Advances in Bioscience and Biotechnology
Vol.4 No.8B(2013), Article ID:35615,8 pages DOI:10.4236/abb.2013.48A2004
Potential of new anti-cancer agents targeting the nuclear translocation signaling of HB-EGF C-terminal fragments during the development of colitis-associated cancer*
![]()
Department of Gastroenterology and Metabolism, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan
Email: #stanida@med.nagoya-cu.ac.jp
Copyright © 2013 Satoshi Tanida et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 29 May 2013; revised 25 June 2013; accepted 13 July 2013
Keywords: Inflammatory Bowel Disease; Ulcerative Colitis; Colitis-Associated Cancer; IL-8; HB-EGF-CTF; PLZF; Telmisaratan
ABSTRACT
In inflammatory bowel diseases (IBD) such as ulcerative colitis (UC) and Crohn’s disease (CD), the duration and severity of inflammation are responsible for the development of colorectal cancer. Inflammatory cytokines such as interleukin (IL)-8 and tumor necrotic factor (TNF)-(, which are released by epithelial and immune cells, are involved in the pathogenesis of colitis-associated cancer. Current treatments for advanced colorectal cancers focus primarily on targeting epidermal growth factor receptor (EGFR) signaling. IL-8 (a G-protein coupled receptor (GPCR) agonist), which is involved in neutrophil recruitment and activation in persistent active colitis, also promotes cleavage of theproheparin-binding epidermal growth factor—like growth factor (proHB-EGF) through a disintegrin and metalloproteinase (ADAM), so that the resulting soluble HB-EGF activates EGFR. In parallel, the carboxy-terminal fragment of proHB-EGF (HB-EGF-CTF) translocates into the inner nuclear membrane, where HB-EGF-CTF binds the nuclear promyelocytic leukemia zinc finger (PLZF) protein, resulting in the nuclear export of the PLZF transcriptional repressor and thereby affecting cell proliferation. Screening for potent chemical inhibitors of the interactions between HB-EGF-CTF and PLZF identified telmisartan (and related compounds in corporating a biphenyl tetrazole moiety) as inhibitors of cell proliferation. Here we focus on the inhibitory effects of these compounds on cell proliferation, demonstrating the potential for targeting the nuclear translocation of HB-EGF-CTF in the treatment of colitisassociated cancer.
1. INTRODUCTION
The incidence of inflammatory bowel diseases such as UC and CD has been increasing on an annual basis. Uncontrolled and excessive host immune responses, during which inflammatory cytokines such as IL-8 and TNF-a mediate inflammatory cell infiltrates in the gut mucosa, are known to induce inflammation and exaggerate mucosal injury in the gut. Long-term high disease activity has been established as a potential risk factor for the development of colitis-associated cancer [1,2]. The cumulative incidence of such malignancies is below 1% in the first 8 - 10 years of UC, but thereafter rises in annual increments of 0.5% - 1.0% to reach 5% - 10% after 20 years and 15% - 20% after 30 years [3]. The perpetuation of active inflammation in the mucosa of patients with UC, which is mediated by IL-8, also is believed to increase the risk of colitis-associated cancer. Thus, IL-8 plays important roles in the perpetuation of inflammation in UC leading to the development of colorectal cancer.
Therapeutic treatments targeted towards advanced colorectal cancers have focused mainly on the design of therapeutic agents that target epidermal growth factor (EGF) signaling, such as the monoclonal antibodies cetuximab and panitumumab, which directly block the EGFR [4-6]. However, less consideration is currently given to the potential for therapeutically targeting the nuclear translocation signaling of HB-EGF-CTF.
This nuclear translocation signaling arises from metalloproteinase cleavage of the precursor of HB-EGF to yield the cleaved extra cellular fragments and CTF. Following cleavage, the extra cellular (soluble) portion of HB-EGF binds to and activates EGFR, whereas the CTF is translocated to the nucleus, where this fragment modulates signaling. Here, we provide an overview of the mechanisms by which IL-8 modulates UC inflammation and induces EGF signaling to regulate cell proliferation. The role of IL-8 emphasizes the potential of inhibiting the nuclear translocation of HB-EGF-CTF as a mechanism for suppressing the development of colitisassociated cancer.
2. IL-8 NOT ONLY MEDIATES THE ACTIVATION OF NEUTROPHILS DURING UC INFLAMMATION BUT ALSO ACTS AS A GROWTH FACTOR
The gut inflammation that occurs in UC is characterized by mucosal and submucosal infiltration by numerous neutrophils and lymphocytes in both moderate and severe UC patients. In patients with UC, the circulating levels of neutrophils also are reported to be up to three times as high as the level in healthy controls [7]. Indeed, granulocyte and monocyteadsorptive apheresis is therapeutic for active UC, confirming that circulating and infiltrating neutrophils are strongly involved in the development of UC [8,9].
Increased levels of IL-8 mRNA expression and IL-8 protein production are observed in the inflamed mucosa of patients with active UC, especially in macrophages, neutrophils and colon epithelial cells [10,11]. Moreover, luminol-dependent chemiluminescence and myeloperoxidase (MPO) activities of neutrophils in the mucosa are markedly increased in active UC compared with the levels observed in patients with inactive UC and in healthy controls. Thus, IL-8 levels correlate with the intensity of chemiluminescence and MPO levels [12]. These results suggested that most neutrophils that infiltrate the inflamed mucosa in UC are activated by IL-8.
In the context of mitogenesis, IL-8 is known to promote angiogenesis [13]. IL-8 also has been implicated in the promotion of cell proliferation mediated by binding to its homologue receptors (CXCR1 and CXCR2: GPCR) in various types of cancer cells [14], thus confirming that IL-8 acts as a growth factor. IL-8 thus has been found to promote both the angiogenesis of vascular endothelial cells and the proliferation of tumor cells. IL-8 is not constitutively expressed in non-cancerous cells, but is upregulated by stimuli such as lipopolysaccharide (LPS) and TNF-a [15]. In contrast, constitutive IL-8 protein expression has been demonstrated in various human malignancies, including leukemia and cancers of the stomach and colon. In addition, IL-8 expression is associated with tumor progression and poor prognosis as well as angiogenesis [16,17].
Thus, IL-8 acts not only as a chemoattractant but also as a growth factor in the inflamed mucosa characteristic of UC, suggesting that this cytokine play a critical role in the development of colitis-associated cancer during chronic UC inflammation.
3. DUAL SIGNALING OF HB-EGF IN IL-8-INDUCED CELL PROLIFERATION
EGF signaling is propagated by a dual pathway that includes both ligand-dependent EGFR transactivation and its nuclear translocation (of the CTF released by cleavage of the pro-protein). Several GPCRs have been demonstrated to activate EGFR (an event known as transactivation), even though GPCR agonists do not directly interact with EGFR. EGFR transactivation by GPCR agonists was first discovered to mediate several critical downstream EGFR signals and activities such as ERK activation, c-fos induction, and cell proliferation [18,19]. EGFR transactivation involves a disintegrin and metalloproteinase (ADAM) that convert the pro-form of EGFR ligands (including EGF, HB-EGF, amphiregulin, betacellulin, epiregulin and epigen) to bioactive forms by inducing shedding of the ectodomain. Blocking of proHBEGF function (using the diphtheria toxin mutant Crm197) or of metalloproteinase activity (using the inhibitor batimastat, BB94), abrogates GPCR-induced phosphorylation of EGFR, Shc, and MAPK [20,21]. Inhibition of metalloproteinase-mediated EGFR ligand shedding also reduces proliferation [22,23].
Numerous GPCR agonists, including angiotensin II, endothelin-1, carbachol, bombesin, phenylephrine, and IL-8 have been reported. IL-8 is the major neutrophil chemoattractant in UC, and has been implicated in the initiation and perpetuation of inflammation in UC. This cytokine promotes cell proliferation in the colon cancer cell line CaCO2 through ADAM-mediated cleavage of pro-HBEGF, as assessed using the incorporation of [3H] thymidine into DNA [22]. Introduction of siRNA targeting ADAM10 dramatically reduces IL-8-induced proHBEGF shedding and EGFR transactivation [24]. Thus, IL-8-induced cell proliferationappears to be mediated by ADAM10-associated proHB-EGF shedding and EGFR transactivation. This hypothesis was confirmed using a dextran sulfate sodium (DSS)-induced murine model of experimental colitis: When the expression of ADAM-17 was reduced (via the exon-induced translational stop method) in DSS-treated mice, colon epithelial cells exhibited defective regeneration and the intestinal barrier showed breakdown, presumably reflecting impaired shedding of EGFR ligands followed by failure to phosphorylate STAT3 [25]. Thus, ADAM-17 mediated cleavage of EGFR ligands also appears to be involved in cell proliferation and tissue regeneration.
HB-EGF-CTF is reportedly translocated into the nucleus following ectodomain cleavage of HB-EGF by specific metalloproteinases. The CTF subsequently exerts effects on the regulation of cell proliferation by binding nuclear promyelocytic leukemia zinc finger (PLZF) protein, a transcriptional repressor, thereby causing nuclear export of PLZF [26]. IL-8 also induces translocation of HB-EGF-CTF into the nucleus after ADAM10-mediated ectodomain shedding of proHB-EGF [27]. Moreover, suppression of HB-EGF-CTF translocation into the nucleus (by use of the ADAM inhibitor KB-R7785) deceleratesmitogenic phenomena through delayed entry into the S-phase of the cell cycle. Inhibition of metalloprotease activityabrogates this process, suggesting that ectodomain processing triggers translocation of HB-EGF-CTF into the nucleus [26,28]. In parallel with HB-EGF-CTF signaling, theshed ectodomain of proHB-EGF promotes G1-phase progression in the cell cycle through binding to and activation of the EGFR, resulting in upregulation of cyclin D expression via the Ras-MAPK signaling cascade, which includes MKK1/2 and ERK [26,28]. Therefore, processing of proHB-EGF, as induced by IL-8, generates two types of mitogenic signals: EGFR transactivation followed by downstream RasMAPK activation, and the release of intracellular HBEGF-CTF leading to nuclear export of PLZF. Thus, IL-8 modulates cell proliferation through dual intracellular signaling pathways.
4. A NEW APPROACH FOR ANTI-CANCER DRUGS TARGETING THE NUCLEAR TRANSLOCATION SIGNALING OF HB-EGF-CTF
When administered together, monoclonal blocking antibodies against the EGFR plus combination chemotherapy provide additive effects on advanced colorectal cancer [5,6]. However, these additive effects are limited in the presence of the KRAS mutation [5,6]. Inhibition of nuclear translocation signaling of HB-EGF-CTF is considered to be a new strategy against the development of colorectal cancer. However, no existing therapeutic agents are known to employ this mechanism of action, that is, to specifically block nuclear translocation of HB-EGFCTF and binding to PLZF.
Inhibitors that block the interaction between HB-EGFCTF and PLZF were recently identified based on high through put screening assay [29]. This assay employed HT1080 cells, which express only low levels of endogenous HB-EGF precursors. Initially, regions of PLZF with affinity for the cytoplasmic domains of pro-EGFR ligands were characterized in HT1080 cells by using a GST pull-down assay. Co-transfection with constructs encoding various FLAG-tagged PLZF derivatives revealed a close interaction between GST-HB-EGF-CTF and zinc finger domains (ZnF) 5 - 8 of PLZF, but not between CTF and a deletion mutant of ZnF 5 - 8 (PLZF/ ΔZnF 5 - 8). These data suggested that the ZnF 5 - 8 region is critical for the interactions between PLZF and HB-EGF-CTF.
Using an AlphascreenÒ technique, a high-through-put assay was developed to screen for inhibitors that block the interaction between ZnF 5 - 8 and CTF. The Alphascreen assay design relies on the fact that a signal can be detected only when streptavidin-coated donor and antiGST antibody-conjugated acceptor beads are in close proximity (within a distance of 200 nm). In this specific assay, the beads are brought into close proximity via the specific interactions between ZnF 5 - 8 and CTF complexes coupled to the beads. An analysis with biotin-ARCTF and GST-Zn 5 - 8 of PLZF validated the inhibitory effects of obtained candidates. A total of 9000 chemical compounds were screened based on the efficacy of blocking the binding of AR-CTF to ZnF 5 - 8 or the binding of HB-EGF-CTF to Zn 5 - 8 of PLZF, yielding twelve candidates (including compounds designated as nos. 8016, 7701 and 7804) (Figure 1(a)). At concentrations of 10 mM, compounds no. 8016, 7701 and 7804 exerted 30.4%, 60.5% and 49.9% inhibition on the interaction between biotin-AR-CTF and GST-ZnF 5 - 8 of PLZF. Notably, although compound no. 8016 did not exhibit the strongest inhibitory effect on the interaction (Table 1), the compound did exhibit the strongest inhibition of cell proliferation among the twelve candidates when assayed via a cell proliferation assay during 12-Otetradecanoyl-phorbor-13-acetate (TPA) stimulation [29]. Based on the cell proliferation results, we selected no. 8016 as a candidate. The half maximal inhibitory concentration (IC50) obtained with compound no. 8016 was 29.9 mM (Figure 1(b)). The presence of the biphenyl tetrazole in the candidate compound suggested the utility of screening structurally similar compounds, including telmisartan, candesartan, olmesartan, and losartan (Figures 1(a)-(c)), which are all angiotensin II type 1 receptor (AT1R) blockers (ARBs). In an assay for binding of AR-CTF to PLZF, these four ARBs exhibited IC50 values as shown in Figures 1(c) and (d). A cell proliferation assay with keratinocytes demonstrated that, of the four ARBs tested, telmisartan exhibited the strongest effect on cell proliferation [29], confirming the proposed correlation between inhibition of CTF-PLZF interaction and inhibition of cell proliferation.
=
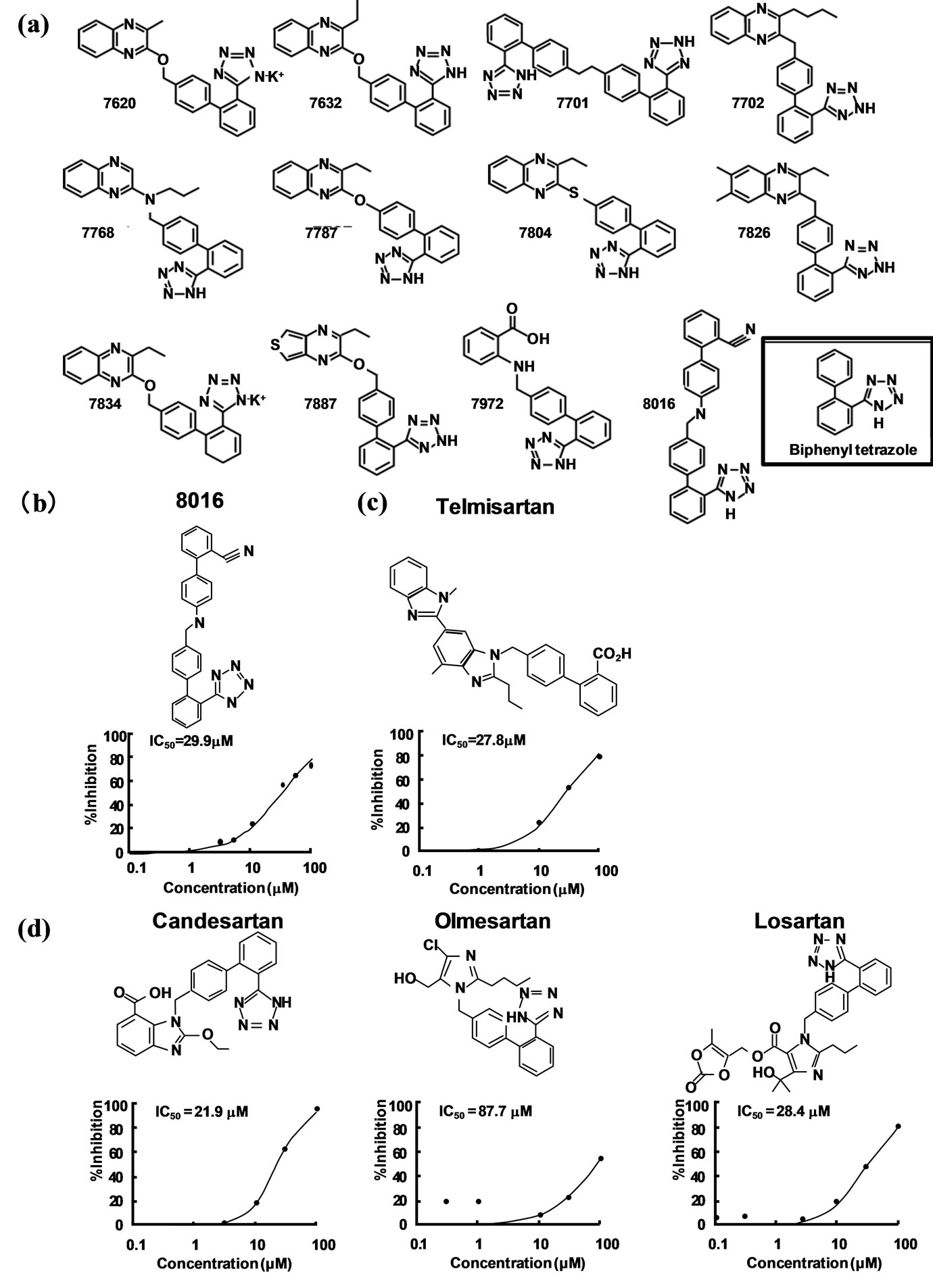
Figure 1. Structures and inhibition of cell proliferation for the most potentinhibitors identified by binding assay. Modifiedfrom reference 25. (a) Structures of the twelve candidate compounds identified in Table 1 binding assay. (b) and (c) The inhibitory effects of the compound no. 8016 and telmisartan on the binding of AR-CTF to PLZF. Plots with the percent inhibition against various concentrations of compound no. 8016 and telmisartan are presented with the IC50 values observed by the Alphascreen system. (d) Inhibitory effects of three ARB candidates on the binding of AR-CTF to PLZF. Plots with the percent inhibition against various concentrations of each inhibitor and inhibition are presented with IC50 values.
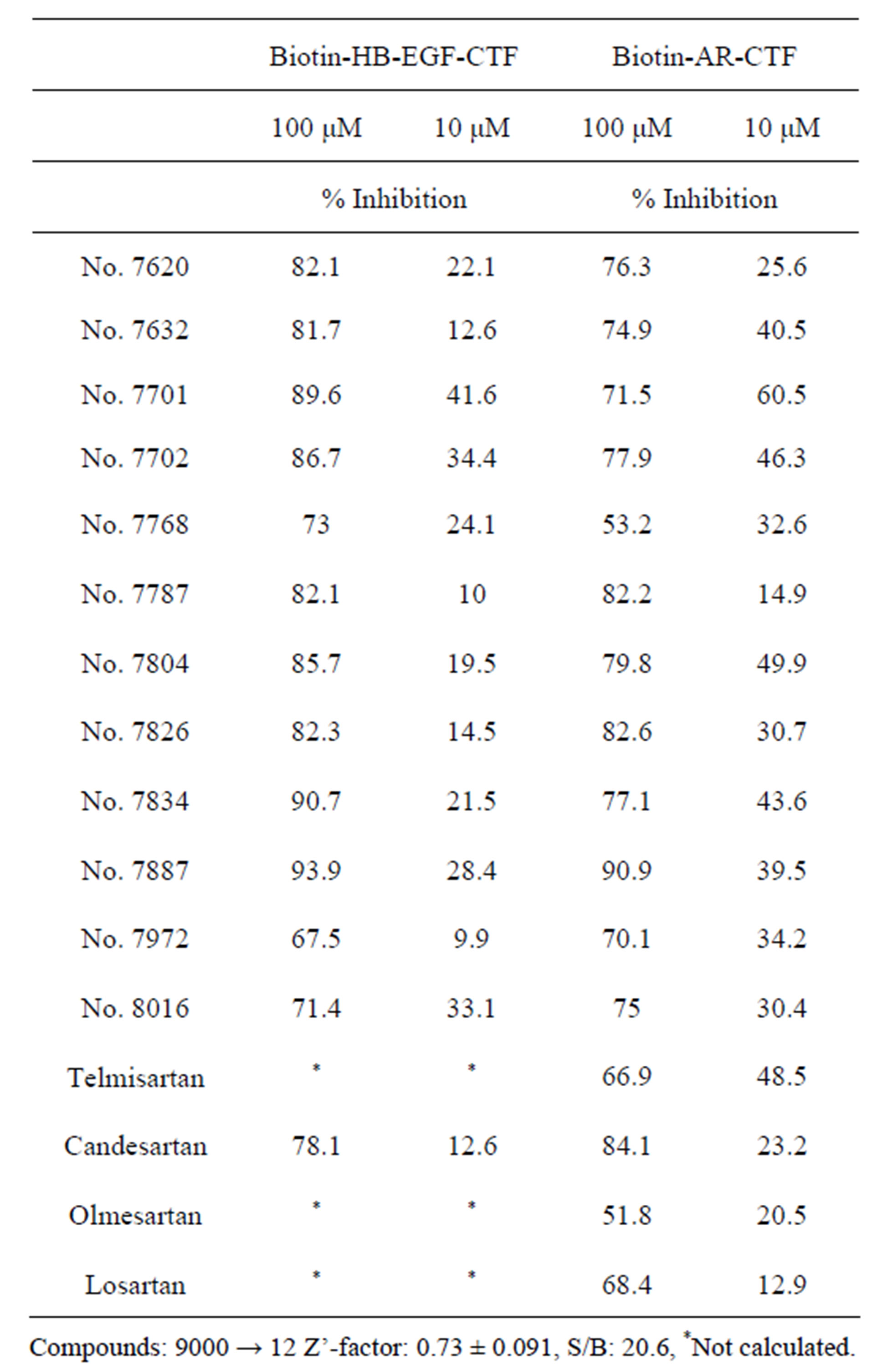
Table 1. Inhibitors of the interactions between the C-terminal fragment (CTF) and zinc finger domains 5 to 8 (ZnF 5 - 8) of the promyelocytic leukemia zinc finger (PLZF) protein. An AlphascreenÒ-based high-through-put screen was used to identify inhibitors of the interaction between HB-EGF-CTF or ARCTF and ZnF 5 - 8 of PLZF. Screening 9000 compounds yielded twelve candidate compounds. Based on structural similarity to compound no. 8016, an additional four angiotensin II type 1 receptor blockers (ARB) also was tested. Values are the mean percent inhibition of binding of the indicated CTF fragment with ZnF 5 - 8, as assayed by the Alpha screen system. The Z’-factor for the screen was shown to be 0.73, demonstrating the reliability of the assay (0.5 < Z’ < 1.0). Table is modified from reference [25].
Compounds: 9000 → 12 Z’-factor: 0.73 ± 0.091, S/B: 20.6, *Not calculated.
5. TELMISARTAN, BUT NOT CANDESARTAN, INHIBITS TPA-INDUCED COLON CANCER CELL PROLIFERATION
TPA, an activator of protein kinase C (PKC)d, is known to induce the ectodomain cleavage of proHB-EGF via ADAM [30]. Using a CCK-8-based cell assay, we demonstrated that TPA-induced proliferation of HT29 cells was inhibited in a dose-dependent fashion by telmisartan but not by candesartan [29]. Using similar assays, we confirmed telmisartan’s ability to inhibit TPA-induced cell proliferation in additional colon cancer cell lines, including HCT116, SW480, and CaCO2 [29].
In addition, the inhibitory effects of telmisartan and candesartan on TPA-induced cell proliferation were assessed using growth curve analyses and cell proliferation assays. Specifically, HT29 and HCT116 cells were continuously cultured for up to 6 days in the presence or absence of TPA and with or without telmisartan or candesartan. The resulting cell counts were 2-fold greater with TPA stimulation compared to a control not exposed to TPA. The TPA induction effects were attenuated (to cell counts approximating those of the control culture) by the additional inclusion of telmisartan, but not by the additional inclusion of candesartan. Notably, in the absence of TPA stimulation, telmisartan and candesartan did not further inhibit cell proliferation (to levels below those of the control) [29].
6. TELMISARTAN, BUT NOT CANDESARTAN, INHIBITS TPA-INDUCED PHENOMENA (NUCLEAR TRANSLOCATION OF HB-EGF-CTF, NUCLEAR EXPORT OF PLZF, AND ASSOCIATION OF HB-EGF-CTF WITH PLZF)
Immunofluorescent staining (IF) in HT29 or HCT116 cells was used to determine the intracellular localization of HB-EGF-CTF and PLZF under various growth conditions. In control (unstimulated) cells, IF demonstrated localization of HB-EGF-CTF to the cell membrane or cytoplasm, and PLZF to the nucleus. In TPA-stimulated cells, IF demonstrated nuclear translocation of HB-EGFCTF and nuclear export of PLZF. The combination of TPA plus telmisartan, but not that of TPA plus candesartan, inhibited the TPA-induced nuclear translocation of HB-EGF-CTF and nuclear export of PLZF. In separate experiments, we used a combination of immunoprecipitation (with anti-HB-EGF-CTF antibody) and immune- (western) blotting (with anti-PLZF antibody) to assay HT29 cells for TPA-induced association between HBEGF-CTF and PLZF. Our results demonstrated that the association between CTF and PLZF peaked at 120 min following treatment with TPA. The additional inclusion of telmisartan, but not that of candesartan, significantly blocked this association [29]. The differences in the inhibitory effects of telmisartan and candesartan on these phenomena can be explained by their lipid solubility. Telmisartan is more lipid soluble than candesartan [31], and thereby, telmisartan can easily pass through the cell membrane into the cytoplasm and block the interaction between HB-EGF-CTF and PLZF.
7. TELMISARTAN AT1R-INDEPENDENTLY INHIBITS NUCLEAR TRANSLOCATION OF HB-EGF-CTF AND CELL PROLIFERATION
Angiotensin II, a GPCR agonist, also induces EGFR transactivation [32]. Both telmisartan and candesartan are antagonists of AT1R (a GPCR), raising the possibility that the TPA-induced nuclear translocation of HBEGF-CTF and nuclear export of PLZF represent crosstalk between the two GPCR pathways. We revised our assay to perform immunoprecipitation with an antiEGFR antibody followed by western blotting with an anti-phosphotyrosine antibody. Our results showed that EGFR phosphorylation during TPA stimulation peaked within 15 min and then gradually decreased to basal levels by 120 min. Notably, EGFR phosphorylation was not inhibited by either telmisartan or candesartan. To address possible cross-talk, we repeated this analysis in cells that had been depleted of AT1R using a gene-specific small interfereing RNA (siRNA). The immunostaining localization of HB-EGF-CTF and PLZF in AT1R-depleted cells revealed that telmisartan, but not that candesartan, inhibited TPA-induced nuclear translocation of HB-EGF-CTF and nuclear export of PLZF, suggesting AT1R signaling [29]. We additionally noted (using a CCK-8-based assay) that TPA-induced cell proliferation was inhibited by telmisartan, but not by candesartan, in both the presence and absence of AT1R depletion. This observation verified that TPA-induced cell proliferation was independent of AT1R signaling. Based on these findings, we hypothesize that telmisartan inhibits cell proliferation during TPA stimulation by specifically blocking nuclear translocation of HB-EGFCTF and nuclear transport of PLZF in colon cancer cells, and note this inhibitory activity proceeds via an AT1Rindpendent mechanism. These results demonstrate a new property of telmisartan.
8. CONCLUSION AND PERSPECTIVES
Extensive knowledge regarding the molecular mechanisms of EGFR ligands and their receptors is indispen-
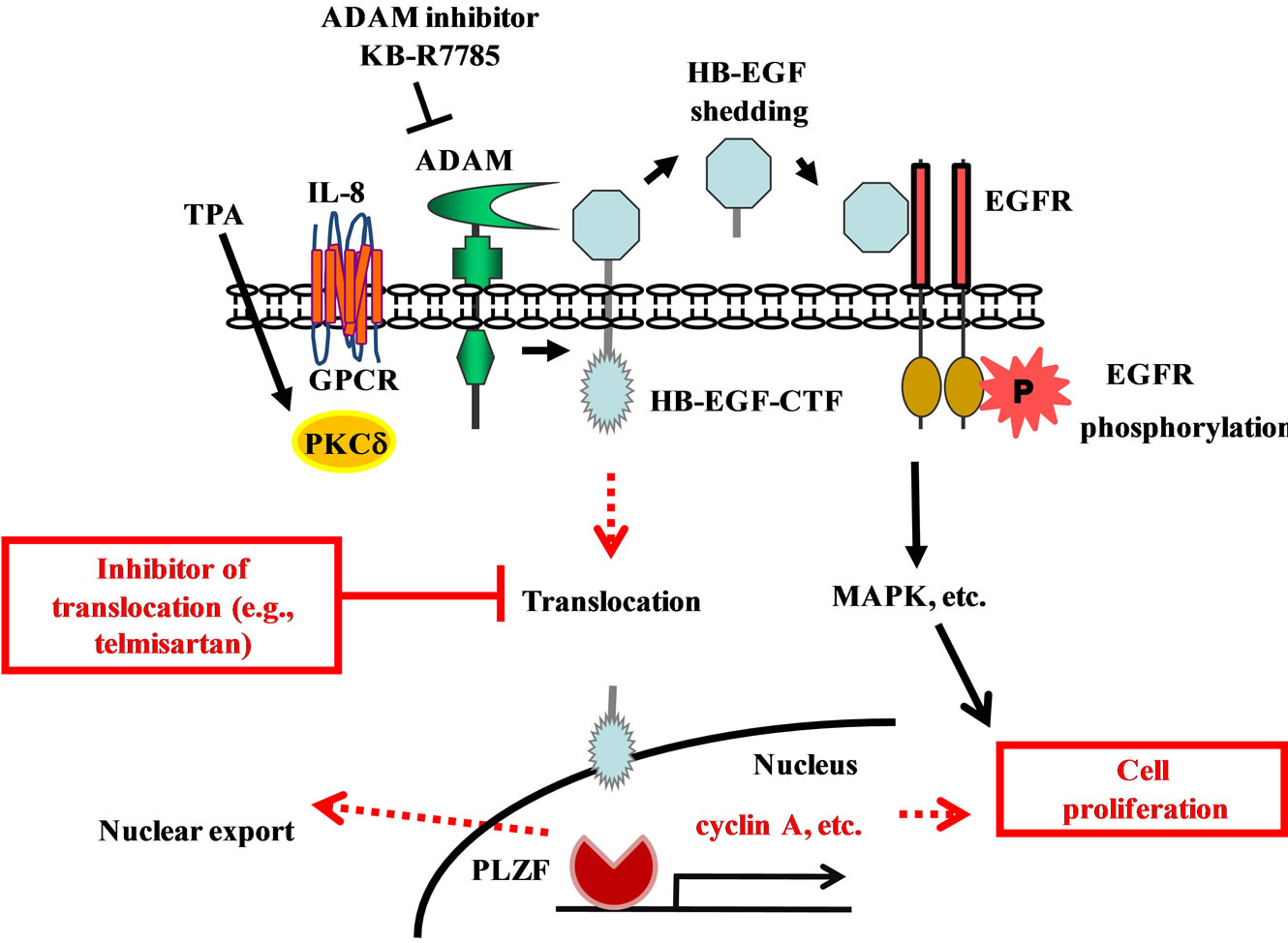
Figure 2. Dual signaling pathways of EGFR phosphorylation and HB-EGF C-terminal fragment nuclear translocation during cell proliferation. IL-8 and TPA induces an ADAM-mediated cleavage of proHB-EGF, and results in the ectodomain shedding of its N-terminal fragment (NTF) and generation of an intracellular C-terminal fragment (CTF). The soluble HB-EGF binds to the EGFR and induces a rapid transient phosphorylation of EGFR. (P indicates phosphorylation). This phosphorylation results in the transcription of various genes. Meanwhile the HB-EGF-CTF translocates into the nucleus, where the CTF subsequently induces the nuclear export of PLZF. This results in the progression of cell cycle. Inhibitors like telmisartan are proposed to act by blocking the nuclear translocation of HB-EGF-CTF.
sable for the design of therapeutic strategies that block EGF signaling to facilitate the treatment of advanced colon cancer. We suggest that the inhibition of HB-EGFCTF with telmisartan and analogs may be a novel strategy for preventing cell proliferation (Figure 2). A recent meta-analysis on the incidence of cancers during treatment with ARBs in 15 large clinical trials found that (compared to controls) patients receiving ARBs exhibited no significant increase in the risk of lung, prostate, or breast cancer nor in overall cancer mortality [33]. In addition, a cell proliferation assay demonstrated that the combination of telmisartan and cetuximab inhibited cell proliferation in a dose-dependent manner, and with efficacy better than that of telmisartan alone (unpublished data). Thus, in patients with colorectal cancer, the use of telmisartan plus combination chemotherapy with anti-EGFR therapy may provide a treatment strategy superior to that of combination chemotherapy with anti-EGFR therapy alone. Future analyses of the detailed molecular mechanisms of these signaling pathways in UC-associated cancer will permit the development of new therapeutic agents that target molecules involved in the development of UC and colorectal cancer.
REFERENCES
- Langholz, E., Munkholm, P., Davidsen, M. and Binder, V. (1992) Colorectal cancer risk and mortality in patients with ulcerative colitis. Gastroenterology, 103, 1444- 1451.
- Gyde, S.N., Prior, P., Allan, R.N., Stevens, A., Jewell, D.P., Truelove, S.C., Lofberg, R., Brostrom, O. and Hellers, G. (1988) Colorectal cancer in ulcerative colitis: A cohort study of primary referrals from three centres. Gut, 29, 206-217. doi:10.1136/gut.29.2.206
- Harpaz, N.T.I. (1996) Colorectal cancer in idiopathic inflammatory bowel disease. Seminars in Diagnostic Pathology, 13, 339-357.
- Cunningham, D., Humblet, Y., Siena, S., Khayat, D., Bleiberg, H., Santoro, A., Bets, D., Mueser, M., Harstrick, A., Verslype, C., Chau, I. and Van Cutsem, E. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. The New England Journal of Medicine, 351, 337-345. doi:10.1056/NEJMoa033025
- Van Cutsem, E., Kohne, C.H., Hitre, E., Zaluski, J., Chang Chien, C.R., Makhson, A., D’Haens, G., Pinter, T., Lim, R., Bodoky, G., Roh, J.K., Folprecht, G., Ruff, P., Stroh, C., Tejpar, S., Schlichting, M., Nippgen, J. and Rougier, P. (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. The New England Journal of Medicine, 360, 1408-1417. doi:10.1056/NEJMoa0805019
- Amado, R.G., Wolf, M., Peeters, M., Van Cutsem, E., Siena, S., Freeman, D.J., Juan, T., Sikorski, R., Suggs, S., Radinsky, R., Patterson, S.D. and Chang, D.D. (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 26, 1626-1634.
- Hanai, H., Takeuchi, K., Iida, T., Kashiwagi, N., Saniabadi, A.R., Matsushita, I., Sato, Y., Kasuga, N. and Nakamura, T. (2004) Relationship between fecal calprotectin, intestinal inflammation, and peripheral blood neutrophils in patients with active ulcerative colitis. Digestive Diseases and Sciences, 49, 1438-1443. doi:10.1023/B:DDAS.0000042243.47279.87
- Sawada, K., Muto, T., Shimoyama, T., Satomi, M., Sawada, T., Nagawa, H., Hiwatashi, N., Asakura, H. and Hibi, T. (2003) Multicenter randomized controlled trial for the treatment of ulcerative colitis with a leukocytapheresis column. Current Pharmaceutical Design, 9, 307-321. doi:10.2174/1381612033391928
- Mizushima, T., Tanida, S., Mizoshita, T., Hirata, Y., Murakami, K., Shimura, T., Kataoka, H., Kamiya, T. and Joh, T. (2011) A complicated case of tacrolimus-induced rapid remission after cesarean section in the early third trimester for refractory severe ulcerative colitis flaring in the initial period of gestation. Case Reports in Gastroenterology, 5, 144-151.
- Mazzucchelli, L., Hauser, C., Zgraggen, K., Wagner, H., Hess, M., Laissue, J.A. and Mueller, C. (1994) Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. American Journal of Pathology, 144, 997-1007.
- Isaacs, K.L., Sartor, R.B. and Haskill, S. (1992) Cytokine messenger RNA profiles in inflammatory bowel disease mucosa detected by polymerase chain reaction amplification. Gastroenterology, 103, 1587-1595.
- Anezaki, K., Asakura, H., Honma, T., Ishizuka, K., Funakoshi, K., Tsukada, Y. and Narisawa, R. (1998) Correlations between interleukin-8, and myeloperoxidase or luminol-dependent chemiluminescence in inflamed mucosa of ulcerative colitis. Internal Medicine, 37, 253-258. doi:10.2169/internalmedicine.37.253
- Koch, A.E., Polverini, P.J., Kunkel, S.L., Harlow, L.A., DiPietro, L.A., Elner, V.M., Elner, S.G. and Strieter, R.M. (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science, 258, 1798-1801. doi:10.1126/science.1281554
- Murphy, P.M., Baggiolini, M., Charo, I.F., Hebert, C.A., Horuk, R., Matsushima, K., Miller, L.H., Oppenheim, J.J. and Power, C.A. (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacological Reviews, 52, 145-176.
- Mukaida, N. (2003) Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. American Journal of Physiology—Lung Cellular and Molecular Physiology, 284, L566-577.
- Yuan, A., Yang, P.C., Yu, C.J., Chen, W.J., Lin, F.Y., Kuo, S.H. and Luh, K.T. (2000) Interleukin-8 messenger ribonucleic acid expression correlates with tumor progression, tumor angiogenesis, patient survival, and timing of relapse in non-small-cell lung cancer. American Journal of Respiratory and Critical Care Medicine, 162, 1957-1963. doi:10.1164/ajrccm.162.5.2002108
- Kitadai, Y., Haruma, K., Mukaida, N., Ohmoto, Y., Matsutani, N., Yasui, W., Yamamoto, S., Sumii, K., Kajiyama, G., Fidler, I.J. and Tahara, E. (2000) Regulation of disease-progression genes in human gastric carcinoma cells by interleukin 8. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 6, 2735-2740.
- Murphy, C., McGurk, M., Pettigrew, J., Santinelli, A., Mazzucchelli, R., Johnston, P.G., Montironi, R. and Waugh, D.J. (2005) Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 11, 4117- 4127.
- Sparmann, A. and Bar-Sagi, D. (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell, 6, 447-458. doi:10.1016/j.ccr.2004.09.028
- Fischer, O.M., Hart, S., Gschwind, A., Prenzel, N. and Ullrich, A. (2004) Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heaprin-binding epidermal growth factor. Molecular and Cellular Biology, 24, 5172-5183. doi:10.1128/MCB.24.12.5172-5183.2004
- Schafer, B., Gschwind, A. and Ullrich, A. (2004) Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene, 23, 991-999. doi:10.1038/sj.onc.1207278
- Itoh, Y., Joh, T., Tanida, S., Sasaki, M., Kataoka, H., Itoh, K., Oshima, T., Ogasawara, N., Togawa, S., Wada, T., Kubota, H., Mori, Y., Ohara, H., Nomura, T., Higashiyama, S. and Itoh, M. (2005) IL-8 promotes cell proliferation and migration through metalloproteinase-cleavage proHB-EGF in human colon carcinoma cells. Cytokine, 29, 275-282. doi:10.1016/j.cyto.2004.11.005
- Joh, T., Kataoka, H., Tanida, S., Watanabe, K., Ohshima, T., Sasaki, M., Nakao, H., Ohhara, H., Higashiyama, S. and Itoh, M. (2005) Helicobacter pylori-stimulated interleukin-8 (IL-8) promotes cell proliferation through transactivation of epidermal growth factor receptor (EGFR) by disintegrin and metalloproteinase (ADAM) activation. Digestive Diseases and Sciences, 50, 2081-2089. doi:10.1007/s10620-005-3011-0
- Tanida, S., Joh, T., Itoh, K., Kataoka, H., Sasaki, M., Ohara, H., Nakazawa, T., Nomura, T., Kinugasa, Y., Ohmoto, H., Ishiguro, H., Yoshino, K., Higashiyama, S. and Itoh, M. (2004) The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology, 127, 559-569. doi:10.1053/j.gastro.2004.05.017
- Chalaris, A., Adam, N., Sina, C., Rosenstiel, P., Lehmann-Koch, J., Schirmacher, P., Hartmann, D., Cichy, J., Gavrilova, O., Schreiber, S., Jostock, T., Matthews, V., Hasler, R., Becker, C., Neurath, M.F., Reiss, K., Saftig, P., Scheller, J. and Rose-John, S. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. The Journal of Experimental Medicine, 207, 1617-1624. doi:10.1084/jem.20092366
- Nanba, D., Mammoto, A., Hashimoto, K. and Higashiyama, S. (2003) Proteolytic release of the carboxy-terminal fragment of proHB-EGF causes nuclear export of PLZF. The Journal of Cell Biology, 163, 489-502. doi:10.1083/jcb.200303017
- Tanida, S., Kataoka, H., Mizoshita, T., Shimura, T., Kamiya, T. and Joh, T. (2010) Intranuclear translocation signaling of HB-EGF carboxy-terminal fragment and mucosal defense through cell proliferation and migration in digestive tracts. Digestion, 82, 145-149. doi:10.1159/000310903
- Shimura, T., Kataoka, H., Ogasawara, N., Kubota, E., Sasaki, M., Tanida, S. and Joh, T. (2008) Suppression of proHB-EGF carboxy-terminal fragment nuclear translocation: A new molecular target therapy for gastric cancer. Clinical Cancer Research, 14, 3956-3965. doi:10.1158/1078-0432.CCR-07-4794
- Ozeki, K., Tanida, S., Morimoto, C., Inoue, Y., Mizoshita, T., Tsukamoto, H., Shimura, T., Kataoka, H., Kamiya, T., Nishiwaki, E., Ishiguro, H., Higashiyama, S. and Joh, T. (2013) Telmisartan inhibits cell proliferation by blocking nuclear translocation of ProHB-EGF C-terminal fragment in colon cancer cells. PloS One, 8, e56770. doi:10.1371/journal.pone.0056770
- Goishi, K., Higashiyama, S., Klagsbrun, M., Nakano, N., Umata, T., Ishikawa, M., Mekada, E. and Taniguchi, N. (1995) Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: Conversion from juxtacrine to paracrine growth factor activity. Molecular Biology of the Cell, 6, 967-980.
- Wienen, W. (2000) A review on telmisartan: A novel, long-acting angiotensin II-receptor antagonist. Cardiovascular Drug Reviews, 18, 127-156. doi:10.1111/j.1527-3466.2000.tb00039.x
- Asakura, M., Kitakaze, M., Takashima, S., Liao, Y., Ishikura, F., Yoshinaka, T., Ohmoto, H., Node, K., Yoshino, K., Ishiguro, H., Asanuma, H., Sanada, S., Matsumura, Y., Takeda, H., Beppu, S., Tada, M., Hori, M. and Higashiyama, S. (2002) Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: Metalloproteinase inhibitors as a new therapy. Nature Medicine, 8, 35-40. doi:10.1038/nm0102-35
- Teo, K.K. (2011) Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138,769 individuals. Journal of Hypertension, 29, 623-635.
NOTES
*Competing Interest: Authors declare that no financial or other conflict of interest exists in relation to the content of the article.
#Corresponding author.