Journal of Diabetes Mellitus
Vol.3 No.3(2013), Article ID:35009,7 pages DOI:10.4236/jdm.2013.33018
Autophagy induced by glibenclamide serves as a defense against apoptosis in INS-1 rat insulinoma cells*
Hua Su1#, Xingyan Liu1#, Ling Su2, Li Zhang1, Xiangguo Liu2, Hong Ji1, Haiqin Rong1†
![]()
1Institute of Endocrine and Metabolic Diseases, Shandong Academy of Medical Sciences. Jinan, China; tosuhua@126.com, liuxingyan0810@126.com, 438609074@qq.com, Jihong993@163.com †Corresponding Author: haiqinrong@126.com
2School of Life Sciences, Shandong University, Jinan, China; suling@sdu.edu.cn, xgliu@sdu.edu.cn
Copyright © 2013 Hua Su et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 7 May 2013; revised 8 June 2013; accepted 15 June 2013
Keywords: Glibenclamide; Pancreatic Beta-Cell; Autophagy; Apoptosis; p70 S6K
ABSTRACT
Glibenclamide, a blocker of ATP-sensitive potassium channels, has been found to induce apoptosis in some cell types, including pancreatic beta-cells. Since autophagy plays doubleedged roles in pancreatic beta-cell survival, frequently through cross-talking with apoptosis, we investigated if glibenclamide induced autophagy in INS-1 rat insulinoma cells and the influence of autophagy on apoptosis. Mammalian target of rapamycin (mTOR) is a negative regulator of autophagy. As one of the substrates of mTOR, p70 S6 kinase (p70 S6K) is phosphorylated upon mTOR activation. Our results showed that glibenclamide induced an elevated protein level of the autophagy marker LC3-II, while decreasing phosphorylated p70 S6K, indicative of inhibition on mTOR signaling in INS-1 cells. Furthermore, inhibiting glibenclamide-induced autophagy via knocking down the autophagy essential gene Atg7 decreased cell viability and increased apoptosis in INS-1 cells. Our results indicate that glibenclamide induces autophagy in INS-1 cells, and that autophagy activation is exerting a protective activity against apoptosis.
1. INTRODUCTION
Autophagy is a naturally occurring degradation process in cells that help removing erroneously folded proteins or protein aggregates, as well as the excessive or defective organelles for reuse of the components [1-3]. It occurs constitutively or in response to internal or external stimuli [4]. Upon nutrient deficiency, autophagy is activated to provide energy and materials for cell survival [1].
Autophagy plays an essential role in cell growth, development, differentiation and homeostasis in various organisms [1,5], and is also implicated in certain diseases, including diabetes and neurodegenerative diseases [6,7].
As the producer of insulin, pancreatic beta-cells are a place where large-scale protein synthesis and degradation occur. Basal level of autophagy is involved in the mechanism underlying dynamic protein turnover [1] and is supposed to be essential for maintaining the architecture and function of normal beta-cells. Besides, autophagy is markedly up-regulated in beta cells of diabetes patients [6,8].
Autophagy is a double-edged sword for the survival and functioning of pancreatic beta-cells [1,2]. It serves as a pro-survival mechanism; But unbalanced autophagy may lead to cell death as in the case of autophagic cell death, also refered to as type II programmed cell death in which extensive autophagic degradation of Golgi apparatus, polyribosomes and endoplasmatic reticulum occur [9].
Palmitate-induced apoptosis was aggravated when autophagy was inhibited in the INS-1 cells, while it was alleviated upon autophagy activation [10]. In autophagy deficient mice with conditional knockout of the essential autophagy gene Atg7 in beta-cells, signs of type 2 diabetes were observed, including beta-cell degeneration, glucose intolerance and reduced insulin secretion, suggesting a protective role of autophagy against diabetes [1]. Jung and Lee also reported a protective role of autophagy by demonstrating that Atg7 deletion in pancreatic beta-cells resulted in increased apoptosis and receded beta-cell mass and insulin secretion [4]. Furthermore, autophagy may also serve as a useful mechanism for eliminating damaged mitochondria and other cellular structures to prevent cell death [11]. Despite the beneficial role of autophagy, deregulated autophagy is proved to be detrimental to beta-cells.
Pancreatic beta-cells in patients with type 2 diabetes present signs of autophagy and cell death without obvious DNA condensation, indicative of autophagic cell death, which may contribute to the loss of beta-cell mass [9].
As blockers of ATP-sensitive potassium (K-ATP) channels, glibenclamide and other sulfonylureas have been used to treat type 2 diabetes via stimulating insulin secreation. But long-term insulin secretory failure has been observed with many patients treated with these drugs [12]. The reason might be that they induce apoptosis of pancreatic beta cells, as indicated in our current work with glibenclamide in rat insulinoma INS-1 cells. But is autophagy involved in glibenclamide-induced cell death? Or is it a protective mechanism against the otherwise severe cell death? In our current work, we tried to figure out if glibenclamide induced autophagy and thus affected apoptosis in INS-1 cells.
Microtubule-associated protein 1A/1B-light chain 3 (LC3) is a soluble protein distributed ubiquitously in cells. During autophagy, autophagosomes engulf cytoplasmic components, including cytosolic proteins and organelles. Concomitantly, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine to form LC3-phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. Autophagosomes fuse with lysosomes to form autolysosomes, and intra-autophagosomal components are degraded by lysosomal hydrolases. At the same time, LC3-II in autolysosomal lumen is degraded. Thus, lysosomal turnover of the autophagosomal marker LC3-II reflects autophagic activity, and detecting LC3 by immunoblotting or immunofluorescence has become a reliable method for monitoring autophagy [1].
Mammalian target of rapamycin (mTOR) is a multifunctional regulator which is also involved in the inhibition of autophagy [13-17]. As one of the substrates of mTOR, p70 S6 kinase (p70 S6K) is phosphorylated upon mTOR activation. Hence elevated protein level of phosphorylated p70 S6K (p-p70 S6K) is regarded as one of the indicators of mTOR signaling [18-21]. In our experiments, we tried to find out if mTOR pathway was involved in glibenclamide-induced autophagy in INS-1 cells.
We also studied the effect of glibenclamide-induced autophagy on apoptosis and cell viability in INS-1 cells via knocking down the autophagy gene Atg7 and then evaluating the extent of apoptosis and cell death upon glibenclamide treatment.
2. MATERIAL AND METHODS
2.1. Cell Culture
INS-1 rat insulinoma cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 50 μM 2-mercaptoethanol, and cultured at 37˚C with 5% CO2. Upon drug treatment, the medium was removed and appropriate amount of drug stocks were added into fresh medium, mixed and added into the plates with cells for further culture at 37˚C with 5% CO2 for another 24 or 48 hours.
2.2. Antibodies and Reagents
LC3B antibody (CST 2775), p70 S6 Kinase antibody (CST 9202), phospho-p70 S6 Kinase (Thr389) antibody (CST 9205), Atg7 antibody (CST 2631) and PARP antibody (CST 9542) were purchased from Cell Signaling Technology (USA). Monoclonal mouse anti-GAPDH antibody (Cw0100) was provided by Tiangen Biotech Co., Ltd. (Beijing, China). LY294002 (Cat No. S1737) was obtained from Beyotime Institute of Biotechnology Co., Ltd. (China). Chloroquine diphosphate salt (C6628) and Bafilomycin A1 (B1793) were provided by Sigma Aldrich (USA).
Glibenclamide standard substance was from National Institutes for Food and Drug Control (Beijing, China) and was dissolved in DMSO to prepare a 200 mM stock solution. Upon cell treatment, the stock solution was diluted with medium to prepare 0.1,1 or 10 μM of work solutions, with the same amount of DMSO diluted in medium as control. Negative control siRNA and siRNA targeting rat Atg7 (5'-GCAUCAUCUUUGAAGUGAA- 3') were provided by GenePharma Co., Ltd. (Shanghai, China). Transfection regent X-tremeGENE was purchased from Roche (Germany).
2.3. LC3 Immunofluorescence Confocal Microscopy
INS-1 cells were seeded and treated with or without glibenclamide on coverslips in 24-well plates. After a four-hour treatment, cells were fixed with 4% formaldehyde in PBS for 15 minutes at room temperature and permeabilized with ice-cold 100% methanol for 10 minutes at −20˚C. After blocked with 1X PBS/5% normal goat serum (Cell Signaling Technology)/0.3% Triton X- 100, cells were incubated with LC3 antibody (Cell Signaling Technology) at 1:400 dilution overnight at 4˚C and Alexa Fluor® 488 Conjugated Anti-rabbit IgG (Cell Signaling Technology) at 1:500 dilution for 1 hour at room temperature in dark thereafter. Nuclei were stained with 4’,6-diamidino-2-phenylindol (DAPI, Sigma-Aldrich) for 5 minutes at room temperature. Cells were rinsed three times in PBS for 5 minutes after each step of the immnunofluorescence staining. The coverslips were mounted on glass slides for confocal laser scanning microscopy using Carl Zeiss LSM700.
2.4. Western Blot Analysis
After drug treatments, cells were washed once in cold PBS and then cellular proteins were lysed on ice in RIPA buffer (P0013C) (Beyotime, China) with 1% Protease Inhibitor Cocktail (Sigma P8340) for at least 30 min, then centrifuged at about 12,000 × g and the supernatants were collected for protein concentration determination. Western blot analysis was carried out as described by Liu XG [22].
Briefly, cell lysates were resolved on SDS-PAGE and then transferred onto PVDF membranes (Millipore). The blots were probed with primary antibodies over night at 4˚C followed by incubation with horseradish peroxidaseconjugated secondary antibodies at room temperature for 2 hours. Finally the blots were detected with Super ECL plus Detection Reagent (P1010) (Applygen, Beijing, China) and exposed to X-ray film (Kodak).
2.5. SRB Viability Assay
Cells were seeded in 96-well plates. After 24 hours, cells were treated with drugs at indicated concentrations or were transfected with siRNAs for specified time durations. Cell viability was evaluated by SRB (Sulforhodamine B) assay following the procedures below: Pour off the medium and fix cells with 100 μl of cold 10% TCA (trichloroacetic acid). Incubate for 1 hour at 4˚C. Discard the supernatant. Wash the plates 5 times with deionized water and leave it to dry. Add 50 μl of 0.4% SRB solution in 1% acetic acid to each well. Shake 5 min on a microplate shaker. Wash the plates 5 times with 1% acetic acid and leave it to dry. Add 100 μl of 10 mM unbuffered Tris base (pH 10.5) to dissolve the bound dye. Mix for 5 min on a microplate shaker and read optical densities at the wavelength of 500 nm using a microplate spectrophotometer.
2.6. Statistical Analysis
Data were presented as means ± SE from at least three independent experiments and analyzed by Student’s t-test with SPSS software. Differences at p < 0.05 were considered statistically significant.
3. RESULTS
3.1. Glibenclamide Induced Autophagy in INS-1 Rat Insulinoma Cells
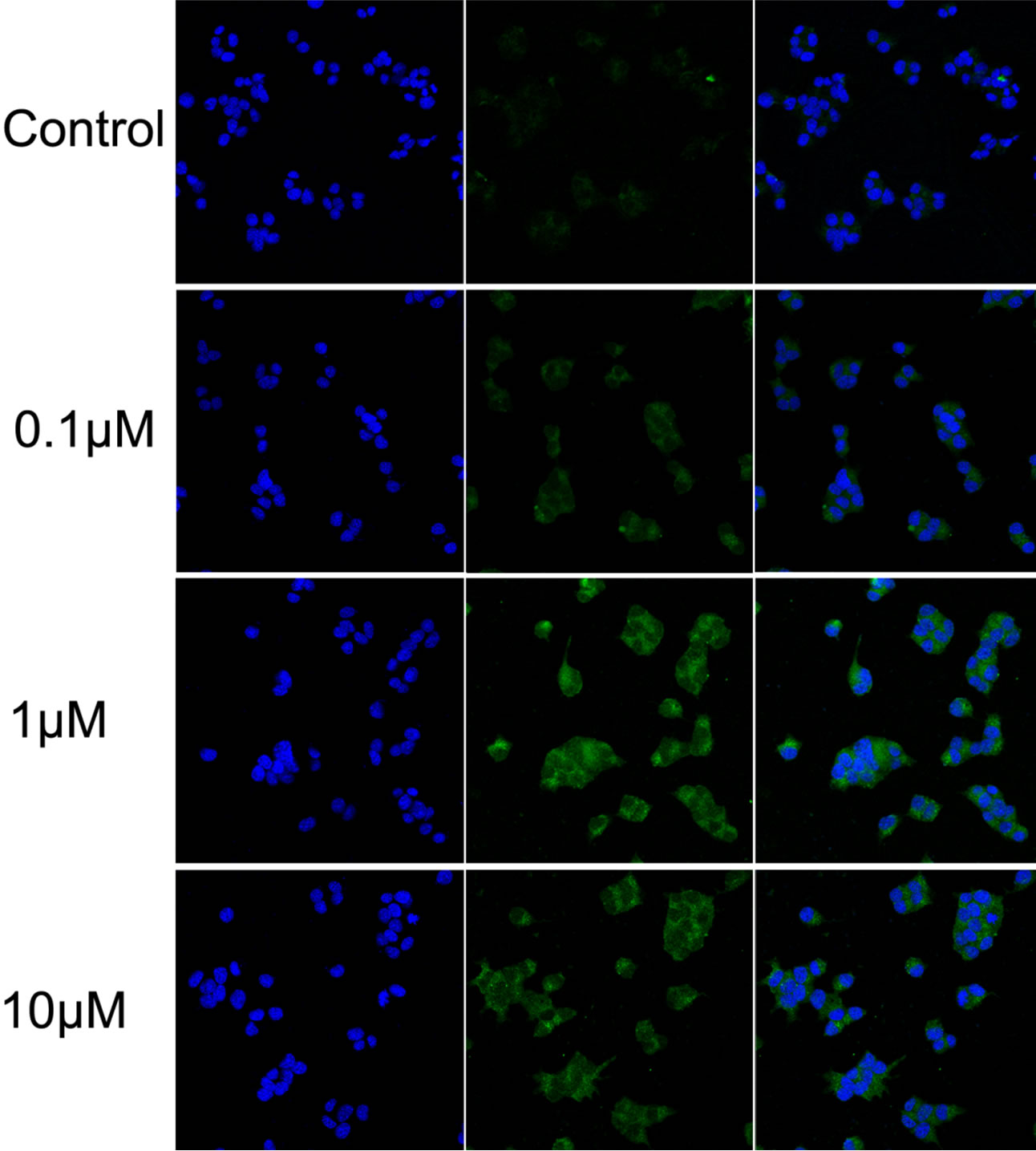
Glibenclamide has been reported to induce apoptosis and we suspected that it might cause autophagy as well in pancreatic beta-cells. Therefore, we performed Immunofluorescence and Western blot analysis to detect autophagy activation. INS-1 cells were treated or left untreated with 0.1, 1 or 10 μM glibenclamide for 24 h, and were then subjected to LC-3 Immunofluorescence experiments and Western blot analysis probing the autophagy marker, LC3-II. As shown by the result, glibenclamide caused more gathered dots of LC3 in the cytoplasm (Figure 1(a)) and elevated protein levels of LC3- II (Figure 1(b) and (c)), indicative of autophagy activation.
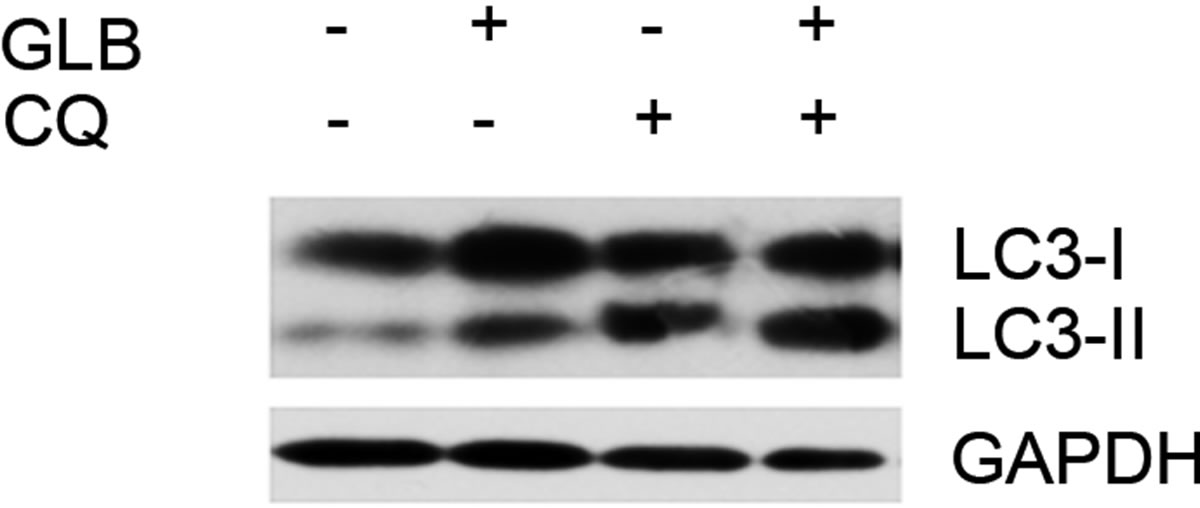
As elevated protein level of LC3-II could reflect upregulated formation of autophagasomes, as well as blocked hydrolysis of the bound LC3-II on the outer membrane of autolysosomes due to retarded fusion of autophagasomes with lysosomes or blocked lysosomal hydrolysis [23], we performed the glibenclamide treatment experiments under autophagy inhibitors to validate the autophagic flux. LY294002 is a specific inhibitor of PI(3)K, which is essential for the formation of autophagosomes [23]. When LY294002 was used in combination with glibenclamide, we found that glibenclamideinduced LC3-II accumulation was inhibited (Figure 2). We also used two downstream inhibitors bafilomycin A1 and chloroquine, respectively, in combination with glibenclamide. Bafilomycin A1 inhibits the fusion of autophagosomes with lysosomes, as well as the acidification inside lysosomes. Chloroquine blocks lysosomal acidification. Both can inhibit the downstream step of autophagy, the degradation of the autophagic cargo, as well as LC3-II bound to the membrane of autolysosomes. The results showed that they caused significant accumulation of LC3-II upon glibenclamide treatment (Figure 3). Taken together, these results demonstrated that glibenclamide triggered an autophagic flux in INS-1 cells.
3.2. Glibenclamide Treatment Resulted in Decreased Phosphorylation of p70 S6K
To determine if the canonical autophagy inhibition pathway, mTOR signaling, was involved in glibenclamide-induced autophagy, we performed Western blot analysis to detect the activated form of mTOR substrate p70 S6K, namely phosphorylated p70 S6K (p-p70 S6K) upon glibenclamide treatment (Figure 4). The results showed a dramatic decrease of the protein level of p-p70 S6K, indicating that an inhibition of the mTOR signaling might be involved in glibenclamide-induced autophagy.
3.3. Glibenclamide-Induced Autophagy Defended INS-1 Cells against Apoptosis
To elucidate the role of glibenclamide-induced autophagy for the survival of INS-1 cells, we inhibited auto-
 (a)
(a)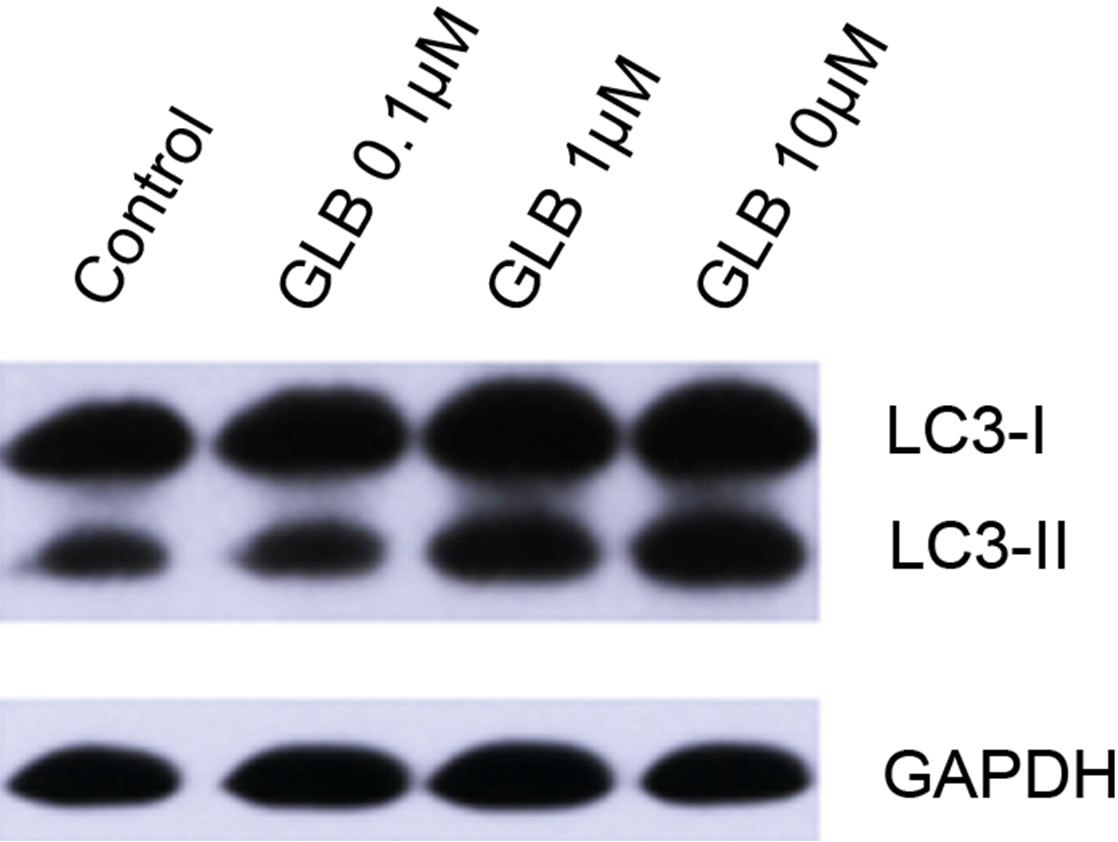 (b)
(b)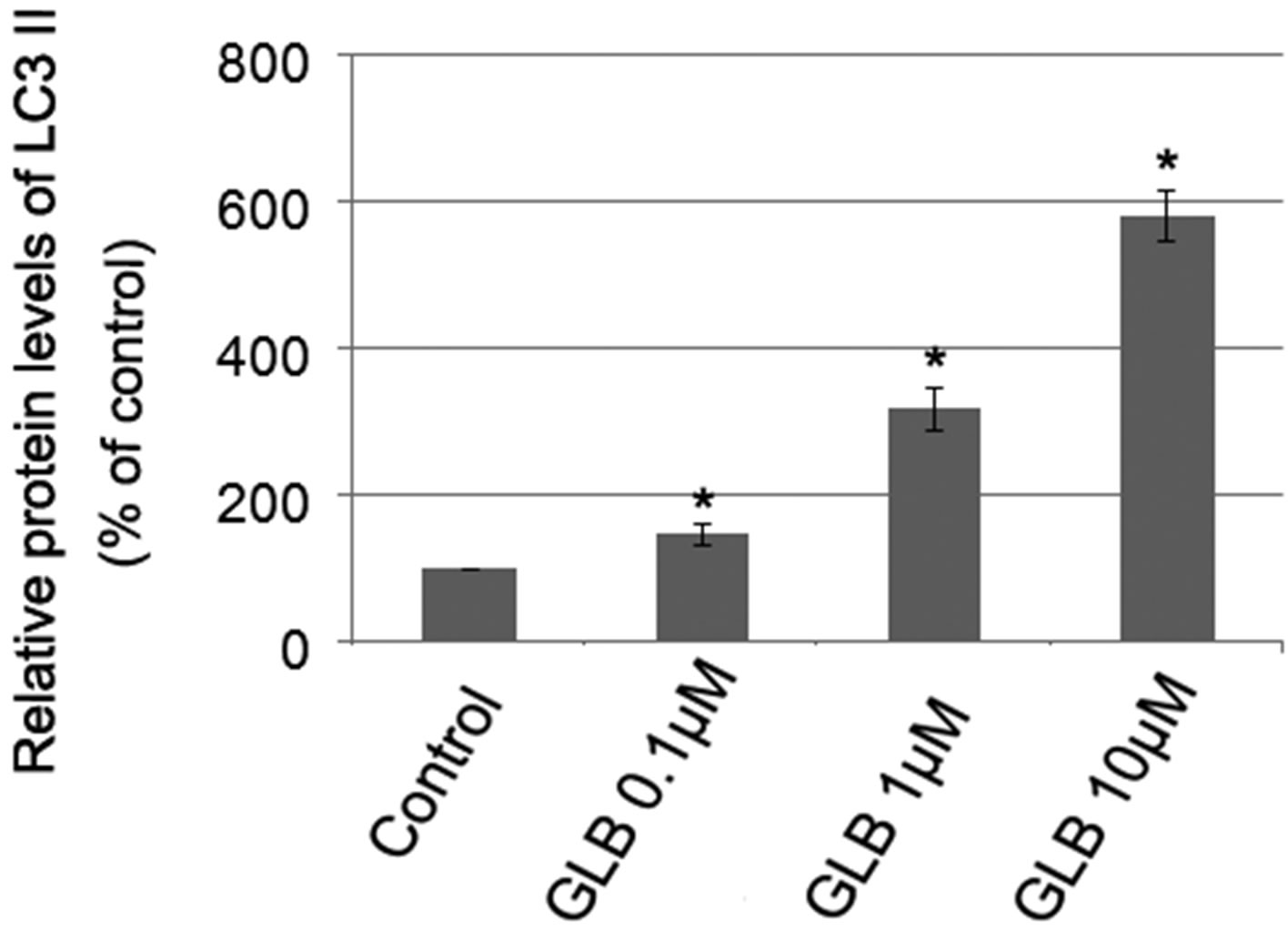 (c)
(c)
Figure 1. Glibenclamide induced autophagy in INS-1 cells. INS-1 cells were left untreated (control) or treated with 0.1, 1 and 10 μM glibenclamide for 24 h, respectively. Then LC-3 Immunofluorescence experiments were performed (a), green: Alexa Fluor® 488; blue: DAPI stained nuclei) and cellular proteins were harvested and subjected to Western blot analysis with LC3B antibody, with GAPDH as the internal control (b), (c). (*: p < 0.05 versus control, n = 3).
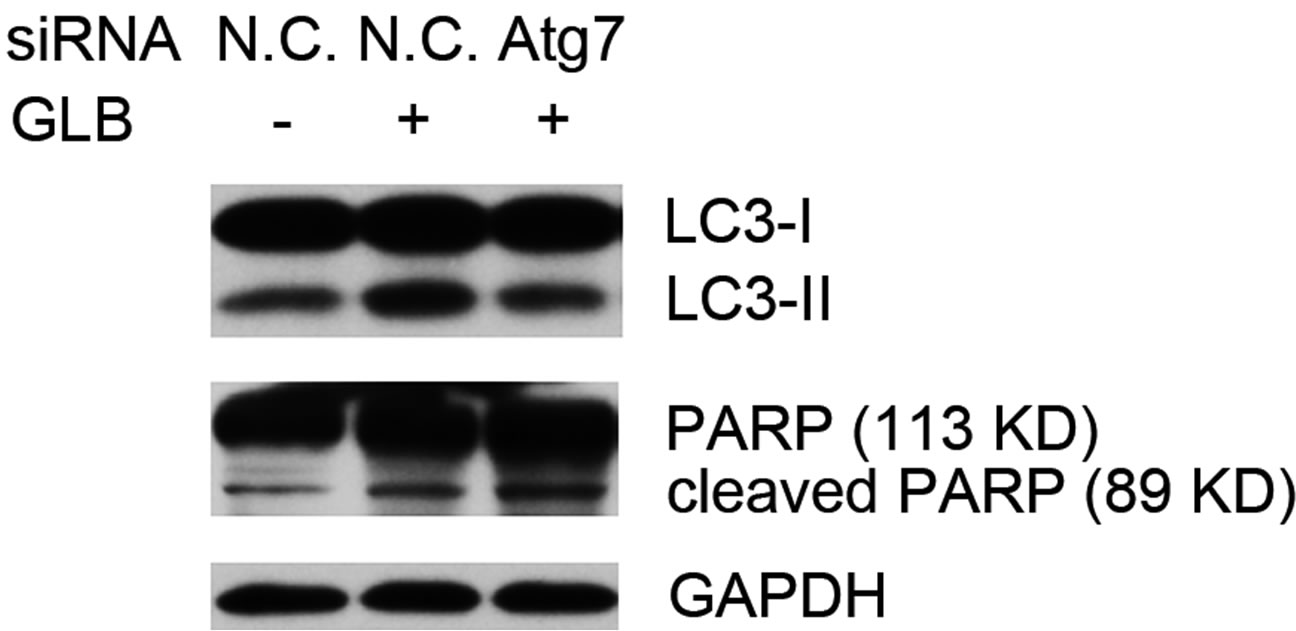
phagy via siRNA-mediated RNAi on the autophagy gene Atg7. Knock down of Atg7 resulted in decreased level of the autophagy indicator LC3-II, while elevating that of cleaved PARP upon glibenclamide treatment (Figure 5), suggesting that glibenclamide-induced autophagy served as a defense against apoptosis.
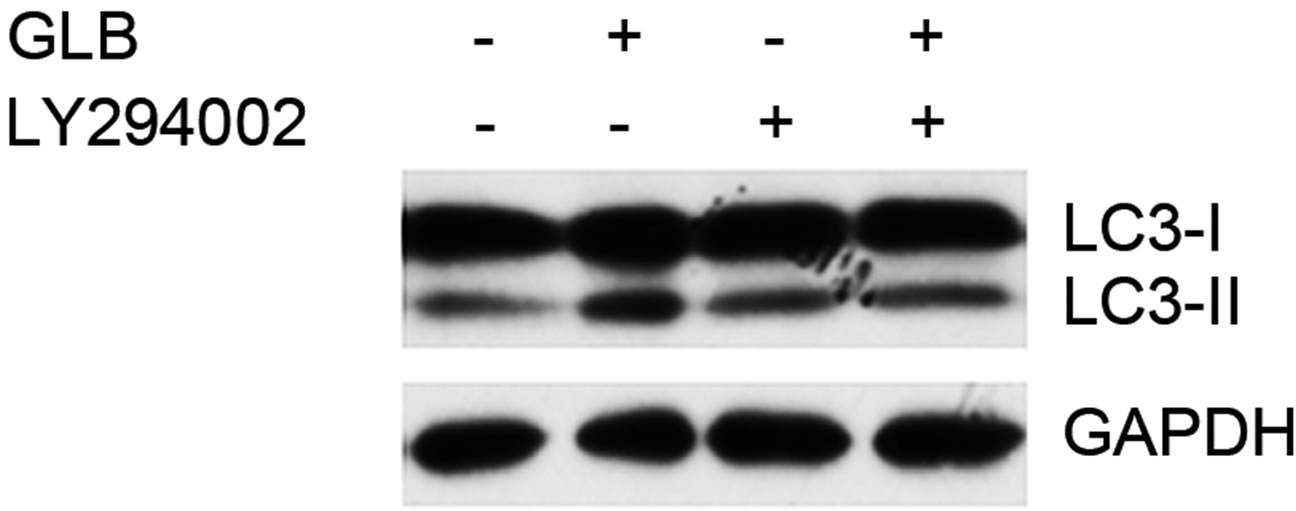
Figure 2. Glibenclamide-induced autophagy was inhibited by LY294002 in INS- 1 cells. INS-1 cells were left untreated or treated with 20 μM LY294002 for 30 min, and were then left untreated or treated with 10 μM glibenclamide for another 24 h. Cellular proteins were harvested and subjected to Western blot analysis with LC3B antibody, with GAPDH as the internal control.
 (a)
(a)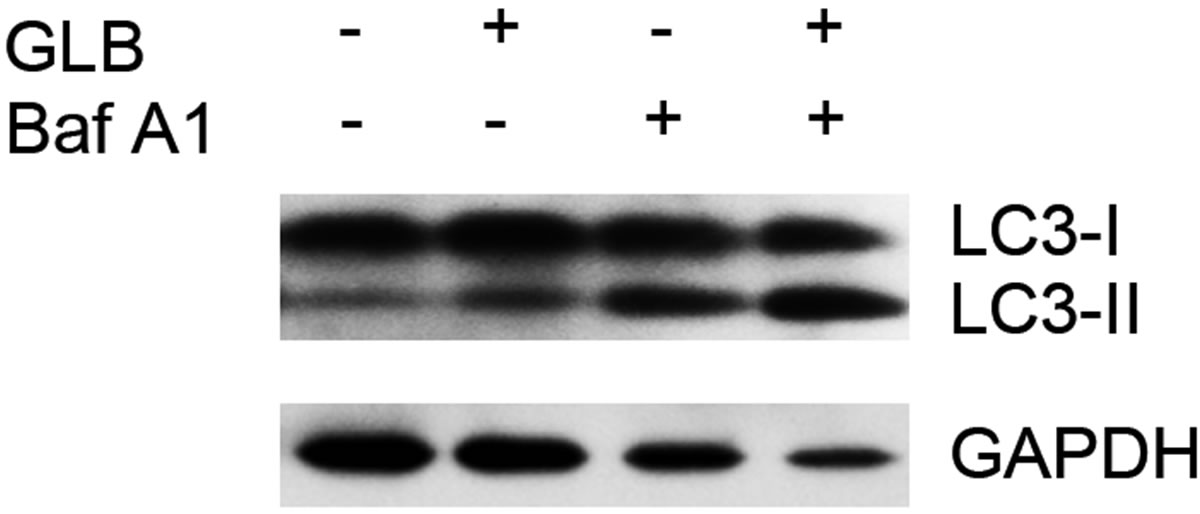 (b)
(b)
Figure 3. Glibenclamide-induced LC3-II accumulation was enhanced by chloroquine or bafilomycin A1 in INS-1 cells. INS-1 cells were left untreated or treated with 10 μM chloroquine (a) or 20 nM bafilomycin A1 (b) for 30 min, and were then left untreated or treated with 10 μM glibenclamide for another 24 h. Cellular proteins were harvested and subjected to Western blot analysis with LC3B antibody, with GAPDH as the internal control.
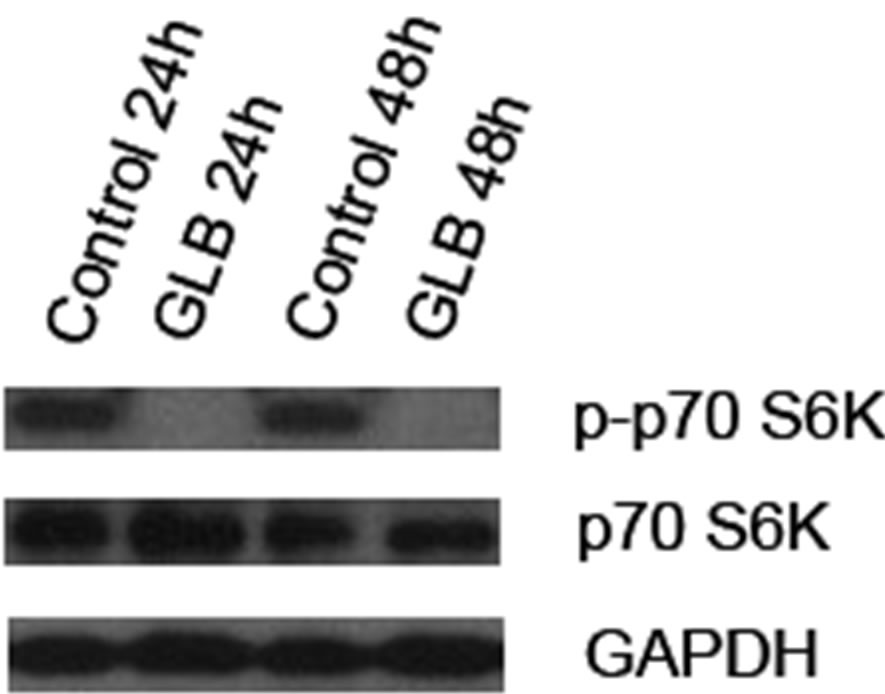
Figure 4. Glibenclamide inhibited phosphorylation of p70 S6K in INS-1 cells. INS-1 cells were left untreated (control) or treated with 10 μM glibenclamide for 24 or 48 h. Then cellular proteins were harvested and subjected to Western blot analysis with p-p70 S6K antibody, with p70 S6K and GAPDH as the internal controls.
 (a)
(a)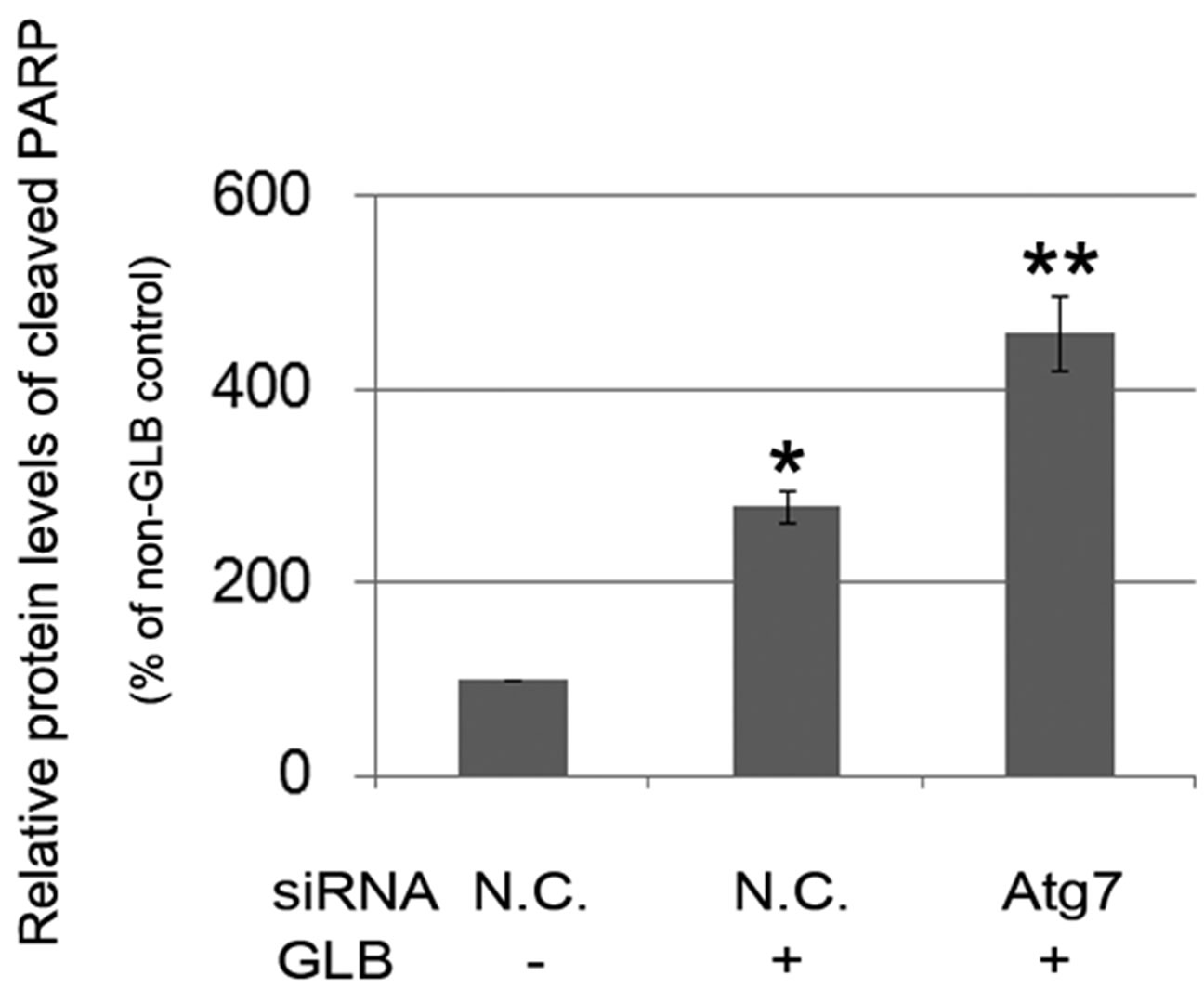 (b)
(b)
Figure 5. Glibenclamide-induced autophagy defended INS-1 cells against apoptosis. INS-1 cells were transfected with negative control siRNA (N.C.) or siRNA targeting rat Atg7 gene. 24 h after transfection, Atg7 protein levels were determined by Western blot and analyzed by Quantity One software (a), or cells were left untreated or treated with 10 μM glibenclamide for another 24 h, then cellular proteins were harvested and subjected to Western blot analysis with LC3B and PARP antibodies, with GAPDH as the internal control (b). (*: p < 0.05 versus GLB non-treated group; **: p < 0.05 versus N.C. siRNA transfected group, n = 3).
To further evaluate the impact of glibenclamide-induced autophagy on cell death, cell viabilities were determined with Atg7 knockdown. Upon glibenclamide treatment, Atg7 siRNA transfection resulted in decreased viability compared with that of the negative control siRNA (p < 0.05) (Figure 6), indicating that autophagy
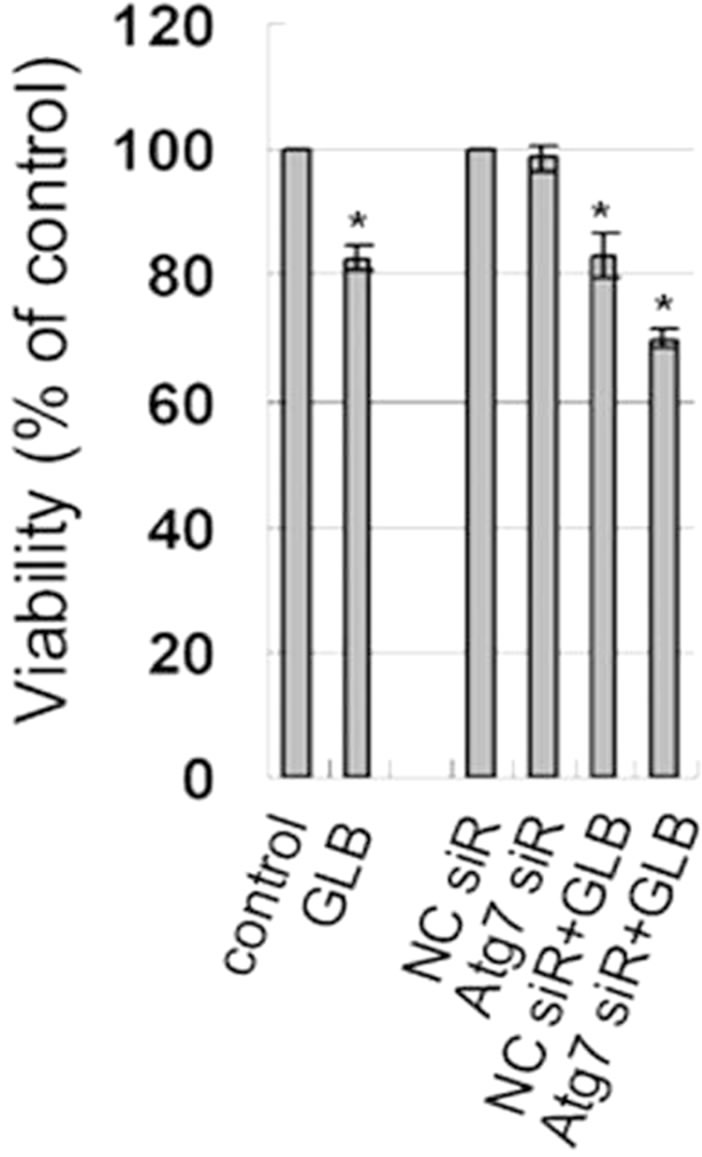
Figure 6. Effect of autophagy inhibition on cellular viability in INS-1 cells. INS-1 cells were transfected with negative control siRNA (N.C.) or siRNA targeting rat Atg7 gene. 24 h after transfection, cells were left untreated or treated with 10 μM glibenclamide for another 24 h. Cellular viabilities were determined by SRB assay. (*: p < 0.05 versus control, n = 3).
protected INS-1 cells from cell death upon glibenclamide treatment.
4. DISCUSSION
Glibenclamide used to be found to induce apoptosis in certain cell lines or animal models [12,24-26]. This may in part explain its side effects on patients taking this drug for diabetes treatment. In the current study, we found that besides causing apoptosis, glibenclamide gave rise to autophagy, which may serve as a protective mechanism against apoptosis in INS-1 cells. This is the first time that a sulfonylurea is reported to be an autophagy inducer. And it’s an intriguing expectation that the side effects of sulfonylureas could be alleviated via properly activating autophagy.
It has been widely observed that a common stimulator could simultaneously activate autophagy and apoptosis [10], as in the case of glibenclamide on INS-1 cells in our experiments. Autophagy plays double-edged roles for the survival and functioning of pancreatic beta-cells. It serves as a defence against apoptosis in some circumstances, but may lead to cell death under other conditions [1,2]. The decisive switch between beneficial and detrimental effects of autophagy has attracted much research interest these years, but still lacks a definite picture.
It seems that the cross talk between autophagy and apoptosis might be bridged by some common regulators [27]. Bcl-2 can interact with the autophagy regulator Beclin 1 and inhibit its function in autophagy activation [28-30]. The cross talk may also be the result of caspase-mediated cleavage of Beclin 1 [31]. Further more, it was recently found that the autophagy protein LC3B is also a positive regulator of apoptosis [32]. Therefore, it is likely that autophagy and apoptosis can act mutually on each other to cooperatively determine cell fate. In our study, glibenclamide-induced autophagy protected INS-1 cells against apoptosis. Though the underlying mechanism remains to be elucidated, it might be used as a model to study the mechanism of the protective roles of autophagy and the molecular links between autophagy and apoptosis.
Glibenclamide is a blocker of ATP-sensitive K+ channels. We need further experiments to investigate whether its effects on autophagy are carried out via blocking ATP-sensitive K+ channels or through a mechanism beyond its channel blocker role.
mTOR serves as an integrator of signals from nutrients, energy, growth factors and stress. Upon nutrient and energy abundance signals, or activation by growth factors including insulin or insulin-like growth factor (IGF), the activities of mTOR are elevated, thereby carrying out its role in promoting protein synthesis, anabolism and proliferation of cell and organisms. Whereas under deprivation of nutrients and energy, insufficient growth factor inputs, or existence of stress, mTOR is inactivated while autophagy is induced. Furthermore, in circumstances where mTOR is over activated and subsequently over phosphorylates and activates p70 S6K by excess food intake, a p70 S6K-dependent feedback loop is activated, which restrains the function of insulin receptor substrate 1 (IRS-1) and ultimately leading to insulin desensitization or adding to insulin resistance [33]. In this sense, glibenclamide-mediated down-regulation of the activities of p70 S6K may play a beneficial role in preventing the onset of insulin resistance in patients with excess food intake. But the degree of inhibition of p70 S6K actitivity must be delicately manipulated to avoid the adverse impact on the normal functions of it in protein synthesis.
An in-depth investigation into the biological roles and molecular mechanisms of autophagy in pancreatic betacells might help elucidating the pathophysiology of diabetes mellitus and exploring novel strategies for fighting this disease. It might be of research interest to use glibenclamide as a tool to study the molecular mechanism of autophagy in the development or prevention of diabetes.
5. CONCLUSION
Besides causing apoptosis of INS-1 rat insulinoma cells, glibenclamide induces autophagy. And autophagy may serve as a protective mechanism against glibenclamide-induced apoptosis in INS-1 cells. Therefore, the side-effect of glibenclamide in causing apoptosis of pancreatic beta cells might be alleviated via stimulating the autophagy machinery.
6. ACKNOWLEDGEMENTS
H. Su, X. Y. Liu, L. Su, L. Zhang, X. G. Liu, H. Q. Rong and H. Ji all contribute to conception and design of the work covered in this manuscript, to drafting and revising the article, and to the final approval of the submitted version of the manuscript.
This work was supported by Shandong Natural Science Foundation (ZR2011CM017) and Shandong Scientific and Technological Project (2009GG20002046 and 2009GG20002049).
REFERENCES
- Chen, Z.F., Li, Y.B., Han, J.Y., Wang, J., Yin, J.J., Li, J.B. and Tian, H. (2010) The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy, 7, 12-16. doi:10.4161/auto.7.1.13607
- Shintani, T. and Klionsky, D.J. (2004) Autophagy in health and disease: A double-edged sword. Science, 306, 990- 995. doi:10.1126/science.1099993
- Gonzalez, C.D., Lee, M.S., Marchetti, P., Pietropaolo, M., Towns, R., Vaccaro, M.I., Watada, H. and Wiley, J.W. (2010) The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy, 7, 2-11. doi:10.4161/auto.7.1.13044
- Jung, H.S. and Lee, M.S. (2010) Role of autophagy in diabetes and mitochondria. Annals of the New York Academy of Sciences, 1201, 79-83. doi:10.1111/j.1749-6632.2010.05614.x
- Pua, H.H., Guo, J., Komatsu, M. and He, Y.W. (2009) Autophagy is essential for mitochondrial clearance in mature T lymphocytes. The Journal of Immunology, 182, 4046-4055. doi:10.4049/jimmunol.0801143
- Ebato, C., Uchida, T., Arakawa, M., Komatsu, M., Ueno, T., Komiya, K., Azuma, K., Hirose, T., Tanaka, K., Kominami, E., et al. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metabolism, 8, 325-332. doi:10.1016/j.cmet.2008.08.009
- Banerjee, R., Beal, M.F. and Thomas, B. (2010) Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends in Neurosciences, 33, 541-59. doi:10.1016/j.tins.2010.09.001
- Jung, H.S., Chung, K.W., Won Kim, J., Kim, J., Komatsu, M., Tanaka, K., Nguyen, Y.H., Kang, T.M., Yoon, K.H., Kim, J.W., et al. (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metabolism, 8, 318-324. doi:10.1016/j.cmet.2008.08.013
- Masini, M., Bugliani, M., Lupi, R., del Guerra, S., Boggi, U., Filipponi, F., Marselli, L., Masiello, P. and Marchetti, P. (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia, 52, 1083-1086. doi:10.1007/s00125-009-1347-2
- Choi, S.E., Lee, S.M., Lee, Y.J., Li, L.J., Lee, S.J., Lee, J.H., Kim, Y., Jun, H.S., Lee, K.W. and Kang, Y. (2009) Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology, 150, 126-134. doi:10.1210/en.2008-0483
- Meijer, A.J. and Codogno, P. (2007) Macroautophagy: Protector in the diabetes drama? Autophagy, 3, 523-526.
- Remedi, M.S. and Nichols, C.G. (2008) Chronic antidiabetic sulfonylureas in vivo: Reversible effects on mouse pancreatic beta-cells. PLOS Medicine, 5, Article ID: e206. doi:10.1371/journal.pmed.0050206
- Koren, I., Reem, E. and Kimchi, A. (2010) Autophagy gets a brake: DAP1, a novel mTOR substrate, is activated to suppress the autophagic process. Autophagy, 6, 1179- 1180. doi:10.4161/auto.6.8.13338
- Vucicevic, L., Misirkic, M., Kristina, J., Vilimanovich, U., Sudar, E., Isenovic, E., Prica, M., Harhaji-Trajkovic, L., Kravic-Stevovic, T., Vladimir ,B., et al. (2010) Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/ mTOR pathway. Autophagy, 7, 40-50. doi:10.4161/auto.7.1.13883
- Zhang, Y. and Ren, J. (2010) Autophagy in ALDH2-elicited cardioprotection against ischemic heart disease: Slayer or savior? Autophagy, 6, 1212-1213. doi:10.4161/auto.6.8.13652
- Zhao, Y., Wang, L., Yang, J., Zhang, P., Ma, K., Zhou, J., Liao, W. and Zhu, W.G. (2010) Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy, 6, 988-990. doi:10.4161/auto.6.7.13289
- Jung, C.H., Ro, S.H., Cao, J., Otto, N.M. and Kim, D.H. (2010) mTOR regulation of autophagy. FEBS Letters, 584, 1287-1295. doi:10.1016/j.febslet.2010.01.017
- Kanazawa, T., Taneike, I., Akaishi, R., Yoshizawa, F., Furuya, N., Fujimura, S. and Kadowaki, M. (20004) Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. The Journal of Biological Chemistry, 279, 8452-8459. doi:10.1074/jbc.M306337200
- Daido, S., Yamamoto, A., Fujiwara, K., Sawaya, R., Kondo, S. and Kondo, Y. (2005) Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Research, 65, 4368-4375. doi:10.1158/0008-5472.CAN-04-4202
- Marchetti, P. and Masini, M. (2009) Autophagy and the pancreatic beta-cell in human type 2 diabetes. Autophagy, 5, 1055-1056. doi:10.4161/auto.5.7.9511
- Kraft, C., Peter, M. and Hofmann, K. (2010) Selective autophagy: Ubiquitin-mediated recognition and beyond. Nature Cell Biology, 12, 836-841. doi:10.1038/ncb0910-836
- Liu, X., Yue, P., Chen, S., Hu, L., Lonial, S., Khuri, F.R. and Sun, S.Y. (2007) The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Research, 67, 4981-4988. doi:10.1158/0008-5472.CAN-06-4274
- Mizushima, N., Yoshimori, T. and Levine, B. (2010) Methods in mammalian autophagy research. Cell, 140, 313-326. doi:10.1016/j.cell.2010.01.028
- Maedler, K., Carr, R.D., Bosco, D., Zuellig, R.A., Berney, T. and Donath, M.Y. (2005) Sulfonylurea induced betacell apoptosis in cultured human islets. The Journal of Clinical Endocrinology & Metabolism, 90, 501-506. doi:10.1210/jc.2004-0699
- Hambrock, A., de Oliveira Franz, C.B., Hiller, S. and Osswald, H. (2006) Glibenclamide-induced apoptosis is specifically enhanced by expression of the sulfonylurea receptor isoform SUR1 but not by expression of SUR2B or the mutant SUR1(M1289T). Journal of Pharmacology and Experimental Therapeutics, 316, 1031-1037. doi:10.1124/jpet.105.097501
- Sawada, F., Inoguchi, T., Tsubouchi, H., Sasaki, S., Fujii, M., Maeda, Y., Morinaga, H., Nomura, M., Kobayashi, K. and Takayanagi, R. (2008) Differential effect of sulfonylureas on production of reactive oxygen species and apoptosis in cultured pancreatic beta-cell line, MIN6. Metabolism, 57, 1038-1045. doi:10.1016/j.metabol.2008.01.038
- Maiuri, M.C., Zalckvar, E., Kimchi, A. and Kroemer, G. (2007) Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nature Reviews Molecular Cell Biology, 8, 741-752. doi:10.1038/nrm2239
- Heneka, M.T., Rodriguez, J.J. and Verkhratsky, A. (2010) Neuroglia in neurodegeneration. Brain Research Reviews, 63, 189-211. doi:10.1016/j.brainresrev.2009.11.004
- Lian, J., Karnak, D. and Xu, L. (2010) The Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy, 6, 1201- 1203. doi:10.4161/auto.6.8.13549
- Oh, S., Pirooz, S.D., Ni, D., Zhao, Z. and Liang, C. (2010) Anti-autophagic Bcl-2: Not just an innocent bystander. Autophagy, 7, 56-57.
- Djavaheri-Mergny, M., Maiuri, M.C. and Kroemer, G. (2010) Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene, 29, 6508. doi:10.1038/onc.2010.514
- Chen, Z.H., Lam, H.C., Jin, Y., Kim, H.P., Cao, J., Lee, S.J., Ifedigbo, E., Parameswaran, H., Ryter, S.W. and Choi, A.M. (2010) Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proceedings of the National Academy of Sciences of the United States of America, 107, 18880-18885. doi:10.1073/pnas.1005574107
- Zoncu, R., Efeyan, A. and Sabatini, D.M. (2010) mTOR: From growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology, 12, 21- 35.
NOTES
*Duality of interest: there is no conflict of interest regarding this work.
#Equal contributors.