Advances in Bioscience and Biotechnology
Vol. 3 No. 7 (2012) , Article ID: 24688 , 12 pages DOI:10.4236/abb.2012.37104
Breast cancer: Small molecules targeting apoptosis, a prospective approach to safe scientific success
![]()
1School of Molecular and Cell Biology, University of the Witwatersrand, Johannesburg, South Africa
2Faculty of Science, University of South Africa, Pretoria, South Africa
3Department of Biochemistry, University of Limpopo, Sovenga, South Africa
4Department of Biology, Morgan State University, Baltimore, USA
Email: *lesetja.motadi@wits.ac.za
Received 12 August 2012; revised 13 September 2012; accepted 17 October 2012
Keywords: Breast Cancer; Apoptosis; p53; Natural Products
ABSTRACT
Breast carcinoma represents the second leading cause of cancer death in developed countries amongst women. Current cytotoxic chemotherapy plays an important role in the management of patients with hormone-insensitive or metastatic breast carcinoma, although most of them ultimately develop recurrences. Therefore, there is a need for novel targets and treatment strategies in patients with advanced breast carcinoma that is refractory to conventional chemotherapy. This paper summarizes current knowledge on breast cancer targets and molecular mechanisms that follows apoptosis induction.
1. INTRODUCTION
Breast cancer is a malignant tumour that starts in the breast tissue; it usually starts in the ducts or in lobules. It can occur in both males and females; however, male breast cancer is rare. It is the second leading cancer in the world following lung cancer, with an estimated 1.15 million cases a year [1]. The American Cancer Society estimated 230,480 cases were diagnosed in women for 2011 in the USA alone, resulting in 39,970 deaths [2]. Breast cancer is primarily a postmenopausal women’s disease, with the American National Cancer Institute’s Cancer Review estimating the average age of women at risk to be 61 years of age. Approximately 0 case have being reported for women below 20, but the figures are gradually increasing with every decade [3].
1.1. Epidemiology
The lifetime risk of an individual developing breast cancer cannot be attributed to one factor alone. A number of variables play a role in the development of breast malignancies. Even though it is fairly difficult to pinpoint one cause as the main risk factor for breast cancer, studies have shown there are certain genetic, reproductive and lifestyle factors that play a major role in its induction and progression. One of these factors is age, with studies finding that the incidence rates double with every 10 years up until menopause, where the rates start to dramatically decline [4]. Exposure to radiation and use of oral contraceptives may be associated with induction of breast cancer. Very small fraction of breast cancers could be attributed to preventable lifestyle behaviour such as obesity, alcohol consumption, lack of physical activity and long term use of hormone replacement therapy. In several studies conducted to determine the role these factors have on breast cancer incidence, the data appears to be inconsistent, hence it is hard to establish the severity these factors play in breast cancer induction that is if they even do at all [5,6]. In developed countries, incidence rates are higher relative to developing countries. However, incidence rates are steadily increasing in developing countries [7]. This increase may be attributed to the adoption of a more westernized lifestyle which encompasses a more sedentary lifestyle, delayed childbearing, high alcohol use and a diet high in fat [8,9].
In a lot of breast malignancies, hormones play a major role. It is known that estrogen starts breast tissue proliferation, and studies have now shown that the longer a woman is exposed to estrogen, the greater the risk of developing breast cancer. Hence, women with an early age at menarche (<12 years), were found to be 2.2 times more likely to develop breast cancer in the future than those with late menarche. In a different study, women who started menopause late i.e. at the age of 55, were found to have 44% higher breast cancer incidence rates than women who started menopause at 45. This may be due to a 10-year increase in the amount of estrogen the women were exposed to. Exposure to excessive estrogen plays a role in hyperestrogenia. Hyperestrogenia could trigger breast cancer in two ways: Either by breast epithelium tumour promotion or by DNA damage which could be caused by estrogen metabolites directly [10,11].
Another important factor which may increase susceptibility to breast cancer is inheritance of susceptibility genes, it has been estimated that 7% - 10% of breast cancers arise due to inherited mutated genes. The major predisposing factors to breast cancer are breast susceptibility genes, known as BRCA1 and BRCA2, which are important for early onset of breast cancer [11,12]. An initial germline mutation in one of these genes followed by a second mutation leading to the loss of function of the second allele is required for induction of tumouriginesis [13]. These genetic factors mostly show an autosomal dominant pattern of inheritance, with germline mutations rendering susceptibility to the following generation [14,15].
1.2. BRCA in Breast Cancer
Defects in DNA repair results in increased genomic instability, which can lead to malignant transformation. Additionally, defects in DNA repair renders cells sensitive to DNA damaging agents. BRCA1 and BRCA2 are important DNA repair proteins that are required for effective repair of DNA double-strand breaks by homologous re combination and DNA replication fidelity [16]. This they do by interacting with a recombinational repair protein Rad51, thereby maintaining genomic integrity [17,18]. In accordance with their role in DNA repair; a cell with a mutant BRCA gene will not be able to have a functional protein product thereby leading to defects in DNA repair mechanisms. Inherited mutations in BRCA1 and BRCA2 increase the risk of developing various cancers, especially breast and ovarian cancers. Tumours that develop in patients with inherited BRCA1/2 mutations are generally believed to be BRCA1/2 deficient. Cancer cells with BRCA1/2 deficiency are defective in DNA repair by homologous recombination and sensitive to interstrand DNA crosslinking (ICL) agents. The BRCA genes are thought to be tumour suppressors since the wild type allele is often lost in tumours of heterozygous people [19]. Though BRCA mutations have been implicated in a lot of breast cancer cases, there are still breast cancers which occur due to deregulation of other genes. A substantial proportion of susceptibility to breast cancer can be attributed to mutations in cell cycle regulation and apoptosis genes.
BRCA and Small Molecule Targets
Defects in DNA repair render cells sensitive to DNA damaging agents. BRCA1 and BRCA2 are important DNA repair proteins, hence, cancer cells with BRCA1/2 deficiency are defective in DNA repair. Several management strategies have been suggested for reduction of the cancer risk in individuals who bear the BRCA1 or BRCA2 mutations. In recent studies, small molecules have been targeted to induce DNA repair. Indole-3-carbinol (I3C) and genistein are naturally occurring chemicals derived from cruciferous vegetables and soy, respectively, with potential cancer prevention activity for hormone-responsive tumours [20]. Fan et al. showed that I3C induced BRCA1 expression and that both I3C and BRCA1 inhibit oestrogen (E2)-stimulated oestrogen receptor (ER-alpha) activity in human breast cancer cells. They further reported that a combination of both I3C and genistein induced the expression of both BRCA1 and BRCA2 genes in breast cancer cells. In another study, tamoxifen, a selective oestrogen receptor regulator, was reported to have reduced the risk of developing breast cancer among women with inherited BRCA1 or BRCA2 mutations [19].
The first human phase I study reported that a single agent therapy with olaparib, an oral selective inhibitor of PARP1, has promising antitumour activity in advanced solid tumours with either BRCA1 or BRCA2 mutations. A very recent multi-centre proof-of-concept phase II trial further demonstrated a positive result with olaparib single agent therapy in women with both advanced breast cancer and BRCA1 or BRCA2 mutations [16]. Iniparib another PARP inhibitor similar to olaparib, has been reported in a phase I study to improve the clinical benefits and survival of patients with metastatic triple-negative breast cancer, which shares clinical and pathological features with hereditary BRCA1-related breast cancer [21]. Increasing knowledge of the roles of BRCA1 and BRCA2 genes in DNA repair and the relations between their mutations and cancer predisposition suggests the therapeutic potential of strategies targeting the BRCA1 and BRCA2 defects.
2. APOPTOSIS AND BREAST CANCER
Apoptosis is a series of highly coordinated and complex processes, which multicellular organisms use to ensure elimination of defective and damaged cells [20]. Defective apoptosis has been implicated in development and progression of malignant phenotypes. Apoptosis is controlled by a number of proteins which require various triggers to be activated resulting in a cascade of signaling modules which eventually lead to cell death [22]. Based on the triggering stimulus, there are at least two main pathways that can activate apoptosis [23]. The first is the intrinsic pathway or the mitochondrial pathway. Apoptotic signals that trigger activation of this pathway will lead to a cascade of events that will result in the release of cytochrome c sequestered in the mitochondrial intermembrane space [24]. The second pathway is the extrinsic pathway, which is triggered through receptors of the tumour necrosis factor (TNF) superfamily. Both these pathways converge to a common pathway which leads to activation of a proteolytic cascade of caspases (Figure 1).
2.1. Small Molecules Targeting Extrinsic Pathway in Breast Cancer
The observation that activation of the extrinsic pathway can amplify apoptotic signals initiated by cytotoxic drugs acting primarily through the induction of mitochondrial pathway has prompted the search for strategies capable of triggering the receptor-related cascade [25]. This has led to the identification of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and its death receptors DR4 and DR5 [26]. Activation of DR4 and DR5, like that of Fas/APO, leads to activation of the initiator caspase,
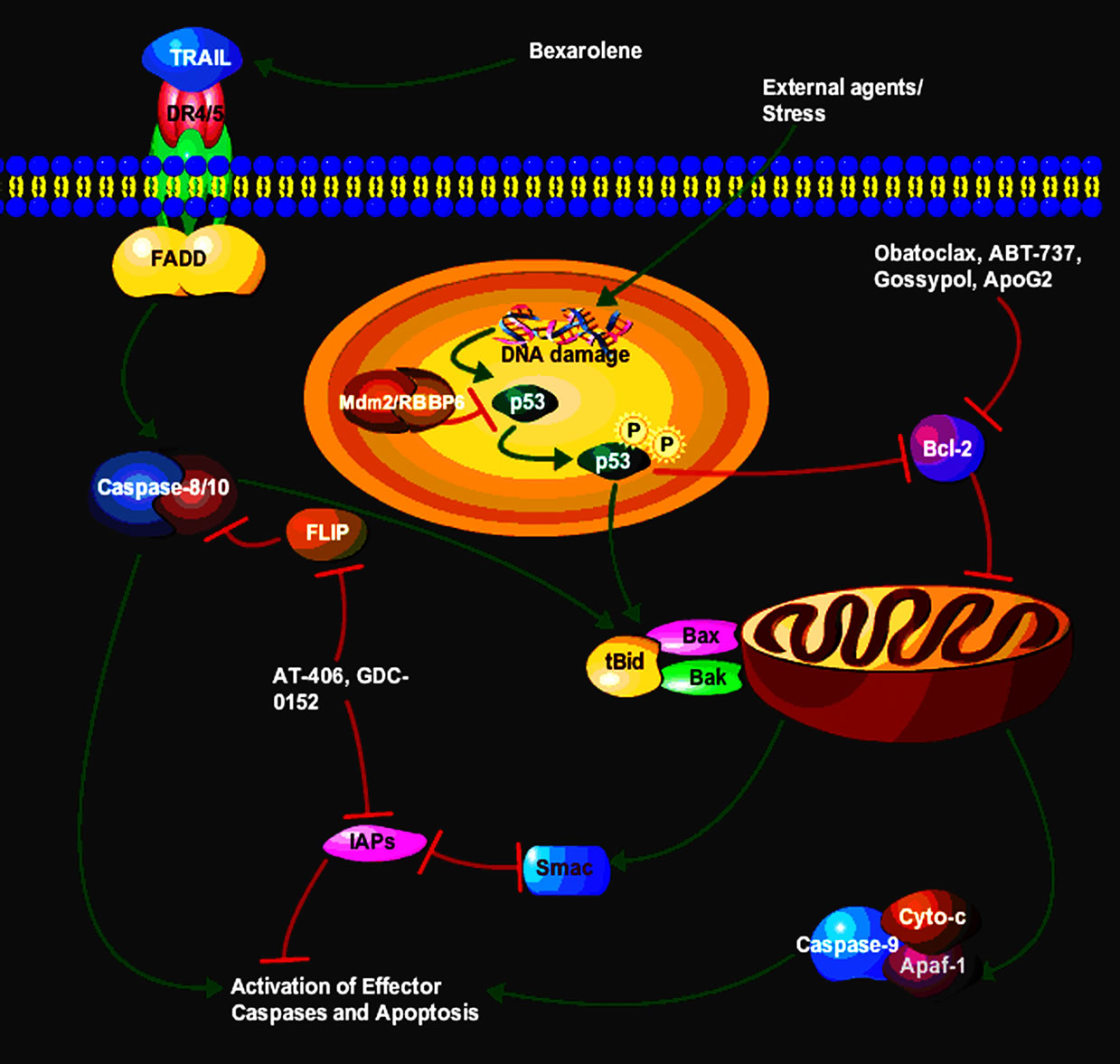
Figure 1. Proposed steps of apoptosis induced by TRAIL ligand. TRAIL ligands bind to and activate their receptors by inducing receptor trimerization. Activated receptors recruit adaptor molecules such as Fas-associating protein with death domain (FADD), which recruit procaspase 8 to the receptor complex, where it undergoes autocatalytic activation. Activated caspase 8 activates caspase 3 through two pathways; the complex one is that caspase 8 cleaves Bcl-2 interacting protein (Bid) and its COOH-terminal domain translocates to the mitochondria where it triggers cytochrome c release. The released cytochrome c binds to apoptosis protease activating factor-1 (Apaf-1) together with dATP and procaspase 9 and activates caspase 9. The caspase 9 cleaves procaspase 3 and activates caspase 3. In another pathway, p53 activation following external agents/stress results in the inhibition of pro-survival protein Bcl-2 and the activation of Bax that leads to activation of the mitochondria to release cytochrome c and caspases that end with apoptosis.
caspase-8, and its downstream targets [27]. Interestingly, TRAIL seems to exert selective toxicity towards neoplastic cells, possibly because of the presence in normal cells of high levels of decoy receptors or the caspase-8 antagonist FLICE-inhibitory protein [26]. In this context, coadministration of TRAIL has been shown to increase the lethal effects of conventional cytotoxic drugs in several cancers [28] (Figure 1).
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces programmed cell death preferentially in tumour cells. TRAIL holds enormous promise as a cancer therapeutic due to its highly selective apoptosis-inducing action on neoplastic versus normal cells Moreover, a recently published Phase I clinical trial revealed that recombinant TRAIL administration is safe and well tolerated, and that dose escalation achieved peak TRAIL serum concentrations equivalent to those associated with preclinical antitumor efficacy [29]. However, to exploit the opportunity to successfully treat cancers with TRAIL, the problems of TRAIL resistance in a variety of tumor cells must first be overcome [30]. In trying to address this setback, several natural compounds have been evaluated as possible solutions. Bexarotene, which is a retinoid specifically selective for retinoid X receptors works by stopping the growth of cancer cells. In exploiting a combinational therapy with bexarotene, Ying et al. (2007) reported that the effect of bexarotene on TRAIL-mediated programmed cell death involved proximal events of the extrinsic pathway; however, downstream signals involved the intrinsic pathway in leukemia cancer cells (Figure 1). In another study by Park et al., they observed that both DR5 and CHOP upregulation were required for γ-T3-induced apoptosis and DR5 was transcriptionally regulated by CHOP after γ-T3 treatment. In another study, apigenin induced extrinsic apoptosis pathway, up-regulating the levels of cleaved caspase-8, and inducing the cleavage of poly (ADP-ribose) polymerase. However, apigenin did not induce apoptosis via intrinsic mitochondrial apoptosis pathway since this compound did not decrease mitochondrial membrane potential maintaining red fluorescence and did not affect the levels of B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein [31]. Moreover, apigenin reduced the tyrosine phosphorylation of HER2 (phospho-HER2 level) in MCF- 7 HER2 cells, and up-regulated the levels of p53, phospho-p53 and p21 in MCF-7 vec and MCF-7 HER2 cells [31]. TRAIL receptor activating agents have been found to exhibit favourable in vitro and in vivo activity in treatment of several malignancies, including breast and gynaecological cancers. Preclinical and early phase I studies have provided some support to the assumption that these novel agents are safe, with increased target specificity for malignant cells. When these targeted agents are combined with conventional chemotherapy drugs or radiotherapy, they appear to increase cell death over single agent modalities.
2.2. Small Molecules Targeting Intrinsic Pathway in Breast Cancer
The mitochondrial pathway of apoptosis is the major mechanism of physiological cell death in vertebrates. In this pathway, proapoptotic members of the Bcl-2 family (contain only a BH3 domain) cause mitochondrial outer membrane permeabilization (MOMP), allowing the release of cytochrome c, which interacts with Apaf-1 to trigger caspase activation and apoptosis. Activation of the intrinsic apoptotic pathway represents a major mechanism for breast cancer regression resulting from anti-estrogen therapy. The BH3-only protein is inducible by estrogenstarvation and anti-estrogen treatment and plays an important role in anti-estrogen induced apoptosis of breast cancer cells [32]. In this section we present current targets of this pathway using small molecules of natural products. Recently, Jin et al. (2010) trying to exploit phytoestrogen to induce apoptosis in MCF-7, they used Daidzein which belongs to the isoflavone family, which is the most commonly ingested and most intensely studied type of phytoestrogen, often found in nuts, fruits, soybeans, and soy-based products. In their studies Jin et al. (2010) demonstrated that daidzein induces apoptosis through the generation of ROS that perturb mitochondrial function, leading to mitochondrial permeability, cytochrome c release, and caspase activation. They further reported the loss of the antiapoptotic mitochondrial Bcl-2 protein as partially responsible for the opening of the mitochondrial permeability transition pore.
In another study using Plumbagin a constituent of species of the plant genera Drosera and Plumbago which displays antineoplastic activity toward various cancers, Kawiak et al. (2012) reported antiproliferative activity of Plumbagin that was associated with apoptosis-mediated cell death, as revealed by caspase activation and an increase in the sub-G1 fraction of the cell cycle. Compound Plumbagin increased the levels of the proapoptotic Bcl-2 family of proteins and decreased the level of the antiapoptotic Bcl-2 protein in SKBR3 and BT474 cells [33]. Thus, these findings indicated that Plumbagin induces apoptosis in Her2-overexpressing breast cancers through the mitochondrial-mediated pathway and suggest its potential for further investigation for the treatment of Her2-overexpressing breast cancer (Figure 1). The mitochondrial pathway of apoptosis was further shown to play a critical role in estradiol-induced apoptosis in long-term estrogen-deprived breast cancer cells. Physiologic concentrations of estradiol could potentially be used to induce apoptosis and tumour regression in tumours that have developed resistance to aromatase inhibitors [34]. Plant-derived polyphenols inhibit cancer cell proliferation and induce apoptosis. Recently, prenylflavonoids and alkyl-phloroacetophenones have been reported for their in vitro antitumor activity [35]. In the same study Cho et al. (2012) reported cytotoxic activity as a result of treating MCF-7 cell line with prenyl (3-PAP) and geranyl (3-GAP) derivatives of phloroacetophenone, and xanthohumol (XN), a prenyl-chalcone. In addition, pro-apoptotic Bax but not B-cell lymphoma 2 (Bcl-2) expression was increased by 3-GAP. In accordance with the Bax increase, 3-GAP induced mitochondrial cytochrome c release and activated caspase-9, an initiator of the caspase cascade. Currently there are several data that support or continue to show that small molecules particularly the ones extracted from natural plants present a supplementary and safer target in many cancers.
3. THE BCL-2 FAMILY (TABLE 1)
The Bcl-2 family of proteins are one of the primary regulators of the intrinsic pathway or the mitochondrial pathway. These proteins can be characterized into proapoptotic members: Bax, Bak, Bik, Bad and Bid, with the anti-apoptotic members being Bcl-2 Mcl-1, Bcl-xl and Bcl-w. The proapoptotic members of this family work by promoting caspase activation and cell death. Some of them work by inactivating death inhibiting members of this family whilst others stimulate the release of cytochrome c from the mitochondria [36]. The anti-apoptotic members inhibit apoptosis by blocking release of cytochrome c from the mitochondria.
As our understanding of the field of apoptosis and its regulatory machinery evolves, it becomes important to identify and exploit new potential targets that restore proapoptotic activities. The most promising recent strategies in the development of anti-cancer drugs involve targeting essential apoptotic genes and proteins directly to induce cell death. Members of the Bcl-2 family of proteins and IAPs have been shown to be very important regulators of caspases. Absence or impairment of caspase activation almost always leads to prevention of cell death. Novel strategies that will overcome caspase inhibition will be of great value in cancer therapeutics that aim to directly restore a tumour cells apoptotic program. Hence, the development of small molecule agents that target IAPs and Bcl-2 appear to be attractive therapeutic target.
The proapoptotic effectors such as Bax and Bak contain BH1, BH2, and BH3, and a C-terminal transmembrane segment that selectively targets these proteins to the membranes of mitochondria and endoplasmic reticulum. The pro-survival (antiapoptotic) members Bcl- 2, Mcl-1, Bcl-xl, Bcl-w contain a BH1, BH2, and BH3, but with the exception of Mcl-1, they also contain a BH4 domain. The last group, which are the proapoptotic members including Bid, Bad, Bik, Bim, Puma, and Noxa only have a BH3 domain [37] (Figure 1).
A number of small molecules antagonists to pro-
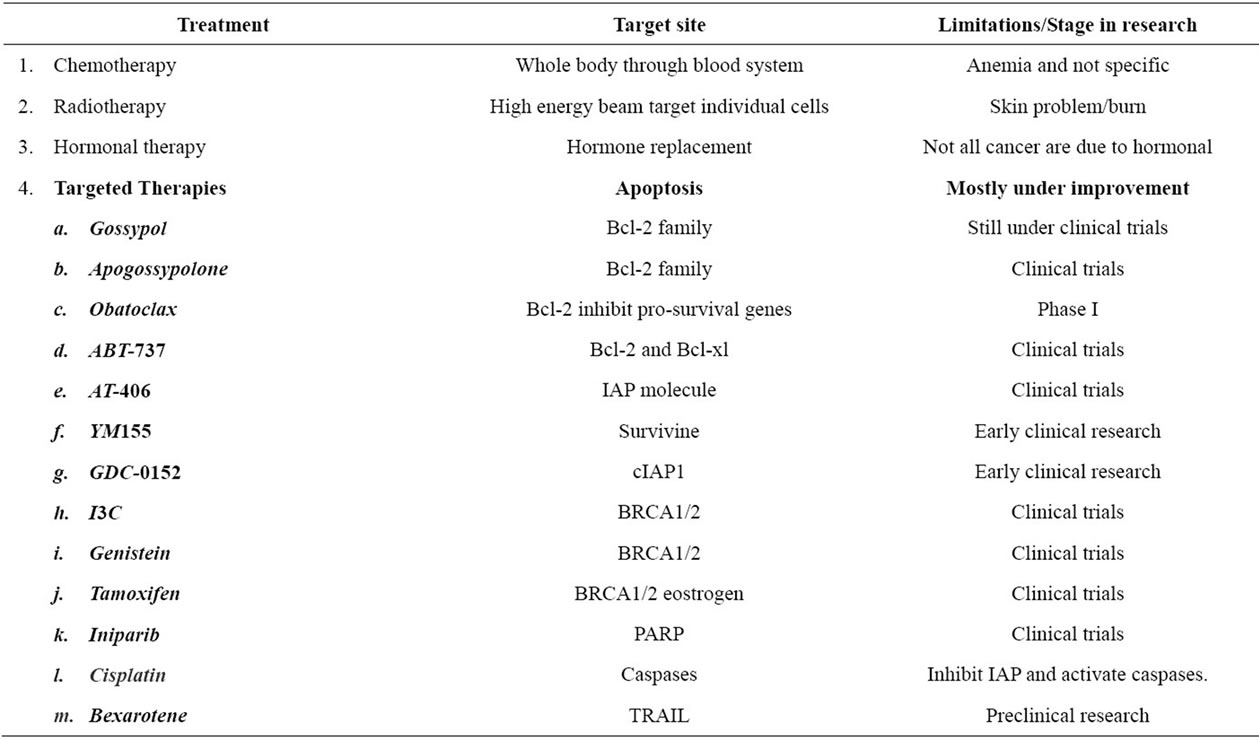
Table 1. Breast cancer current treatment strategies.
survival Bcl-2 have been developed and some are still undergoing clinical trials. Some of these include:
3.1. Gossypol
Gossypol is a naturally occurring polyphenolic compound with BH3 mimetic property. It induces apoptosis in cells with high Bcl-2 expression. It was found to have a high affinity to multiple Bcl-2 family proteins, and thus it binds to and blocks the binding of antiapoptotic Bcl-2 to pro-apoptotic Bcl-2 family proteins [38]. Gossypol has demonstrated anticancer activity against a wide range of malignancies including prostate cancer and human colorectal carcinoma [39,40]. Gossypol is currently under clinical trials.
3.2. Apogossypolone (ApoG2)
Apogossypolone is a semi synthesized analogue of gossypol. It was created to reduce the toxicity of gossypol. Apogossypolone was designed by removing two aldehyde groups on poly-phenolic rings of gossypol [41]. It was due to these reactive two aldehyde groups that gossypol was thought to exhibit toxicity [20]. Similarly to gossypol, ApoG2 binds to Bcl-2 family proteins. In a study, ApoG2 was found to exhibit better stability and was also better tolerated by mice than gossypol. It showed significant inhibition to diffuse large-cell lymphoma, a type of non-Hodgkin’s lymphoma, and was not toxic to normal peripheral blood lymphocytes [42]. Recently, a number of ApoG2 derivatives have been created such as Bl97D6. Bl97D6 inhibits binding of the BH3 peptide to the antiapoptotic Bcl-2 family proteins. It has been shown to be a potent inhibitor of human prostate cancer and human lung cancer [43].
3.3. Obatoclax
Obatoclax is a small molecule novel drug designed to inhibit prosurvival Bcl-2 family proteins and thereby increase cellular threshold for apoptosis in cancer cells. It acts as a BH3 mimetic and binds to prosurvival Bcl-2 proteins and neutralizes them [44]. In a study, obatoclax showed significant cell viability reductions in human pancreatic cell lines [44]. In phase I trials obatoclax exhibited antitumour activity in patients with refractory leukemia and myelodysplasia [45].
3.4. ABT-737
ABT-737 is a small molecule inhibitor of the anti-apoptotic proteins Bcl-2 and Bcl-xl but not Mcl-1. A study by Oltersdorf (2005) found that ABT-737 does not directly induce apoptosis but sensitizes cells to death signals, showing synergistic cytotoxicity with chemotherapeutic treatment and radiation. ABT-737 exhibits cytotoxicity against lymphoma, multiple myeloma and small-cell lung carcinoma lines [46,47].
4. CASPASE IN BREAST CANCER
Caspases are a class of cysteine-aspartyl proteases that are synthesized as inactive zymogens. They lie dormant in a healthy cell then due to cell death stimuli, they are proteolytically cleaved at two aspartate residues, generating mature active enzymes [48,49]. The generation of active caspases forms a cascade whereby initiator caspases interact with a specialized adapter molecule which results in its own activation and eventually activation of execution caspases which are responsible for the final pathway of apoptosis [35,48]. Since all apoptotic pathways converge on caspases it renders them crucial effecttors of apoptosis. Hence, it is very crucial that expression of caspases is tightly controlled. Deregulation of caspases has been implicated in a number of diseases. Overexpression has been linked to certain neurodegenerative disease and ischemia-associated injury, whilst their suppression has been linked to a number of cancers.
4.1. Caspase 3 in Breast Cancer
The role of caspase-3 in the response of breast cancer cells to chemotherapeutic drugs remains a controversial issue. The loss of caspase-3 expression as well as defaults in cytochrome c release from the mitochondria, which is required in most apoptotic pathways to activate caspase-3 through caspase-9 activation, are associated with multidrug resistance. Accordingly, expression of caspase-3 in the human MCF-7 breast tumor cell line (which is deficient for caspase-3) restores the apoptotic response to the topoisomerase II inhibitor, etoposide [50]. Caspase-3 was also involved in breast cancer cell apoptosis upon exposure to anthracyclines and cisplatin [27,51]. Its role in tumor cell response to paclitaxel has been challenged [11].
4.2. Initiator Caspases (8 and 9) in Breast Cancer
The initiator caspases appear to display some specificity according to the type of apoptotic signal. Two main activation cascades for apoptosis induction have been described [52]. Fas receptor-ligand interactions use caspase-8 activation to trigger the downstream executioner caspases. An alternative mitochondrial pathway, which is triggered by various anticancer agents, involves activetion of caspase-9 upon recruitment to the mitochondria by cytochrome c and apoptosis protease activation factor-1(APAF-1) [47]. Mitomycin c which is one of the anticancer agents, led to an exclusive activation of caspase-8 but not of caspase-9 in comparison to various other drugs with differing mechanisms of action at concentrations formerly established as equitoxic in the NCI/ADR-RES cell line [47]. In a contrary study by Lee at al using Styrylpyrone derivative (SPD) which is a pharmacologically active compound extracted from the plant Goniothalamus sp. of the Annonaceae family have shown that procaspase-8 was not activated in MCF-7 cells but caspase-9 activation was detected in response to SPD treatment, with the release of cytochrome c into the cytosol. In another study, prodigiosin which is a red pigment produced by Serratia marcescens with proapoptotic activity was potently cytotoxic in both estrogen receptor positive (MCF-7) and negative (MDA-MB-231) breast cancer cell lines. It lead to cytochrome c release, activation of caspases-9, -8 and -7 and cleavage of poly (ADPribose) polymerase protein typified the apoptotic event and caspase inhibition revealed that PG acts via the mitochondrial pathway [53].
5. P53 IN BREAST CANCER
The p53 tumor suppressor gene prevents tumorigenesis in response to physiological and environmental stress and plays a role in cell cycle progression, apoptosis and repair of DNA damage. p53 has be involved in transcriptional regulation of pro-apoptotic genes associated with intrinsic and extrinsic pathways [54]. The activation of the p53 pathway leads either to cell cycle arrest or to apoptosis. Activated p53 increases the expression of p21 in DNA damaged cells and affects expression of p27 [42]. The up-regulation of cyclin dependent kinase inhibitors (CDKI), such as p21 and p27, is related to cell cycle arrest and contributes to down-regulation of cyclin D1, cyclin E, cyclin-dependent kinases (cdks) [55]. The activation of p53 induces up-regulation of pro-apoptotic Bax but not anti-apoptotic Bcl-2. Also, p53-mediated apoptosis involves the activation of Fas and the intrinsic mitochondrial pathway, which results in activation of caspase-8 and -9 (Figure 1) [56]. So, targeting p53 in cancer not only holds a promise to cancer eradication but also guarantees a safe elimination of those cells.
Many observations have implicated p53 signaling pathway in the transcriptional activation of Bax in apoptosis, but also in the alteration of the Bcl-xL/Bax ratio, thereby indicating that the ratio between these proand anti-apoptotic proteins is one of the important factors deciding the fate of a cell [52,57]. Like many new plant extracts, the role of curcumin a naturally occurring phytochemical responsible for the yellow colour of the commonly used spice turmeric in regulating the balance between these proand anti-apoptotic factors was tested and it revealed that curcumin induced apoptosis with an increase in p53 level in MCF-7 cell lines. Interestingly, the curcumin-induced increase in p53 expression precedes that of Bax thereby leading to believe that p53 transactivates Bax expression [52]. In the same study, the Bcl-xl levels remained almost unchanged thereby shifting the Bcl-xl/Bax ratio towards apoptosis. In recent times several small molecules have been identified e.g., Nutlin-3a and MI-219 that disrupt MDM2-p53 interaction resulting in inhibition of tumor growth, however they are less effective in cancer cells that express high levels of MDMX. Another molecule called benzofuroxan derivative following treatments of MCF-7 cells with this smallmolecule MDMX inhibitor activated p53, resulting in elevated expression of proapoptotic genes (e.g., PUMA, BAX, and PIG3). Importantly, this novel small-molecule p53 activator caused MCF-7 cells to undergo apoptosis, and acted additively with Nutlin-3a to activate p53 and decrease the viability of cancer cells [58]. Table 2 shows other small molecules that are involved in p53-induced apoptosis.
6. INHIBITORS OF APOPTOSIS PROTEINS (IAP)
The IAP’s were discovered in baculoviruses and these proteins are known to biochemically function by suppressing the host’s cell death response to a viral infection thereby allowing viral propagation in host insect cells [47,59]. Several other cellular homologous IAP’s were also identified in a range of species such as Drosophila to vertebrates. Neuronal Apoptosis Inhibitory Protein (NAIP) was the first mammalian IAP to be identified during positional cloning in an effort to identify the causative gene for Spinal Muscular Atrophy (SMA) [47]. Other members of mammalian inhibitor of apoptosis family of proteins, include the X chromosome-linked IAP (XIAP), cellular IAP 1 (cIAP1), cellular IAP II (cIAP2), melanoma IAP (ML-IAP), Survivin, Bruce, ILP-2 and Livin to name but a few. These are overexpressed in cancer cells thereby conferring cancer cell protection against a variety of proapoptotic stimuli by

Table 2. List of small molecule agents that target Bcl-2 and mode of action.
functionally regulating signal transduction pathways associated with malignancy [60,61].
Structurally, the IAPs are similar with one or more 70 - 80 amino acid baculovirus IAP repeat (BIR) domains with the core domain consiting of the variable sequence C(X)2C(X)6W(X)3D(X)5H(X)6C with the X representing any amino acid [46]. A typical mammalian IAP family member contains a carboxy-terminal RING (Really Interesting New Gene) zinc finger possessing a E3 ubiquitin ligase activity which directly regulates self-ubiquitination and protein degradation and the cellular IAP (cIAP 1 and cIAP 2) uniquely possesses the caspase recruitment domain (CARD) which mediates oligomerization with other CARD containing proteins. Although the mechanism of action is not known, the CARD in IAP may form protein-protein interactions with protease activating factor-1 (Apaf-1), and with some of the death domain (DD) or death effector domain (DED) containing proteins [35] (Figure 2).
The fact that this family of proteins, when over-expressed in human cells protects cells from stimuli that induces apoptosis provides the mechanism and way to explain treatment resistance in cancer cells [61]. One strategy to overcome this is to silence IAPs and this in turn can sensitize cells to apoptotic stimuli. Other efforts involve targeting the IAP proteins by designing small molecules that can mimic the binding of the endogenous IAP antagonist second mitochondria-derived activator of caspases/direct IAP-binding protein, with low pI (Smac/ DIABLO) to a shallow groove on the surface of select IAP baculovirus repeat (BIR) domain, thereby inhibiting the inhibitor of apoptosis (IAP) [47,59].
IAP in Apoptosis and Cancer
Cancer cells thus temper with the equilibrium of cell death and cell growth in order to survive. Apoptosis regulators such as IAPs play a fundamental role in oncogenesis because they suppress the intrinsic and extrinsic apoptosis pathway. This they do via the central mechanisms of IAP apoptotic suppression through direct caspase and pro-caspase inhibition (primarily caspase 3 and 7) in a cascade manner and modulation of and by the transcription factor NF-kappaB [62]. In many cancers IAP’s are over-expressed or over-activated, conferring cells to no longer be able to die in resistance to standard chemo and radiation therapies which in turn have a negative effect on normal healthy cells because DNA is damaged chemically and physically [47,63]. Novel yet effecttive and “safe” therapeutic targets are therefore required for the treatment of cancer.
In search for possible targeted markers that inhibit expression of IAPs, Smac/DIABLO was discovered in 2000, as a protein released from the mitochondria into the
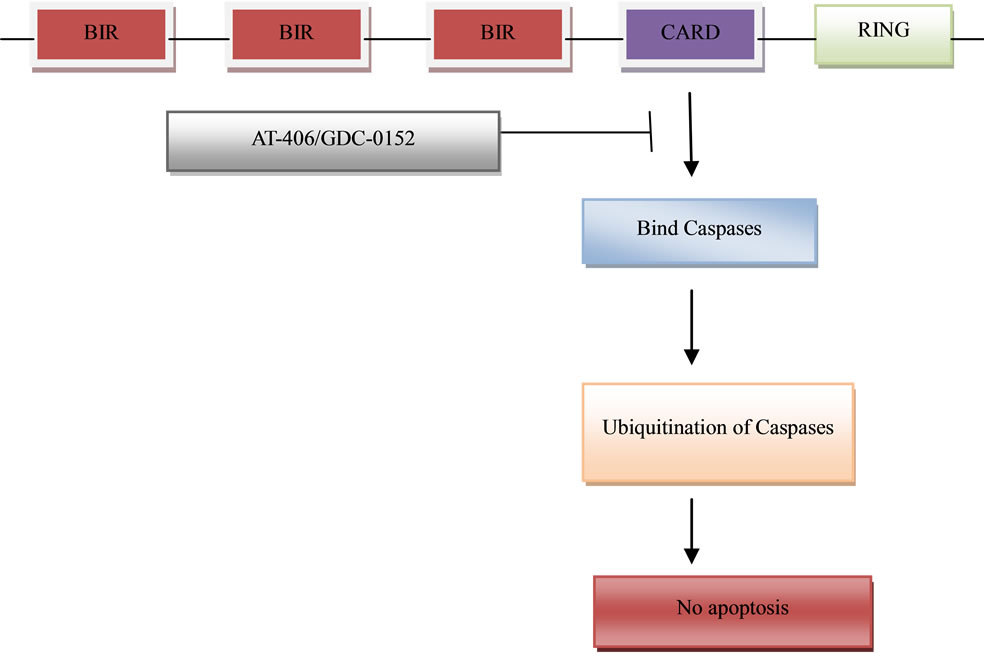
Figure 2. Domain structure of IAP proteins. Pro-survival proteins bear up to three domains BIR, CARD and RING. The IAP family mediated caspase-ubiquitination pathway that then results in the pro-survival of the cell and its escape to cell death by apoptosis. The BIR domain is necessary for IAP protein interaction with a number of proapoptotic factors, including invertebrate death inducers. Several small molecules have been identified that result in the inhibition of the binding of caspases to the CARD region on the IAP thereby resulting in activation of apoptosis.
cytosol in response to apoptotic stimuli. It functions as an endogenous antagonist of several inhibitors of apoptosis proteins through direct binding. The interaction between Smac and IAPs involves the AVPI tetrapeptide binding motif on the N-terminus of Smac and a well-defined groove on the surface of these IAP proteins, providing an ideal site for the design of small-molecule Smac mimetics [13,64]. Potent and cell-permeable small-molecule Smac mimetics have provided powerful pharmacological tools for study of the regulation of apoptosis by IAP proteins, and several such compounds are now in early clinical trials as new anticancer agents. One focus has been on the generation of molecules that mimic the aminoterminus of mature Smac. AT-406 which is an orally-active, small molecule drug was designed to promote programmed cell death (apoptosis) in tumour cells by blocking the activity of IAPs thereby creating conditions in which apoptosis can proceed. AT-406 is considered a multi-IAP antagonist.
IAPs are key components of the complex cascade of protein signaling that activates enzymes called caspases to initiate the breakdown of cancer cells [64,65]. AT-406 is thought to mimic the activity of Smac by binding to XIAP and preventing it from inhibiting caspase activetion. Upon binding to cIAP1 and cIAP2, AT-406 induces rapid degradation of these proteins and promotes apoptosis through activation of the death-receptor complex and caspase 8 [13] The other molecule, GDC-0152 has the best profile of these compounds; it binds to the XIAP BIR3 domain, the BIR domain of ML-IAP, and the BIR3 domains of cIAP1 and cIAP2. GDC-0152 achieves apoptosis induction by promoting degradation of cIAP1, inducing activation of caspase-3/7, and leads to decreased viability of breast cancer cells without affecting normal mammary epithelial cells [64]. The second member of the IAP that has also been targeted is survivin. Survivin protein was found to be expressed in all the most common human carcinomas but not in normal tissue. YM155, a small-molecule survivin suppressor resulted in suppression of survivin expression, caspase-3/7 activation, induction of spontaneous apoptosis, and growth inhibition. In another study by Zhang et al. (2012), valproic acid which is a broad-spectrum inhibitor of class I and II histone deacetylases and shows great anticancer activity in a variety of human cancers including breast cancer was found to significantly inhibit cell migration but not proliferation of human breast cancer MDA-MB-231 cells. Ribonuclease expression was also found to promote apoptosis in human breast cells MDA-MB-231 by inhibiting the expression of Survivin and activating caspase-3 [66]. The novel findings of this review is that multiple IAPs are concomitantly expressed in breast cancers, and that, in combination with clinically relevant IAP antagonists promote apoptosis and reduce the cell turnover index of breast cancers.
7. FUTURE FOCUS ON BREAST CANCER
It is now recognized that the mechanism of action of chemotherapeutic drugs often involves the induction of cancer cell apoptosis, and that apoptosis resistance is a major contributing factor in chemotherapeutic drug resistance. Therefore, restoring apoptosis signalling in cancer cells with targeted therapeutics has enormous potential to improve the outcome of cancer chemotherapy by reversing a major mechanism of drug resistance. In this review, we assessed the outlook for improving the outcome of cancer therapy by targeting different apoptotic genes using small molecules and exploring the possibility of using plant extracts as a target to induce apotosis. Novel modalities of cancer therapies that improves the efficacy of apoptosis as well as chemotherapeutic drugs and lessen the toxicity of these agents by targeting specific genes would be an improvement to the current status of available drugs. Moreover, upregulation of the pro-apoptotic signaling proteins or suppression of specific anti-survival signaling pathways by agents directed at increasing pro-apoptotic proteins may acutely induce chemosensitization of resistant cancer cells.
8. CONCLUSION
Although the mechanism that used to triggers apoptosis in breast cancer using small molecules is still unclear, the identified mechanism of cell death provides a convenient window for further defining the upstream events and elucidating the mechanisms. Finally, the fact that the proapoptotic action are independent of p53 further emphasizes that small molecule analogues are potentially useful to the development of new therapeutics that induce apoptosis in breast cancer.
9. ACKNOWLEDGEMENTS
This works was supported by grant from the National Research Foundation, Carnaige and URC.
![]()
![]()
REFERENCES
- Parkin, D.M., Sitas, F., Chirenje, M., Stein, L., Abratt, R. and Wabinga, H. (2008) Part I: Cancer in indigenous Africans—Burden, distribution and trends. Lancet Oncology, 9, 683-692. doi:10.1016/S1470-2045(08)70175-X
- Park, S.J., Wu, C.H., Choi, M.R., Najafi, F., Emami, A. and Safa, A.R. (2006) P-glycoprotein enhances trailtriggered apoptosis in multidrug resistant cancer cells by interacting with the death receptor DR5. Biochemical Pharmacology, 72, 293-307. doi:10.1016/j.bcp.2006.04.024
- Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J., Murray, T. and Thun, M.J. (2008) Cancer statistics. A Cancer Journal for Clinicians, 58, 71-96. doi:10.3322/CA.2007.0010
- Nachmias, B., Ashhab, Y. and Ben-Yehuda, D. (2004) The inhibitor of apoptosis protein family (IAPs): An emerging therapeutic target in cancer. Seminars in Cancer Biology, 14, 231-243. doi:10.1016/j.semcancer.2004.04.002
- Chen, T., Pengetnze, Y. and Taylor, C.C. (2005) Src inhibition enhances paclitaxel cytotoxicity in ovarian cancer cells by caspase-9-independent activation of caspase-3. Molecular Cancer Therapeutics, 4, 217-224.
- McPherson, K., Steel, C.M. and Duion, J.M. (2000) Breast cancer-epidemiology, risk factors and genetics. British Medical Journal, 321, 624-628. doi:10.1136/bmj.321.7261.624
- Kitada, S., Leone, M., Sareth, S., Zhai, D., Reed, J.C. and Pellecchia, M. (2003) Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-Cell lymphocyte/leukemia-2 proteins. Journal of Medicinal Chemistry, 46, 4259-4264. doi:10.1021/jm030190z
- Bray, F., McCarron, P. and Parkin, D.M. (2004) The changing global patterns of female breast cancer incidence and mortality. Breast Cancer Research, 6, 229-239. doi:10.1186/bcr932
- Shelley, M.D., Hartley, L., Groundwater, P.W. and Fish, R.G. (2000) Structure-activity studies on gossypol in tumour cell lines. Anticancer Drugs, 11, 209-216. doi:10.1097/00001813-200003000-00009
- Chen, M. and Wang, J. (2002) Initiator caspases in apoptotis signaling pathways. Apoptosis, 7, 313-319. doi:10.1023/A:1016167228059
- JunWei, J., Stebbins, J.L., Kitada, S., Dash, R., Zhai, D., Placzek, W.J., Wu, B., Rega, M.F., Zhang, Z., Barile, E., Yang, L., Dahl, R., Fisher, P.B., Reed, J.C. and Pellecchia, M. (2011) An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic Bcl-2 family proteins. Cancer Molecular Targets and Therapeutics, 1, 1-14.
- Ghobrial, I.M., Witzig, T.E. and Adjei, A.A. (2005) Targeting apoptosis pathways in cancer therapy. A Cancer Journal for Clinicians, 55, 178-194. doi:10.3322/canjclin.55.3.178
- Key, T.J., Verkasalo, P.K. and Banks, E. (2001) Epidemiology of breast cancer. The Lancet Oncology, 2, 133- 140. doi:10.1016/S1470-2045(00)00254-0
- Lewis, J.S., Meeke, K., Osipo, C., Ross, E.A., Kidawi, N., Li, T., Bell, E., Chandel, N.S. and Jordan, V.C. (2005) Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. Journal of the National Cancer Institute, 97, 1746-1759. doi:10.1093/jnci/dji400
- Tomek, S., Koestler, W., Horak, P., Grunt, T., Brodowicz, T., Pribill, I., Halaschek, J., Haller, G., Wiltschke, C., Zielinski, C.C. and Krainer, M. (2003) Trail-induced apoptosis and interaction with cytotoxic agents in soft tissue sarcoma cell lines. European Journal of Cancer, 39, 1318-1329. doi:10.1016/S0959-8049(03)00227-2
- Tutt, A., Robson, M., Garber, J.E., Domchek, S.M., Audeh, M.W., Weitzel, J.N., Friedlander, M., Arun, B., Loman, N., Schmutzler, R.K., Wardley, A., Mitchell, G., Earl, H., Wickens, M. and Carmichael, J. (2010) Oral poly(ADPribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet, 376, 235-244. doi:10.1016/S0140-6736(10)60892-6
- Welsch, P.L. and King, M. (2001) BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Human Molecular Genetics, 10, 705-713. doi:10.1093/hmg/10.7.705
- Wu, C.H., Kao, C.H. and Safa, A.R. (2008) TRAIL recombinant adenovirus triggers robust apoptosis in multidrug-resistant Hl-60/Vinc cells preferentially through death receptor DR5. Human Gene Therapy, 19, 731-143. doi:10.1089/hum.2008.001
- Soto-Cerratoa, V., Llagosteraa, E., Montanera, B., Schefferb, G.L. and Perez-Tomas, R. (2004) Mitochondria-mediated apoptosis operating irrespective of multidrug resistance in breast cancer cells by the anticancer agent prodigiosin. Biochemical Pharmacology, 68, 1345-1352. doi:10.1016/j.bcp.2004.05.056
- Park, S.K., Sanders, B.G. and Kline, K. (2010) Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Research and Treatment, 124, 361-375. doi:10.1007/s10549-010-0786-2
- Foster, F.M., Owens, T.W., Tanianis-Hughes, J., Clarke, R.B., Brennan, K., Bundred, N.J. and Streuli, C.H. (2009) Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Research, 11, R41.
- Lorincz, A.M. and Sukumar, S. (2006) Molecular links between obesity and breast cancer. Endocrine-Related Cancer, 13, 279-292. doi:10.1677/erc.1.00729
- Loubser, F., Edge, J. and Fieggen, K. (2008) Epidemiology, risk factors and genetics of breast cancer. Continuing Medical Education, 26, 497-501.
- Boatright, K.M. and Salvesen, G.S. (2003) Mechanisms of caspase activation. Current Opinion in Cell Biology, 15, 725-731. doi:10.1016/j.ceb.2003.10.009
- Turner, N., Tutt, A. and Ashworth. A. (2005) Targeting the DNA repair defect of BRCA tumours. Current Opinion in Pharmacology, 5, 388-393. doi:10.1016/j.coph.2005.03.006
- Tutt, A. and Ashworth, A. (2002) The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends in Molecular Medicine, 8, 571-576. doi:10.1016/S1471-4914(02)02434-6
- Erickson, R.I., Tarrant, J., Cain, G., Lewin-Koh, G.-C., Dybdal, N., Wong, H., Blackwood, E., West, K., Steigerwalt, R., Mamounas, M., Flygare, J.A, Amemiya, K., Dambach, D., Fairbrother, W.J. and Diaz, D. (2012) Toxicity profile of small-molecule IAP antagonist GDC- 0152 is linked to TNF-alpha pharmacology. Toxicological Sciences, 1-44.
- Hedau, S., Jain, N., Husain, S.A., Mandal, A.K., Ray, G., Shahid, M., Kant, R., Gupta, V., Shukla, N.K., Deo, S.S.V. and Das, B.C. (2004) Novel germline mutations in breast cancer susceptibility genes BRCA1, BRCA2 and p53 gene in breast cancer patients from India. Breast Cancer Research and Treatment, 88, 177-186. doi:10.1007/s10549-004-0593-8
- Volate, S.R., Kawasaki, B.T., Hurt, E.M., Milner, J.A. Kim, Y.S., White, J. and Farrar, W.L. (2010) Gossypol induces apoptosis by activating p53 in prostate cancer cells and prostate tumor-initiating cells. Molecular Cancer Therapeutics, 9, 1-17. doi:10.1158/1535-7163.MCT-09-0507
- Kasibhatla, S. and Tseng, B. (2003) Why target apoptosis in cancer treatment? Molecular Cancer Therapeutics, 2, 573-580.
- Seo, S.B., Hur, J.G., Kim, M.J., Lee, J.W., Kim, H.B., Bae, J.H., Kim, D.W., Kang, C.D. and Kim, S.H. (2010) TRAIL sensitizes MDR cells to MDR-related drugs by down-regulation of P-glycoprotein through inhibition of DNA-PKcs/Akt/GSK-3β pathway and activation of caspases. Molecular Cancer, 3, 1658.
- Warr, M.R. and Shore, G.C. (2008) Small-molecule Bcl-2 antagonists as targeted therapy in oncology. Current Oncology, 15, 256-261.
- Bender, C.E., Fitzgerald, P., Tait, S.W.G., Llambi, F., McStay, G.P., Tupper, D.O., Pellettieri, J., Alvarado, A.S., Salvesen, G.S. and Green, D.R. (2012) Mitochondrial pathway of apoptosis is ancestral in metazoans. Proceedings of the National Academy of Sciences, 109, 1-6.
- Porter, P. (2008) “Westernizing” women’s risks? Breast cancer in lower-income countries. The New England Journal of Medicine, 358, 213-216. doi:10.1056/NEJMp0708307
- Satoh, K., Kaneko, K., Hirota, M., Masamune, A., Satoh, A. and Shimosegawa, T. (2001) Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer, 92, 271-278. doi:10.1002/1097-0142(20010715)92:2<271::AID-CNCR1319>3.0.CO;2-0
- Schimmer, A.D., O’Brien, S., Kantarjian, H., Brandwein, J., Cheson, B.D., Minden, M.D., Yee, K., Ravandi, F., Giles, F., Schuh, A., Gupta, V., Andreeff, M., Charles Koller, C., Chang, H., Kamel-Reid, S., Berger, M., Viallet, J. and Borthakur, G. (2008) A Phase I study of the panBcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clinical Cancer Research, 14, 8295-8301. doi:10.1158/1078-0432.CCR-08-0999
- Fan, S., Meng, Q., Auborn, K., Carter, T. and Rosen, E.M. (2006) BRCA1 and BRCA2 as molecular targets for phytochemicals indole-3-carbinol and genistein in breast and prostate cancer cells. British Journal of Cancer, 94, 407-426. doi:10.1038/sj.bjc.6602935
- Powell, S.N. and Kachnic, L.A. (2003) Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene, 22, 5784-5791. doi:10.1038/sj.onc.1206678
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. (2002) Molecular biology of the cell: Programmed cell death. 4th Edition, Garland Science, New York.
- Kishimoto, S., Kawazoe, Y., Ikeno, M., Fukushima, S. and Takeuchi, Y. (2005) Continuous exposure to low-dose cisplatin and apoptosis. Biological & Pharmaceutical Bulletin, 28, 1954-1957. doi:10.1248/bpb.28.1954
- King, M.C., Wieand, S., Hale, K., Lee, M., Walsh, T., Owens, K., Tait, J., Ford, L., Dunn, B.K., Costantino, J., Wickerham, L., Wolmark, N. and Fisher, B. (2001) Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) breast cancer prevention trial. The Journal of the American Medical Association, 286, 2251-2256. doi:10.1001/jama.286.18.2251
- Peeters, P.H.M., Verbeek, A.L.M., Krol, A., Matthyssen, M.M.M. and Waard, F. (1994) Age at menarche and breast cancer risk in nulliparous women. Breast Cancer Research and Treatment, 33, 55-61. doi:10.1007/BF00666071
- Imyanitov, E.N. and Hanson, K.P. (2004) Mechanisms of breast cancer. Discovery Today: Disease Mechanisms, 1, 235-245. doi:10.1016/j.ddmec.2004.09.002
- Cho, M.Y., Park, S.Y., Park, S., Lee, Y.R., Han, G.D. and Kim, J.A. (2012) Geranyl derivative of phloroacetophenone induces cancer cell-specific apoptosis through Bax-mediated mitochondrial pathway in MCF-7 human breast cancer cells. Biological & Pharmaceutical Bulletin, 35, 98-104. doi:10.1248/bpb.35.98
- Flygare, J.A., Beresini, M., Budha, N., Chan, H., Chan, I. T., Cheeti, S., Cohen, F., Deshayes, K., Doerner, K., Eckhardt, S.G., Elliott, L.O., Feng, B., Matthew, C., Franklin, M.C., Reisner, S.F., Gazzard, L., Halladay, J., Hymowitz, S.G., La, H., LoRusso, P., Maurer, B., Murray, L., Plise, E., Quan, C., Jean-Philippe Stephan, J.P., Young, S.G., Tom, J., Tsui, V., Um, J., Varfolomeev, E., Vucic, D., Wagner, A.J., Wallweber, H.J.A., Wang, L., Ware, J., Wen, Z., Wong, H., Wong, J.M., Wong, M., Wong, S., Yu, R., Zobel, K. and Fairbrother, W.J. (2012) Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). Journal of Medicinal Chemistry, 55, 4101-4113. doi:10.1021/jm300060k
- Herbst, R.S., Eckhardt, S.G., Kurzrock, R., Ebbinghaus, S., O’Dwyer, P.J., Gordon, M.S., Novotny, W., Goldwasser, M.A., Tohnya, T.M., Lum, B.L., Ashkenazi, A., Jubb, A.M. and Mendelson, D.S. (2010) Phase I doseescalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. Journal of Clinical Oncology, 28, 2839- 2846. doi:10.1200/JCO.2009.25.1991
- Gartner, E.M., Burger, A.M. and Lorusso, P.M. (2010) Poly (adp-ribose) polymerase inhibitors: A novel drug class with a promising future. The Cancer Journal, 16, 83- 90. doi:10.1097/PPO.0b013e3181d78223
- Liggins, J., Mulligan, A., Runswick, S. and Bingham, S.A. (2002) Daidzein and genistein content of cereals. European Journal of Clinical Nutrition, 56, 961-966. doi:10.1038/sj.ejcn.1601419
- Huang, S., Okumura, K. and Sinicrope, F.A. (2009) BH3 mimetic obatoclax enhances TRAIL-mediated apoptosis in human pancreatic cancer cells. Clinical Cancer Research, 15, 150-159. doi:10.1158/1078-0432.CCR-08-1575
- Jin, S., Zhang, Q.Y., Kang, X.M., Wang, J.X. and Zhao, W.H. (2010) Daidzein induces MCF-7 breast cancer cell apoptosis via the mitochondrial pathway. Annals of Oncology, 21, 263-268. doi:10.1093/annonc/mdp499
- Dean, E.M., Ranson, M., Blackhall, F., Holt, S.V. and Dive, C. (2007) Novel therapeutic targets in lung cancer: Inhibitor of apoptosis proteins from laboratory to clinic. Cancer treatment reviews, 33, 203-212. doi:10.1016/j.ctrv.2006.11.002
- Hougardy, B.M.T., Maduro, J.H., van der Zee, A.G.J., Willemse, P.H.B., de Jong, S. and de Vries, E.G.E. (2005) Clinical potential of inhibitors of survival pathways and activators of apoptotic pathways in treatment of cervical cancer: Changing the apoptotic balance. The Lancet, 6, 589-598. doi:10.1016/S1470-2045(05)70281-3
- Zhou, J.H., Tang, X.Y., Zhao, R., Wang, H. and Xia, J. (2012) Effects of ribonuclease inhibitor on apoptosis and invasion of human breast cancer MDA-MB-231 cells. Chinese Journal of Cellular and Molecular Immunology, 28, 260-264.
- Kawiak, A., Zawacka-Pankau, J. and Lojkowska, E. (2012) Plumbagin induces apoptosis in her2-overexpressing breast cancer cells through the mitochondrial-mediated pathway. Journal of Natural Products, 75, 747-751. doi:10.1021/np3000409
- von Haefen, C., Wieder, T., Essmann, F., Schulze-Osthoff, K., Dorken, B. and Daniel, P.T. (2003) Paclitaxelinduced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8-driven mitochondrial amplification loop. Oncogene, 22, 2236-2247. doi:10.1038/sj.onc.1206280
- Altekruse, S.F., Kosary, C.L., Krapcho, M., Neyman, N., Aminou, R., Waldron, W., Ruhl, J., Howlader, N., Tatalovich, Z., Cho, H., Mariotto, A., Eisner, M.P., Lewis, D.R., Cronin, K., Chen, H.S., Feuer, E.J., Stinchcomb, D.G. and Edwards, B.K (2009) SEER cancer statistics review, 1975-2007. National Cancer Institute.
- Gonzalez de Aguilar, J.L., Gordon, J.W., Rene, F., de Tapia, M., Lutz-Bucher, B., Gaiddon, C. and Loeffler, J.P. (2000) Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: Evidence for the implication of the p53 signaling pathway. Neurobiology of Disease, 7, 406-415. doi:10.1006/nbdi.2000.0295
- Hunter, A.M., LaCasse, E.C. and Korneluk, R.G (2007) The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis, 12, 1543-1568. doi:10.1007/s10495-007-0087-3
- Ambrosini, G., Adida, C., Sirugo, G. and Altieri, D.C. (1998) Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting. Journal of Biological Chemistry, 273, 11177-11182. doi:10.1074/jbc.273.18.11177
- [61] Balabhadrapathruni, S., Thomas, T.J., Yurkow, E.J., Amenta, P.S. and Thomas, T. (2000) Effects of genistein and structurally related phytoestrogens on cell cycle kinetics and apoptosis in MDA-MB-468 human breast cancer cells. Oncology Reports, 7, 3-12.
- [62] Bodet, L., Gomez-Bougie, P., Touzeau, C. Dousset, C., Descamps, G., Maïga, S., Avet-Loiseau, H., Bataille, R., Moreau, P., Le Gouill, S., Pellat-Deceunynck, C. and Amiot, M. (2011) ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood, 118, 3901-3910. doi:10.1182/blood-2010-11-317438
- [63] Butler, L.M., Potischman, N.A., Newman, B., Millikan, R.C., Brogan, D., Gammon, M.D., Swanson, C.A. and Brinton, L.A. (2000) Menstrual risk factors and earlyonset breast cancer. Cancer Causes and Control, 11, 451- 458. doi:10.1023/A:1008956524669
- [64] Choudhuria, T., Pala, S., Agwarwalb, M.L., Dasa, T. and Saa, G. (2002) Curcumin induces apoptosis in human breast cancer cells through p53-dependent bax induction. FEBS Letters, 512, 334-340. doi:10.1016/S0014-5793(02)02292-5
- [65] Kita, A., Nakahara, T., Yamanaka, K., Nakata, M., Kaneko, N., Koutoku, N. and Sasamata, N. (2009) YM155: A small molecule survivin suppressant with potent antitumor effect in human breast cancer models. Cancer Research, 69, 3.
- [66] Cummings, B.S. and Schnellmann, R.G. (2002) Cisplatininduced renal cell apoptosis: Caspase-3-dependent and -independent pathways. Journal of Pharmacology and Experimental Therapeutics, 302, 8-17. doi:10.1124/jpet.302.1.8
- [67] Zhou, H., Zhang, Y., Yu, F., Chan, L. and Lee, A.S. (2011) Novel mechanism of anti-apoptotic function of 78-kDa Glucose-Regulated Protein (GRP78) endocrine resistance factor in breast cancer, through release of b-cell lymphoma 2 (bcl-2) from bcl-2-interacting killer (bik). The Journal of Biological Chemistry, 286, 25687-25696. doi:10.1074/jbc.M110.212944
NOTES
*Corresponding author.