International Journal of Otolaryngology and Head & Neck Surgery
Vol.1 No.3(2012), Article ID:24785,6 pages DOI:10.4236/ijohns.2012.13015
Applicability of PCR-DGGE and 16S rDNA Sequencing for Microbiological Analysis of Otitis Media with Effusion
1Department of Otorhinolaryngology, Tartu University Hospital, Tartu, Estonia
2Institute of Microbiology, University of Tartu, Tartu, Estonia
Email: priit.kasenomm@kliinikum.ee
Received September 3, 2012; revised November 3, 2012; accepted November 15, 2012
Keywords: Otitis Media with Effusion; 16S rDNA Targeted PCR; DGGE Fingerprinting
ABSTRACT
Background: The aim of the study was to analyze the performance of PCR-DGGE based assay and its applicability as a tool for the identification of bacteria in the middle ear of children with otitis media with effusion (OME). Methods: The middle ear effusions from 20 children with OME were analyzed both by bacterial culture and by 16S rDNA-gene-targeted PCR assay, DGGE fingerprinting and sequencing analysis. Results: In bacterial culture assay, only three middle ear effusions (15%) showed bacterial growth. None of the samples were positive for anaerobic culture. The PCR assay with 16S rDNA-gene-targeted universal primers was positive in 10 (50%) cases. The subsequent DGGE fingerprinting and 16S rDNA sequencing analysis revealed that the most commonly encountered bacteria in the middle ear effusions of children with OME are Haemophilus influenzae, Alloiococcus otitidis and Bacteroides spp. Conclusions: The present study demonstrated the applicability of PCR-DGGE based assay and 16S rDNA sequencing for analyzing of bacterial diversity in the middle ear effusion of children OME. The results of our study may contribute to a better understanding of the etiology of OME.
1. Introduction
Otitis media with effusion (OME) is defined as the presence of fluid in the middle ear cleft without clinical symptoms or signs of acute ear infection [1]. Although OME is one of the most common diseases of childhood, its etiology and pathogenesis are still under debate. OME was considered a sterile condition for a long time; however, recent studies have demonstrated bacterial growth in 21% to 70% of the middle ear effusions (MEE) [2-6]. The most frequently found pathogens are S. pneumoniae, H. influenzae and M. catarrhalis. Less frequently cultured aerobic bacteria have been A. otitidis, S. aureus, S. epidermidis, S. pyogenes and Corynebacterium spp. In some studies, different anaerobic bacteria have also been cultured from MEEs of OME patients, such as Peptostreptococcus spp., Prevotella spp., Bacteroides spp., Fusobacterium spp. and Propionibacterium spp. [4-6]. During the last decade, several studies have applied more sensitive molecular methods, most often PCR-based assays, for detection of middle ear pathogens in OME. The overall rate of PCR-positive effusions from patients with OME has been significantly higher than by conventional culture, varying from 46% to 100% [3,4,7,8]. The bacteria found by PCR-based assays are usually the same as listed above. The advantage of PCR-based assays over conventional culture is their higher sensitivity and the possibility to detect fastidious and difficult to culture microorganisms, like intracellular microbes or those entrapped within the biofilm [9,10]. Compared to conventional culture, recovery rate of A. otitidis, Chlamydia pneumoniae, Mycoplasma pneumoniae, several anaerobes and various viruses is far superior by PCR [8,11,12].
However, the PCR-based assay is time consuming and requires prior knowledge of microbial composition on particular microbiota. PCR approach combined with denaturing gradient gel electrophoresis (PCR-DGGE) allows assessing the structure of complex biological systems using single step PCR. It enables to simultaneously detect broad range of different species of bacteria, as well as different strains of the single species, and is suggested for its rapidity and reliability for diseases of polymicrobial nature [13]. The general principle of DGGE is the separation of individual rRNA genes based on differences in chemical stability of genes. These methods separate multitemplate PCR products as bands on gels according to GC content, dependent on melting behaveiors of the amplicons as they migrate thorough the gels. [14]. PCR-DGGE may be used for whole community analysis, or for the investigation of specific populations or groups within the sample. This method has been widely used for analysing human faecal microbiota [15], monitoring dynamic changes in mixed bacterial populations over time [16], assessing the effect of antibiotic therapy, assessment of the microbial composition in mouth [17], stomach [18] and cerebrospinal fluid [19].
In the present study, the bacterial composition of MEEs of children with OME was investigated by means of PCR using 16S rRNA-targeted universal primers, followed by denaturing gradient gel electrophoresis (DGGE) and 16S rRNA sequencing analysis. The performance of PCR-DGGE based assay and its applicability as a tool for the identification of bacteria in the middle ear of children with OME was analyzed.
2. Material and Methods
2.1. Study Design and Diagnosis of OME
The study group consisted of 20 children (12 boys and 8 girls) with OME, who were referred to the Department of Otolaryngology, Tartu University Hospital, for tympanostomy tube insertion. The age of the patients ranged between 1 to 6 years, mean 4.3 ± 2.8 years. The diagnosis of OME was made by finding effusion in the middle ear cleft, without symptoms and signs of acute infection. The diagnosis was supported with type B tympanogram and audiogram that indicated conductive hearing loss. The lack of response to medical treatment with oral antihistamines or topical decongestants for at least 3 months was an indication for ventilation tube insertion. OME was bilateral in all cases. Children with purulent MEE, with a systemic disease, those who received antibiotic treatment during the previous month were excluded. Informed consent from the parents of all children and approval from the local ethics committee were obtained for the use of the specimens.
MEEs were obtained by myringotomy under the general anesthesia with the help of an operating microscope. The ear canal was first cleaned and myringotomy was performed in the anteroinferior part of the tympanic membrane. A MEE sample was aspirated under sterile conditions with an electric suction device into a TymTap collector (Juhn Tym-Tap, Xomed Inc., Jacksonville, Florida, USA). MEE was collected randomly from one of the middle ear clefts of the each child. Some amount of the fluid was removed by a streile cotton probe, placed into the Stuart transport medium and taken to the Microbiology Laboratory for aerobic and anaerobic culture.
2.2. Bacteriological Analysis
The samples were seeded on a Columbia agar base supplemented with 7% horse blood and chocolate agar with with Vitox supplement for aerobic bacteria, and on Wilkins-Chalgren agar for anaerobic bacteria. The plates were incubated at 37˚C in a microaerobic or anaerobic atmosphere for a maximum of one week. All the media and supplements were from Oxoid Ltd., UK.
2.3. DNA Extraction
DNA was extracted from all MEEs by the cetyltrimethylammonium bromide (CTAB) method with slight modifications [20]. All samples were first lyophilized using freeze dryer (Christian Martin Ltd., Germnany). The lyophilized powder was mixed with 1 ml of lysis buffer (200 mM Tris-HCl (pH 8.0), 25 mM EDTA, 300 mM NaCl, 1.2% sodium dodecyl sulfate) and 20 µl of proteinase K (400 µg/ml) was added. The mixture was incubated at 37˚C for 24 h. Thereafter, 200 µl of 5 M NaCl was added and samples were vortexed for few seconds; 160 µl of CTAB/NaCl solution was added, followed by incubation for 10 min at 65˚C. The lysate was extracted with an equal volume of phenol-chloroform and precipitated with ethanol. A DNA pellet was collected by centrifugation, washed with 70% ethanol and finally resuspended in TE buffer (10 MM Tris-HCl (pH 8.0), 0.1 mM EDTA).
2.4. PCR Amplification and DGGE Analysis
The PCR was performed in a reaction volume of 50 µl containing 10 mM deoxyribonucleotide triphosphate each, 1.25 U Taq polymerase (Invitrogen, USA), 10x reaction buffer, 10 µmol of the each primer and 200 ng (1 µl) of DNA solution. The set of primers (968-GC-f, 5’-AACGCGAAGAACCTTA-3’; and 1401-r, 5’-GGTGTGTACAAGACCC-3’) was used to amplify V6 to V8 regions of the 16S rRNA gene [14,21]. The PCR mixture was subjected to 35 amplification cycles (30 s at 94˚C, 20 s at 55˚C, and 40 s at 72˚C). DGGE analysis on PCR products was performed using a DcodeTM System apparatus (Bio-Rad, Hercules, CA). Polyacrylamide gels (8% w/v) acrylamide-bisacrylamide (37.5:1) in 0.5 x Tris-acetic acid-EDTA buffers with a denaturing gradient was prepared with a gradient mixer and Econopump (Bio-Rad). Gradients from 30% to 60% were employed for the separation of the products amplified.
2.5. Cloning of PCR Products
The PCR was performed with primers 8f (5’-CACGGCGGATCCAGAGTTTGAT(C/T)(A/C)TGGCTCACAG-3’) and 1501r (5’-GTGAAGCTTACGG(C/T)TACCTTGTTACGACTT-3’) to amplify the bacterial 16S rRNA [15,21]. The PCR amplicons were purified and concentrated with the QIAquick PCR purification kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and cloned in Escherichia coli JM109 using the pGEM-T vector system (Promega, Madison, WI). Colonies for sequencing were selected according to the migration position of the PCR fragment of the clone in DGGE in comparison with the fragments in the original DGGE profile. The plasmid DNA of the selected transformants was isolated using the QIAprep spin miniprep kit (Qiagen).
2.6. Sequence Analysis
Sequencing of the cloned PCR fragments was carried out using purified plasmid DNA and the sequencing primers SP6 and T7 (Promega). Sequencing reactions were performed with Sequenase sequencing kit (Amersham, Slough, UK) according to the manufacturer’s instructions. The sequences were analyzed with automatic LI-COR DNA Sequencer 4000 L (Lincoln, USA) and corrected manually. Sequence alignment of the complementary strands was carried out using the DNASTAR SEQMAN program (Madison, USA). Similarity searches for the 16S rRNA gene sequences were performed in the GenBank database using BLAST algoritm.
2.7. Statistical Analysis and Calculation of Similarity Indices
DGGE gels were scanned and analysed by using the software of Bionumetrics 2.5 (Applied Maths, Belgium) [22]. The statistical analyses were performed using SigmaStat 2.0 (Jandel Scientific, USA) software programs, Chi-square test. The differences were considered statistically significant if the p < 0.05.
3. Results
A total of 20 MEE samples from children with OME were analyzed using bacteriological and 16S rDNA denaturing gradient gel electrophoresis (DGGE) followed by sequence analysis. The molecular analysis of MEEs was more sensitive than bacteriological (10 of 20 (50%) vs 3 of 20 (15%); p = 0.043). H. influenzae was isolated in one and M. catarrhalis (b-lactamase positive) in two MEEs. All cultures for anaerobic bacteria were negative.
Following DGGE analysis of PCR amplicons revealed that each MEEs consisted of multiple 16S rDNA gene sequences, each assumed to represent a unique bacterial DNA (Figure 1). The number of clearly recognizable DNA fragments in DGGE profiles varied between 2 to 5, forming a unique DGGE profile for each MEE sample. In further analysis, the most dominating DNA fragments from DGGE profiles were isolated and sequenced. The obtained sequences were compared with the sequences in GenBank database (Table 1). The most common bacteria were H. influenzae and Bacteroides spp., each found in 4 MEE samples. A. otitidis was found in two samples (Table 1). Those three bacteria were predominating in all 10 PCR positive MEEs. Sequence analysis also revealed that MEEs may contain not only different species of bacteria but also different strains of single bacteria as in case for H. influenzae, A. otitidis and Bacteroides spp. (patients 5, 6, 8 and 14).
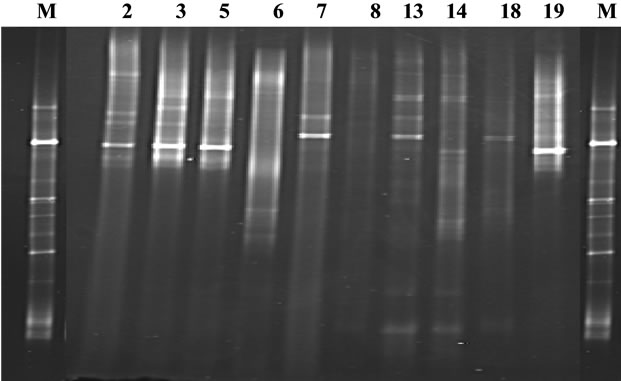
Figure 1. PCR-DGGE profile of amplified V6-V8 regions of the 16S rDNA gene of middle ear effusions. Lane M—indicates the marker for DGGE, constructed from 16S rRNA amplicons; lanes 2, 3, 5, 6, 7, 8, 13, 14, 18, 19—DGGE profile of PCR positive patients.


Table 1. The results of sequence analysis of the dominant 16S rDNA gene amplicons found by PCR-DGGE fingerprinting.
4. Discussion
During the last decade, both conventional culture and PCR-based methods have widely been applied to assess the role of different microorganisms in the etiology of OME. The advantage of PCR-based methods over bacterial culture is the higher sensitivity and the possibility to detect fastidious and difficult to culture microorganisms. On the other hand, the positive culture is the only definite proof of the presence of viable bacteria, but PCR may indicate bacterial DNA rather than the bacteria itself. In the present study, the recovery rate of bacteria in MEEs of children with OME by culture was 15%, yielding M. catarrhalis and H. influenzae in one and two samples, respectively. These two microorganisms, together with S. pneumoniae, are among the most common infectious agents implicated in both AOM and OME [2,6]. The incidence of bacterial growth in the MEE of our patients was lower than in many previous studies, where the rate varies from 21% to 70% [2-6]. However, such lower rate of positive culture for OME has similarly been reported previously [8,23].
In accordance with other studies, the PCR with 16S rDNA targeted universal primers proved to be far more sensitive than conventional culture [2,3,7,8]. The presence of bacterial DNA was demonstrated 50% of MEEs of children with OME. However, the limitation of PCRbased assays is that they target only one or in some studies up to four pathogens simultaneously, and depend on prior knowledge of the microbial composition of the particular microbiota [7]. Moreover, the role of detected microorganisms in the etiology of OME is impossible to assess based solely on the colonization. Therefore, PCR amplicons were further analyzed by DGGE fingerprinting. DGGE is a technique which separates DNA fragments of the same length but with different base pair sequences, according to the point at which they denature. It also enables to assess the predominating microbes as the intensity of the fragments in DGGE corresponds semi-quantitatively with the abundance of the particular microorganism in the sample. We suggest that the predominating bacteria could be among those which are critical or even involved in the etiology of OME. The PCR-DGGE assay for detection of bacteria in MEE has not been used in previous studies. DGGE is the method of choice when the desired information does not have to be as phylogenetically exhaustive as that provided by cloning, but still relatively precise to determine the dominant members of a microbial community with medium phylogenetic resolution.The advantage of PCRDGGE approach is that bands of interest can be excised and sequenced to obtain information about the species that they represent. The limitation of DGGE is that heterologous sequences may migrate similarly, and thus bands at the same position in the gel are not necessarily phylogenetically related (13,15). However, application of sequencing of 16S rRNA quite solves this problem.
In the present study, PCR-DGGE fingerprinting demonstrated that each MEE from OME patients contained at least 2 to 5 predominating 16S rDNA gene sequences, each assumed to represent a unique bacterial DNA. Subsequent cloning and sequence analysis of DGGE fragments revealed that the most predominating bacteria in our patients with OME were H. influenzae, A. otitidis and Bacteroides spp. Those three bacteria were predominating in all 10 PCR positive MEEs. Moreover, sequence analysis also revealed that particular MEEs may contain not only different species of bacteria, but also different H. influenzae, A. otitidis and Bacteroides strains. The role of H. influenzae, A. otitidis and Bacteroides spp. in the etiology of OME is not completely understood. H. influenzae is a part of normal bacterial flora of the upper respiratory tract. Most H. influenzae strains are opportunistic pathogens, causing a disease only when host-dependent factors create an opportunity, such as in the case of AOM. The incidence of H. influenzae in OME has been from 4% to 22% by culture, and from 18% to 56% by PCR [2,6,7]. A. otitidis is another fastidious, slowly growing aerobic gram-positive diplococcus frequently encountered in the upper respiratory tract. The recovery rate of A. otitidis from MEEs of children with OME has been up to 5% by culture and up to 50% by PCR [7,8]. It was considered as an important middle ear pathogen after its first recovery from OME, but its etiological role in OME has been doubted in more recent studies [11,24]. Bacteroides spp. is anaerobic bacteria representing one of the most important groups of human commensals in the gastrointestinal, genitourinary and respiratory tracts [25]. Bacteroides spp. and other anaerobes could be recovered up to one-third of MEEs of OME patients [4,6]. Anaerobic bacteria have been associated with chronic and recurrent forms of otitis media [4]. All those three groups of bacteria are generally considered as the commensals in OME, which have possibly been translocated from the nasopharynx through the Eustachian tube to the middle ear. Although their exact role in the etiology of OME is unclear, their presence in the MME of OEM patients may have an impact on the treatment of once the acute recurrence of middle ear infection occurs. For example, Haemophilus influenzae and Bacteroides spp. strains are well-known beta-lactamase producers, which may help to survive pathogens in the same microbiota despite the presence of beta-lactams, the most commonly used antimicrobial agents in respiratory tract infections in childhood.
5. Conclusion
The present study demonstrated the applicability of PCRDGGE based assay and 16S rDNA sequencing for analyzing of bacterial diversity in the middle ear effusion of children OME. This method may have general usefulness in characterizing bacterial populations at the site of infection and may indicate microorganisms that are candidates for further investigation to gain a better understanding of the etiology of OME.
6. Acknowledgements
We are grateful for the critical review of the manuscript by Marika Mikelsaar and for the help and contribution by Maris Suurna. This research was supported by SF109870 from Estonian Science Foundation.
REFERENCES
- R. M. Rosenfeld, L. Culpepper, K. J. Doyle, K. M. Grunfast, A. Hoberman, M. A. Kenna, A. S. Lieberthal, M. Mahoney, R. A. Wahl, C. R. Woods and B. Yawn, “Clinical Practice Quideline: Otitis Media with Effusion,” Otolaryngology—Head and Neck Surgery, Vol. 130, No. 5, 2004, pp. S95-S118. doi:10.1016/j.otohns.2004.02.002
- J. C. Post, R. A. Preston, J. J. Aul, M. Larkins-Pettigrew, J. Rydquist-White, K. W. Anderson, R. M. Wadowsky, D. R. Reagan, E. S. Walker, L. A. Kingsley, A. E. Magit and G. D. Ehrlich, “Molecular Analysis of Bacterial Pathogens in Otitis Media with Effusion,” Journal of the American Medical Association, Vol. 273, No. 23, 1995, pp. 1598-1604. doi:10.1001/jama.273.20.1598
- U. Gok, Y. Bulut, E. Keles, S. Yalcin and M. Ziya Doymaz, “Bacteriological and PCR Analysis of Clinical Material Aspirated from Otitis Media with Effusions,” International Journal of Pediatric Otorhinolaryngology, Vol. 60, No. 1, 2001, pp. 49-54. doi:10.1016/S0165-5876(01)00510-9
- I. Brook, P. Yocum and K. Shah, “Aerobic and Anaerobic Bacteriology of Concurrent Chronic Otitis Media with Effusion and Chronic Sinusitis in Children,” Archives of Otolaryngology—Head and Neck Surgery, Vol. 126, No. 2, 2000, pp. 174-176.
- I. Brook, P. Yocum, K. Shah, B. Feldman and S. Epstein, “Microbiology of Serous Otitis Media in Children: Correlation with Age and Length of Effusion,” Annals of Otology, Rhinology, and Laryngology, Vol. 110, No. 1, 2001, pp. 87-90.
- I. Brook, P. Yocum, K. Shah, B. Feldman and S. Epstein, “Aerobic and Anaerobic Bacteriological Features of Serous Otitis Media in Children,” American Journal of Otolaryngology, Vol. 4, No. 6, 1983, pp. 389-392. doi:10.1016/S0196-0709(83)80044-1
- P. H. Hendolin, L. Paulin and J. Ylikoski, “Clinically Applicable Multiplex PCR for Four Middle Ear Pathogens,” Journal of Clinical Microbiology, Vol. 38, No. 1, 2000, pp. 125-132.
- A. J. Beswick, B. Lawley, A. P. Fraise, A. L. Pahor and N. L. Brown, “Detection of Alloiococcus Otitis in Mixed Bacterial Populations from Middle-Ear Effusions of Patients with Otitis Media,” Lancet, Vol. 354, No. 9176, pp. 386-389. doi:10.1016/S0140-6736(98)09295-2
- L. Hall-Stoodley, Z. H. Hu, A. Gieseke, L. Nistico, D. Nguyen, J. Hayes, M. Forbes, D. P. Greenberg, B. Dice, A. Burrows, P. A. Wackym, P. Stoodley, J. C. Post, G. D. Ehrlich and J. E. Kerschner, “Direct Detection of Bacterial Biofilms on the Middle-Ear Mucosa of Children with Chronic Otitis Media,” Journal of the American Medical Association, Vol. 296, No. 2, 2006, pp. 202-211. doi:10.1001/jama.296.2.202
- H. Coates, R. Thornton, J. Langlands, P. Filion, A. D. Keil, S. Vijayasekaran and P. Richmond, “The Role of Chronic Infection in Children with Otitis Media with Effusion: Evidence for Intracellular Persistence of Bacteria,” Otolaryngology—Head and Neck Surgery, Vol. 138, No. 6, 2008, pp. 778-781. doi:10.1016/j.otohns.2007.02.009
- K. Leskinen, P. Hendolin, A. Virolainen-Julkunen, J. Ylikoski and J. Jero, “The Clinical Role of Alloiococcus otitidis in Otitis Media with Effusion,” International Journal of Pediatric Otorhinolaryngology, Vol. 66, No. 1, 2002, pp. 41-48. doi:10.1016/S0165-5876(02)00186-6
- M. Storgaard, B. Tarp, T. Ovesen, B. Vinther, P. L. Andersen, N. Obel and J. S. Jensen, “The Occurrence of Chlamydia pneumoniae, Mycoplasma pneumoniae, and Herpesviruses in Otitis Media with Effusion,” Diagnostic Microbiology and Infectious Disease, Vol. 48, No. 2, 2004, pp. 97-99. doi:10.1016/j.diagmicrobio.2002.03.001
- X. Wang, S. P. Heazlewood, D. O. Krause and M. Florin, “Molecular Characterization of the Microbial Species That Colonize Human Ileal and Colonic Mucosa by Using 16 rDNA Sequence Analysis,” Journal of Applied Microbiology, Vol. 95, No. 3, 2003, pp. 508-520. doi:10.1046/j.1365-2672.2003.02005.x
- G. Muyzer, E. C. De Waal and A. G. Uitterlinder, “Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction Amplified Genes Coding for 16S rRNA,” Applied and Environmental Microbiology, Vol. 59, No. 3, 1993, pp. 695-700.
- E. G. Zoetendal, A. D. L. Akkermans and W. M. de Vos, “Molecular Characterization of Microbial Communitiesbased on 16 rRNA Sequence Diversity,” In: L. Dijkhoorn, K. J. Towner and M. Struelens, Eds., New Approaches for Generation and Analysis of Microbial Typing Data, Elsevier Science, Amsterdam, 2001, pp. 267-298. doi:10.1016/B978-044450740-2/50012-5
- C. F. Favier, E. V. Vaughan, W. M. De Vos and A. D. L. Akkermans, “Molecular Monitoring of Succession of Bacterial Communities in Human Neonates,” Applied and Environmental Microbiology, Vol. 68, No. 1, 2002, pp. 219-226. doi:10.1128/AEM.68.1.219-226.2002
- J. Maukonen, M.-L. Mätto, M. Suihko and M. Saarela, “Intra-Individual Diversity and Similarity of Salivary and Faecal Microbiota,” Journal of Medical Microbiology, Vol. 57, No. 12, 2008, pp. 1560-1568. doi:10.1099/jmm.0.47352-0
- H. J. Monstein, A. Tiveljung, C. H. Kraft, K. Borch and J. Jonasson, “Profiling of Bacterial Flora in Gastric Biopsies from Patients with Helicobacter pylori-Associated Gastritis and Histologically Normal Control Individuals by Temperature Gradient Gel Electrophoresis and 16S rDNA Sequence Analysis,” Journal of Medical Microbiology, Vol. 49, No. 9, 2000, pp. 817-822.
- B. E. Ley, C. J. Linton, S. Longhurst, H. Jalal and M. R. Millar, “Eubacterial Approach to the Dianosis of Bacterial Infection,” Archives of Disease in Childhood, Vol. 77, No. 2, 1997, pp. 148-149. doi:10.1136/adc.77.2.148
- K. Wilson, “Preparation of Genomic DNA from Bacteria,” In: F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman and J. A. Smith, Eds., Current Protocols in Molecular Biology, Greene Publishing Associates/Wiley Interscience, New York, 1987.
- D. J. Lane, “16S/23S rRNA Sequencing,” In: E. R. Stackebrandt and M. Goodfellow, Eds., Nucleic Acid Techniques in Bacterial Systematics, John Wiley & Sons, New York, 1991, pp. 115-147.
- N. Fromin, J. Hamelin, S. Tarnawski, D. Roesti, K. Jourdain-Miserez, N. Forestier, S. Teyssier-Cuvelle, F. Gillet, M. Aragno and P. Rossi, “Statistical Analysis of Denaturating Gel Electrophoresis (DGGE) Fingerprinting Patterns,” Environmental Microbiology, Vol. 4, No. 11, 2002, pp. 634-643. doi:10.1046/j.1462-2920.2002.00358.x
- M. Saffer, J. F. L. Neto, O. B. Piltcher and V. F. Petrillo, “Chronic Secretory Otitis Media: Negative Bacteriology,” Acta Otolaryngologica, Vol. 116, No. 2, 1996, pp. 836- 839. doi:10.3109/00016489609137936
- K. Tano, R. von Essen, P. O. Erikson and A. Sjöstedt, “Alloiococcus otitidis—Otitis Media Pathogen or Normal Bacterial Flora?” APMIS, Vol. 116, No. 9, 2008, pp. 785- 790. doi:10.1111/j.1600-0463.2008.01003.x
- H. R. Jousimies-Somer, P. H. Summanen, H. Wexler, S. M. Finegold, S. E. Gharbia and H. N. Shah, “Bacteroides, Porphyromonas, Prevotella, Fusobacterium, and Other Anaerobic Gram-Negative Bacteria,” In: P. R. Murray, E. J. Baron, J. H. Jorgensen, M. A. Pfaller and R. Y. Yolken, Eds., Manual of Clinical Microbiology, 8th Edition, ASM Press, Washington DC, 2003, pp. 880-901.