fatty acid concentrations in type 2 diabetics correlated with homeostasis model assessment ratio, index of insulin resistance [54]. Moreover, fatty acid composition in adipose TG depends on anatomical site, and the dependency disappears in insulin-resistant, high-fat fed rats [55]. Matsuzaka et al. demonstrated that hepatic fatty acid composition was changed by inhibition of production of longchain fatty acid elongase, Elovl6, and insulin resistance was improved in obese mice [56]. Therefore, improvement of insulin resistance by LPL activation may contribute to normalize of fatty acid composition in blood and various tissues.
4. CONCLUSION
The main LPL synthetic tissues are adipose tissue and skeletal muscle. LPL in adipose tissue has a role in fat storage, where LPL in skeletal muscle has a role in fat oxidation. LPL activator NO-1886 improved obesity by elevating skeletal muscle LPL activity. Therefore, if skeletal muscle-specific LPL activators are developed, we may be able to design more beneficial antiobesity drugs. We expect further evolution of tissue-specific LPL activators may also show a clinically relevant benefit in the treatment of lipid-associated and non-lipid-associated diseases.
REFERENCES
- Spiegelman, B.M. and Flier, J.S. (1996) Adipogenesis and obesity: Rounding out the big picture. Cell, 87, 377-389. doi:10.1016/S0092-8674(00)81359-8
- Goldberg, I.J. (1996) Lipoprotein lipase and lipolysis: Central roles in lipoprotein metabolism and atherogenesis. Journal of Lipid Research, 37, 693-707.
- Hara, T., Cameron-Smith, D., Cooney, G.J., Kusunoki, M., Tsutsumi, K., et al. (1998) The actions of a novel lipoprotein lipase activator, NO-1886, in hypertriglyceridemic fructose-fed rats. Metabolism, 47, 149-153. doi:10.1016/S0026-0495(98)90211-6
- Peterson, J., Fujimoto, W.Y. and Brunzell, J.D. (1992) Relationship of activity, heparin affinity, and conformation as studied with monoclonal antibodies. Journal of Lipid Research, 33, 1165-1170.
- Sparkes, R.S., Zollman, S., Klisak, I., Kirchgessner, T.G., Komaromy, M.C., et al. (1987) Human genes involved in lipolysis of plasma lipoproteins: Mapping of loci for lipoprotein lipase to 8p22 and hepatic lipase to 15q21. Genomics, 1, 138-144. doi:10.1016/0888-7543(87)90005-X
- Wion, K.L., Kirchgessner, T.G., Lusis, A.J., Schotz, M.C. and Lawn, R.M. (1987) Human lipoprotein lipase complementary DNA sequence. Science, 235, 1638-1641. doi:10.1126/science.3823907
- Senda, M., Oka, K., Brown, W.V., Qasba, P.K. and Furuichi, Y. (1987) Molecular cloning and sequence of cDNA coding for bovine lipoprotein lipase. Proceedings of the National Academy of Sciences of the United States of America, 84, 4369-4373. doi:10.1073/pnas.84.13.4369
- Clarke, A.R., Luscombe, M. and Holbrook, J.J. (1983) The effect of the chain length of heparin on its interaction with lipoprotein lipase. Biochimica et Biophysica Acta, 747, 130-137. doi:10.1016/0167-4838(83)90131-0
- Pedersen, M.E., Cohen, M. and Schotz, M.C. (1983) Immunocytochemical localization of the functional fraction of lipoprotein lipase in the perfused heart. Journal of Lipid Research, 24, 512-521.
- Brunzell, J.D. (1995) Familial lipoprotein lipase deficiency and other causes of chylomicromia syndrome. In: Scriver, C.R., Baudette, A.L., Sly, W.S. and Valle, D., Eds., The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, New York, 1913-1932. doi:10.1016/S0307-4412(96)80019-7
- Nikkilä, E.A., Taskinen, M.R. and Kekki, M. (1978) Relation of plasma high-density lipoprotein cholesterol to lipoprotein-lipase activity in adipose tissue and skeletal muscle of man. Atherosclerosis, 29, 497-501.
- Kusunoki, M., Tsutsumi, K., Sato, D., Nakamura, A., Habu, S., et al. (2011). Activation of lipoprotein lipase increases serum high density lipoprotein 2 cholesterol and enlarges high density lipoprotein 2 particles in rats. European Journal of Pharmacology, 668, 337-339. doi:10.1016/j.ejphar.2011.06.040
- Tsutsumi, K., Inoue, Y., Shima, A., Iwasaki, K., Kawamura, M., et al. (1993) The novel compound NO-1886 increases lipoprotein lipase activity with resulting elevation of high density lipoprotein cholesterol, and long-term administration inhibits atherogenesis in the coronary arteries of rats with experimental atherosclerosis. Journal of Clinical Investigation, 92, 411-417. doi:10.1172/JCI116582
- Raynolds, M.V., Awald, P.D., Gordon, D.F., GutierrezHartmann, A., Rule, D.C., et al. (1990) Lipoprotein lipase gene expression in rat adipocytes is regulated by isoproterenol and insulin through different mechanisms. Molecular Endocrinology, 4, 1416-1422. doi:10.1210/mend-4-9-1416
- Tsutsumi, K., Inoue, Y., Shima, A. and Murase, T. (1995) Correction of hypertriglyceridemia with low high-density lipoprotein cholesterol by the novel compound NO-1886, a lipoprotein lipase-promoting agent, in STZ-induced diabetes rats. Diabetes, 44, 414-417. doi:10.2337/diabetes.44.4.414
- Miesenböck, G., Hölzl, B., Föger, B., Brandstätter, E., Paulweber, B., et al. (1993) Heterozygous lipoprotein lipase deficiency due to a missense mutation as the cause of impaired triglyceride tolerance with multiple lipoprotein abnormalities. Journal of Clinical Investigation, 91, 448-455. doi:10.1172/JCI116222
- Katzel, L.I., Busby-Whitehead, M.J., Rogus, E.M., Krauss, R.M. and Goldberg, A.P. (1994) Reduced adipose tissue lipoprotein lipase responses, postprandial lipemia, and low high-density lipoprotein-2 subspecies levels in older athletes with silent myocardial ischemia. Metabolism, 43, 190-198. doi:10.1016/0026-0495(94)90244-5
- Reymer, P.W., Gagné, E., Groenemeyer, B.E., Zhang, H., Forsyth, I., et al. (1995) A lipoprotein lipase mutation (Asn291Ser) is associated with reduced HDL cholesterol levels in premature atherosclerosis. Nature Genetics, 10, 28-34. doi:10.1038/ng0595-28
- Gaziano, J.M., Hennekens, C.H., O’Donnell, C.J., Breslow, J.L. and Buring, J.E. (1997) Fasting triglycerides, high-density lipoprotein, and risk of myocardial infarction. Circulation, 96, 2520-2525. doi:10.1161/01.CIR.96.8.2520
- Fan, J., Unoki, H., Kojima, N., Sun, H., Shimoyamada, H., et al. (2001) Overexpression of lipoprotein lipase in transgenic rabbits inhibits diet-induced hypercholesterolemia and atherosclerosis. The Journal of Biological Chemistry, 276, 40071-40079. doi:10.1074/jbc.M105456200
- Shimada, M., Shimano, H., Gotoda, T., Yamamoto, K., Kawamura, M., et al. (1993) Overexpression of human lipoprotein lipase in transgenic mice. Resistance to dietinduced hypertriglyceridemia and hypercholesterolemia. The Journal of Biological Chemistry, 268, 17924-17929.
- Shimada, M., Ishibashi, S., Inaba, T., Yagyu, H., Harada, K., et al. (1996) Suppression of diet-induced atherosclerosis in low density lipoprotein receptor knockout mice overexpressing lipoprotein lipase. Proceedings of the National Academy of Sciences of the United States of America, 93, 7242-7246. doi:10.1073/pnas.93.14.7242
- Tsutsumi, K., Inoue, Y., Hagi, A. and Murase, T. (1997) The novel compound NO-1886 elevates plasma highdensity lipoprotein cholesterol levels in hamsters and rabbits by increasing lipoprotein lipase without any effect on cholesteryl ester transfer protein activity. Metabolism, 46, 257-260. doi:10.1016/S0026-0495(97)90250-X
- Shimokawa, H. and Vanhoutte, P.M. (1989) Impaired endothelium-dependent relaxation to aggregating platelets and related vasoactive substances in porcine coronary arteries in hypercholesterolemia and atherosclerosis. Circulation Research, 64, 900-914. doi:10.1161/01.RES.64.5.900
- Lakatta, E.G. and Yin, F.C. (1982) Myocardial aging: Functional alterations and related cellular mechanisms. The American Journal of Physiology, 242, H927-H941.
- Moritoki, H., Tanioka, A., Maeshiba, Y., Iwamoto, T., Ishida, Y., et al. (1988) Age-associated decrease in histamine-induced vasodilation may be due to reduction of cyclic GMP formation. British Journal of Pharmacology, 95, 1015-1022. doi:10.1111/j.1476-5381.1988.tb11734.x
- Hara, T., Kusunoki, M., Tsutsumi, K., Okada, K., Sakamoto, S., et al. (1998) A lipoprotein lipase activator, NO-1886, improves endothelium-dependent relaxation of rat aorta associated with aging. European Journal of Pharmacology, 350, 75-79. doi:10.1016/S0014-2999(98)00230-1
- Kusunoki, M., Tsutsumi, K., Hara, T., Ogawa, H., Nakamura, T., et al. (2002) A lipoprotein lipase activator, NO-1886 prevents impaired endothelium-dependent relaxation of aorta caused by exercise in aged rats. Experimental Gerontology, 37, 891-896. doi:10.1016/S0531-5565(02)00023-2
- Ohta, T., Takata, K., Horiuchi, S., Morino, Y. and Matsuda, I. (1989) Protective effect of lipoproteins containing apoprotein A-I on Cu2+-catalyzed oxidation of human low density lipoprotein. FEBS Letters, 257, 435-438. doi:10.1016/0014-5793(89)81590-X
- Chiba, T., Miura, S., Sawamura, F., Uetsuka, R., Tomita, I., et al. (1997) Antiatherogenic effects of a novel lipoprotein lipase-enhancing agent in cholesterol-fed New Zealand white rabbits. Atherosclerosis, Thrombosis, and Vascular Biology, 17, 2601-2608. doi:10.1161/01.ATV.17.11.2601
- Yin, W., Tsutsumi, K., Yuan, Z. and Yang, B. (2002) Effects of the lipoprotein lipase activator NO-1886 as a suppressor agent of atherosclerosis in aorta of mild diabetic rabbits. Arzneimittelforschung, 52, 610-614. doi:10.1055/s-0031-1299939
- White, R.T., Damm, D., Hancock, N., Rosen, B.S., Lowell, B.B., et al. (1992) Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. The Journal of Biological Chemistry, 267, 9210-9213.
- Friedman, J.M. (2000) Obesity in the new millennium. Nature, 404, 632-634. doi:10.1038/35017505
- Shimomura, I., Funahashi, T., Takahashi, M., Maeda, K., Kotani, K., et al. (1996) Enhanced expression of PAI-1 in visceral fat: Possible contributor to vascular disease in obesity. Nature Medicine, 2, 800-803. doi:10.1038/nm0796-800
- Steppan, C.M., Bailey, S.T., Bhat, S., Brown, E.J., Banerjee, R.R., et al. (2001) The hormone resistin links obesity to diabetes. Nature, 409, 307-312. doi:10.1038/35053000
- Hotamisligil, G.S. (1999) The role of TNFα and TNF receptors in obesity and insulin resistance. Journal of Internal Medicine, 245, 621-625. doi:10.1046/j.1365-2796.1999.00490.x
- Scherer, P.E., Williams, S., Fogliano, M., Baldini, G. and Lodish, H.F. (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. The Journal of Biological Chemistry, 270, 26746-26749. doi:10.1074/jbc.270.45.26746
- Kern, P.A., Ong, J.M., Saffari, B. and Carty, J. (1990) The effect of weight loss on the activity and expression of adipose-tissue lipoprotein lipase in very obese humans. The New England Journal of Medicine, 322, 1053-1059. doi:10.1056/NEJM199004123221506
- Sadur, C.N., Yost, T.J. and Eckel, R.H. (1984) Insulin responsiveness of adipose tissue lipoprotein lipase is delayed but preserved in obesity. The Journal of Clinical Endocrinology and Metabolism, 59, 1176-1182. doi:10.1210/jcem-59-6-1176
- Shimada, M., Ishibashi, S., Yamamoto, K., Kawamura, M., Watanabe, Y., et al. (1995) Overexpression of human lipoprotein lipase increases hormone-sensitive lipase activity in adipose tissue of mice. Biochemical and Biophysical Research Communications, 211, 761-766. doi:10.1006/bbrc.1995.1878
- Jensen, D.R., Schlaepfer, I.R., Morin, C.L., Pennington, D.S., Marcell, T., et al. (1997) Prevention of diet-induced obesity in transgenic mice overexpressing skeletal muscle lipoprotein lipase. The American Journal of Physiology, 273, R683-R689.
- Ferrano, R.T., Eckel, R.H., Larson, D.E., Fontvieille, A.M., Rising, R., et al. (1993) Relationship between skeletal muscle lipoprotein lipase activity and 24-hour macronutrient oxidation. The Journal of Clinical Investigation, 92, 441-445. doi:10.1172/JCI116586
- Kusunoki, M., Hara, T., Tsutsumi, K., Nakamura, T., Miyata, T., et al. (2000) The lipoprotein lipase activator, NO-1886, suppresses fat accumulation and insulin resistance in rats fed a high-fat diet. Diabetologia, 43, 875- 880. doi:10.1007/s001250051464
- Boss, O., Muzzin, P. and Giacobino, J.P. (1998) The uncoupling proteins, a review. European Journal of Endocrinology, 139, 1-9. doi:10.1530/eje.0.1390001
- Schrauwen, P. and Hesselink, M. (2002) UCP2 and UCP3 in muscle controlling body metabolism. The Journal of Experimental Biology, 205, 2275-2285.
- Doi, M., Kondo, Y. and Tsutsumi, K. (2003) Lipoprotein lipase activator NO-1886 (ibrolipim) accelerates the mRNA expression of fatty acid oxidation-related enzymes in rat liver. Metabolism, 52, 1547-1550. doi:10.1016/j.metabol.2003.07.007
- Kusunoki, M., Tsutsumi, K., Iwata, K., Yin, W., Nakamura, T., et al. (2005) NO-1886 (ibrolipim), a lipoprotein lipase activator, increases the expression of uncoupling protein 3 in skeletal muscle and suppresses fat accumulation in high-fat diet-induced obesity in rats. Metabolism, 54, 1587-1592. doi:10.1016/j.metabol.2005.06.005
- Spiegelman, B.M. (1998) PPAR-γ: Adipogenic regulator and thiazolidinedione receptor. Diabetes, 47, 507-514. doi:10.2337/diabetes.47.4.507
- Miyazaki, Y. and Defronzo, R.A. (2008) Rosiglitazone and pioglitazone similarly improve insulin sensitivity and secretion, glucose tolerance and adipocytokines in type 2 diabetic patients. Diabetes, Obesity & Metabolism, 10, 1204-1211. doi:10.1111/j.1463-1326.2008.00880.x
- Boden, G. and Zhang, M. (2006) Recent findings concerning thiazolidinediones in the treatment of diabetes. Expert Opinion on Investigational Drugs, 15, 243-250. doi:10.1517/13543784.15.3.243
- Hermansen, K. and Mortensen, L.S. (2007) Bodyweight changes associated with antihyperglycemic agents in type 2 diabetes mellitus. Drug Safety, 30, 1127-1142. doi:10.2165/00002018-200730120-00005
- Hallakou, S., Doaré, L., Foufelle, F., Kerqoat, M., Guerre-Millo, M., et al. (1997) Pioglitazone induces in vitro adipocyte differentiation in the obese Zucker fa/fa rat. Diabetes, 46, 1393-1399. doi:10.2337/diabetes.46.9.1393
- Kusunoki, M., Tsutsumi, K., Sato, D., Nakamura, A., Habu, S., et al. (2011) Pioglitazone-induced body weight gain is prevented by combined administration with the lipoprotein lipase activator NO-1886. European Journal of Pharmacology, 668, 486-491. doi:10.1016/j.ejphar.2011.07.030
- Kusunoki, M., Tsutsumi, K., Nakayama, M., Kurokawa, T., Nakamura, T., et al. (2007) Relationship between serum concentrations of saturated fatty acids and unsaturated fatty acids and the homeostasis model insulin resistance index in Japanese patients with type 2 diabetes mellitus. The Journal of Medical Investigation, 54, 243-247. doi:10.2152/jmi.54.243
- Sato, D., Nakamura, T., Tsutsumi, K., Shinzawa, G., Karimata, T., et al. (2012) Site dependency of fatty acid composition in adipose triacylglycerol in rats and its absence as a result of high-fat feeding. Metabolism, 61, 92-98. doi:10.1016/j.metabol.2011.05.012
- Matsuzaka, T., Shimano, H., Yahagi, N., Kato, T., Atsumi, A, et al. (2007) Crucial role of a long-chain fatty acid elongase, Elovl6, in obesity-induced insulin resistance. Nature Medicine, 13, 1193-1202. doi:10.1038/nm1662