Journal of Biophysical Chemistry
Vol.3 No.3(2012), Article ID:21943,5 pages DOI:10.4236/jbpc.2012.33027
Insilico studies of 2-methylheptyl isonicotinate produced by Streptomyces sps. 201 against dihydrodipicolinate synthase enzyme of Mycobacterium tuberculosis
![]()
Bioinformatics Infrastructure Facility, Biotechnology Division, North-East Institute of Science & Technology, Jorhat, India; *Corresponding Author: salampradeep@gmail.com
Received 24 July 2011; revised 20 February 2012; accepted 28 May 2012
Keywords: DHDPS; Molecular Docking; Hydrogen Bonding
ABSTRACT
Tuberculosis is thought to have infected onethird of the world’s population and antibiotic resistance is a growing problem in multi-drugresistant tuberculosis which is caused by Mycobacterium tuberculosis (MTB). It has been reported that Mycobacterial cell walls are characterized by high DAP (diaminopimelic acid) content—an intermediate of the (S)-lysine biosynthetic pathway. Hence, the Lysine/DAP biosynthetic pathway is a promising target because of its role in cell wall and amino acid biosynthesis. In this study we performed a molecular docking analysis of a novel antibacterial isolated from Streptomyces sps. 201 against dihydrodipicolinate synthase (DHDPS) enzyme of Mycobacterium tuberculosis. The docking studies suggest that the novel molecule binds at active site LYS 171 forming a cleft and at other potential ligand binding site exhibiting all the major interactions such as hydrogen bonding, hydrophobic interaction and electrostatic interaction with (THR55, TYR143, ARG148, LYS171, VAL257 and GLY256) residues.
1. INTRODUCTION
Tuberculosis is common and in many cases lethal and infectious disease caused by various strains of mycobacteria, usually Mycobacterium tuberculosis [1]. Tuberculosis typically attacks the lungs but can also affect other parts of the body. It is spread through the air when people who have an active TB infection cough, sneeze, or otherwise transmit their saliva through the air. One-third of the world’s population is thought to be infected with Mycobacterium tuberculosis [2] and new infections occur at a rate of one per second [3]. In 2007, an estimated 13.7 million people had active TB disease, with 9.3 million new cases and 1.8 million deaths [4]. Most infections are asymptomatic and latent, but about one in ten latent infections eventually progresses to active disease which, if left untreated, kills more than 50% of those so infected. Hence there is an urgent need to design or develop a novel or potent antitubicular agents.
The three-dimensional crystal structures of DHDPS enzyme of Mycobacterium tuberculosis (PDB ID: 1XXX) [5], is available at Protein Data Bank (http://www.rcsb.org/). There have been reports of experimental procedures for designing of inhibitors against DHDPS but no potent inhibitors have been reported till date [6-8].
The crystal structure of Mtb-DHDPS enzyme (PDBID: 1XXX) is a tetramer comprising of four identical subunits arranged in D2 symmetry. Each monomer comprises an N-terminal (b/a) 8-barrel domain and a C-terminal domain consisting of three a-helices. The residues responsible for substrate binding and catalysis are located in the (b/a) 8-barrel domain. The crystallographic asymmetric unit contains two tetramers of the enzyme. Each tetramer can be described as a dimer of dimers, with the two monomers tightly bound to each other to form the tight dimer, and weaker interactions between the dimeric units [5].
The present study mainly focused on molecular docking studies of a novel compound 2-methylheptyl isonicotinate isolated from Streptomyces sps. 201 [9-12] against dihydrodipicolinate synthase (DHDPS) enzyme of MTB at the active site residue (LYS-171 which is responsible for substrate binding and catalysis), which results in the formation of hydrogen bonds using Molegro Virtual Docker [13]. A few work of virtual screening of pyruvate analogs against DHDPS inhibitors has been reported [14], here we have reported the molecular docking analysis of 2-methylheptyl isonicotinate against DHDPS enzyme of MTB.
2. MATERIALS AND METHOD
The 2D structure of 2-methylheptyl isonicotinate (2MHI) isolated form Streptomyces sp. 201 was generated using Cambridge Soft ChemOffice 2008 [15] shown in Figure 1. The energy of the generated structure of 2MHI was converted to 3D structure using ChemOffice 2008 [15] and minimized to 17.9850 kcal/mol using MM2 force field methods [16] and save as SYBL mol2 files for docking purposes.
The physio-chemical properties of 2-methylheptyl isonicotinate were predicted using Chemoffice 2008 [15] for Lipinski rule of five filters [17]. 2-methyheptyl isonicotinate was again screened for possible side effects and toxic effects using PASS prediction (Prediction of Activity Spectra for Substances) [18].
The three-dimensional crystal structure of MTB DHDPS (PDB ID: 1XXX) retrieved from Protein Databank was imported in the Molegro Virtual Docker [13] and for docking purposes, all the 1587 water molecules, eight DTT molecules, eight Mg2+ and eight Cl– ions were removed. MTB DHDPS enzyme consists of 8 chains (A-H), considering that the chains are identical and independent of each other, only chain A from the enzyme was selected and imported in the MVD to perform the docking simulation.
The potential ligand binding sites of the MTB DHDPS were detected using the cavity detection program in MVD 5.0 Molegro Virtual Docker. A cavity having a volume of 43.5 and a surface area of 162.56 was detected which is shown in Figure 2. The amino acid residues that lie in the potential ligand binding site are THR54, THR55, TYR143, ILE145, GLY147, ARG148, LYS171, ALA173, GLY194-ALA197, ILE211-ILE214 and CYS248-VAL 257.
The methodology adopted in this work to determine the potential binding sites is a grid-based cavity prediction algorithm. First, a discrete grid with a resolution of 0.8 Å covering the proteinis created with a sphere of radius 1.4 Å is placed and checked whether the sphere will overlap with any of the spheres determined by the Van der Waals radii of the protein atoms. Second, each accessible grid point is checked for whether it is part of a cavity or not. The final step is to determine the connected regions. Two grid points are connected if they are neighbours. The cavities found are then ranked according to their volume [19].
Molecular docking was carried out using Molegro Virtual Docker (MVD) (Molegro APS: MVD 5.0). MVD is molecular visualization and molecular docking software which is based on a differential evolution algorithm; the solution of the algorithm takes into account the sum of

Figure 1. 2D structure of 2-methylheptyl isonicotinate.

Figure 2. Potential ligand binding sites predicted using Molegro Virtual Docker.
the intermolecular interaction energy between the ligand and the protein and the intramolecular interaction energy of the ligand. The docking energy scoring function is based on the modified piecewise linear potential (PLP) with new hydrogen bonding and electrostatic terms included. Full description of the algorithm and its reliability compared to other common docking algorithm can be found in reference [19,20].
3. RESULTS AND DISCUSSION
Molecular docking was carried out and the docking score and protein-ligand interaction energy are shown in Table 1 which shows that there has been a strong inter action between the ligand and the receptor indicating a
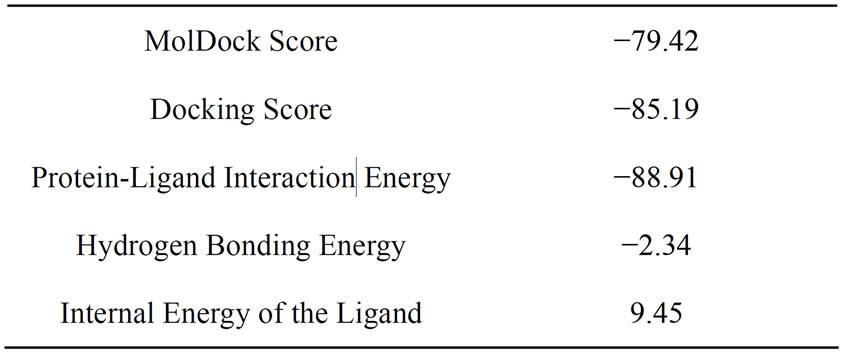
Table 1. Docking Score of 2MHI against MTB DHDPS enzyme.
binding affinity. It is also observed that 2MHI was docked into the active site of MTB DHDPS (shown in Figure 3). Molegro Virtual Docker uses the MolDock docking engine to predict protein-lignad interactions. MolDock is based on a new hybrid search algorithm, called guided differential evolution [20]. The guided differential evolution algorithm combines the differential evolution (DE) optimization technique with a cavity prediction algorithm which is dynamically used during the docking process [19].
The docking results shows 2-methylheptyl isonicotinate bound tightly at the active site of MTB DHDPS. 2MHI was found lying deep into the cavity/potential ligand binding site exhibiting molecular interactions such as hydrogen bonding, hydrophobic interaction (Figure 4) and electrostatic interactions (Figure 5).

Figure 3. Predicted bonded interactions (green dashed lines) between 2MHI and Thr55, Tyr143, Tyr148, Lys171, Gly256 and Gly257 residues of M. tuberculosis DHDPS enzyme.

Figure 4. Predicted hydrophobic interaction between 2MHI and the residues at the active site region.

Figure 5. Predicted electrostatic interaction between 2MHI and the residues at the active site region.
The docking analysis also shows the formation of eight interactions with six amino acid residues THR55, TYR143, ARG148, LYS171, VAL257 and GLY256 respectively of MTB DHDPS (shown in Figure 3).
In the present study, the MolDock docking score were used. The MolDock scoring function (MolDock Score) used in MVD 5.0 is derived from the PLP scoring functions originally proposed by Gehlhaar et al. [21,22] and later extended by Yang et al. [23]. The MolDock scoring function further improves these scoring functions with a new hydrogen bonding term and new charge schemes. The docking scoring function, Escore, is defined by the following energy terms:
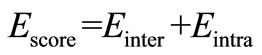
where Einter is the ligand protein interaction energy.
We have also carried out a detailed analysis of docking in terms of protein-ligand interaction energy. The ligandprotein interaction energy analysis (both electrostatic and H-bond) were calculated in order to get a better understanding of the variations between the binding mode of 2MHI and the molecular factors responsible for the activity. Table 2 enlists the protein-ligand interaction energy calculation including the residues present, interacting atom of the protein and the ligand, protein-ligand interaction distances, ligand atom energies, Hbond and electrostatic energy and Epair which is shown in Table 2.
Additionally, the physio-chemical properties predicted using Chemoffice 2008 [19] showed that 2MHI does not violate Lipinski rule of five [21] having a molecular weight of 235.52, 3 hydrogen bond acceptor atom, no hydrogen bond donor atom and LogP of −3.41 which is good enough to be an orally active drug. Moreover the molecule shows a drug likeness of 0.722 from the PASS prediction [22].
Thus the novel isolate form Streptomyces sp. 201 could provide a structural leads for DHDPS inhibitor or a novel class of anti-tubercular agent.

Table 2. Protein-ligand interaction analysis.
4. CONCLUSION
The insilico studies of novel isolate 2-methylheptyl isonicotinate from Streptomyces sps. 201 showed that 2MHI is a good inhibitor of MTB DHDPS enzmye forming both bonded and non-bonded interactions at the active site of the enzyme. The compound showed a strong binding affinity to MTB DHDPS in terms of docking score and hydrogen bonding energy. Additionally the compound does not violate Lipinski rule of five to be an orally active drug and showed a drug likeness of 0.722 using PASS prediction. 2MHI could be a lead molecule or a novel class of potential antitubercular agent. Hence the present study concludes that 2MHI could be an anti-tubercular agent and supports for experimental testing of the compounds.
5. ACKNOWLEDGEMENTS
SPS thanks Director, NEIST, Jorhat for providing financial support. The authors also thank Department of Biotechnology, Govt. of India for providing Bioinformatics Infrastructure Facility in the entire North-East India and CSIR for providing support.
REFERENCES
- Kumar, V., Abbas, A.K., Fausto, N. and Mitchell, R.N. (2007) Robbins basic pathology (8th Edtion). Saunders Elsevier, 516-522.
- Jasmer, R.M., Nahid, P. and Hopewell P.C. (2002) Clinical practice. Latent tuberculosis infection. The New England Journal of Medicine, 347, 1860-1866. doi:10.1056/NEJMcp021045
- World Health Organization (2007) Tuberculosis. http://who.int/mediacentre/factsheets/fs104/en/index.html
- World Health Organization (2009) Global tuberculosis control: epidemiology, strategy, financing. Epidemiology, 6-33. http://who.int/entity/tb/publications/global_report/2009/pdf/chapter1.pdf
- Kefala, G., Evans, G.L., Griffin, M.D., Devenish, S.R., Pearce, F.G., Perugini, M.A., Gerrard J.A., Weiss, M.S. and Dobson, R.C. (2008) Crystal structure and kinetic study of dihydrodipicolinate synthase from Mycobacterium tuberculosis. Biochemical Journal, 411, 351-360. doi:10.1042/BJ20071360
- Turner, J.J., Gerrard, J.A. and Hutton, C.A. (2005) Heterocyclic inhibitors of dihydrodipicolinate synthase are not competitive. Bioorganic & Medicinal Chemistry, 13, 2133-2140. doi:10.1016/j.bmc.2005.01.001
- Turner, J.J., Healy, J.P., Dobson, R.C., Gerrard, J.A. and Hutton, C.A. (2005) Two new irreversible inhibitors of dihydrodipicolinate synthase: Diethyl(E,E)-4-oxo-2,5-heptadiene-dioate and diethyl(E)-4-oxo-2-heptenedioate. Bioorganic & Medicinal Chemistry, 15, 995-998. doi:10.1016/j.bmcl.2004.12.043
- Turner, J.J., Gerrard, J.A. and Hutton, C.A. (2005) Conformationally constrained diketopimelic acid analogues as inhibitors of dihydrodipicolinate synthase. Bioorganic & Medicinal Chemistry, 13, 2133-2140. doi:10.1016/j.bmc.2005.01.001
- Bordoloi, G.N., Babita, K., Arijit, G., Manobjyoti, B., Yadav, R.N.S., Manoj, K.R. and Bora, T.C. (2001) Isolation and structure elucidation of a new antifungal and antibacterial antibiotic produced by Streptomycin sp. 201. Bioscience, Biotechnology, and Biochemistry, 65, 1856- 1858.
- Bordoloi, G., Kumari, B., Bordoloi, M., Roy, M.K. and Bora, T.C. (2001) A process for the production of 2-methylheptyl isonicotinate. Indian Patent NF-9/2000.
- Bordoloi, G., Kumari, B., Bordoloi, M., Roy, M.K. and Bora, T.C. (2000) 2-Methylheptyl isonicotinate as novel antibiotic. Indian Patent NF-116/2001.
- Bordoloi, G., Kumari, B., Bordoloi, M., Roy, M.K. and Bora, T.C. (2000) A process for the production of 2-methylheptyl isonicotinate. US Patent 009 NF 2000, 10/027913.
- Molegro APS: MVD 5.0 Molegro Virtual Docker, DK- 8000 Aarhus C, Denmark.
- Aarti, G., Rupinder, T. and Gajendra, P.S.R. (2010) Virtual screening of potential drug-like inhibitors against Lysine/DAP pathway of Mycobacterium tuberculosis. BMC Bioinformatics, 11, S53.
- CambridgeSoft Corporation: Chemoffice (2008) USA.
- Ulrich B. and Allinger, N. L. (1982) Molecular mechanics. Journal of the American Chemical Society.
- Lipinski, C.A., Lombardo, F., Dominy B.W. and Feeney P.J. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23, 3-25. doi:10.1016/S0169-409X(96)00423-1
- Filimonov, D.A. and Poroikov, V.V. (1996) PASS Computerized prediction of biological activity spectra for chemical substances, Bioactive compound design: Possibilities for industrial use. BIOS Scientific Publishers, Oxford, 47-56.
- Thomsen, R. and Christensen, M.H. (2006) MolDock: A new technique for high-accuracy molecular docking. Journal of Medicinal Chemistry, 49, 3315-3321. doi:10.1021/jm051197e
- Storn, R. and Price, K. (1995) Differential evolution—A simple and efficient adaptive scheme for global optimization over continuous spaces. International Computer Science Institute, Berkley.
- Gehlhaar, D.K., Verkhivker, G., Rejto, P.A., Fogel, D.B., Fogel, L.J. and Freer, S.T. (1995) Docking conformationally flexible small molecules into a protein binding site through evolutionary programming. Proceedings of the Fourth International Conference on Evolutionary Programming, Morgan Kaufmann, San Mateo, 615-627.
- Gehlhaar, D. K., Bouzida, D. and Rejto, P.A. (1998) Fully automated and rapid flexible docking of inhibitors covalently bound to serine proteases. Proceedings of the Seventh International Conference on Evolutionary Programming, Morgan Kaufmann, San Mateo, 449-461.
- Yang, J.M. and Chen, C.C. (2004) Gemdock: A generic evolutionary method for molecular docking. Proteins, 55, 288-304. doi:10.1002/prot.20035