American Journal of Analytical Chemistry
Vol.06 No.03(2015), Article ID:53725,11 pages
10.4236/ajac.2015.63020
Stereoselective High-Performance Liquid Chromatography Tandem Mass Spectrometry Method for Determination of R- and S-Acenocoumarol in Human Plasma and Its Application in a Pharmacokinetics Study
International Pharmaceutical Research Center (IPRC), Amman, Jordan
Email: *dr.salem@iprc.com.jo
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 December 2014; accepted 30 January 2015; published 2 February 2015
ABSTRACT
A stereoselective, sensitive and rapid liquid chromatography-tandem mass spectrometry (LC- MSMS) method for the determination of R-acenocoumarol and S-acenocoumarol in human plasma was developed and validated at IPRC bioanalytical labs. The procedure involved solid phase extraction of both enantiomers and their corresponding internal standard. The chromatographic separation was accomplished employing a chiral column and proper mobile phase. Detection was carried out using Waters Micromass® Quattro Premier mass spectrometer in multiple reaction monitoring (MRM) mode using turbo ion spray with negative ionization. The method was validated over a linear range of 0.40 - 40.00 ng/ml for R-acenocoumarol and 0.20 - 20.00 ng/ml for the S-acenocoumarol. Method validation covered different parameters such as linearity, accuracy, precision and stability. The method was successfully applied for the determination of R and S- acenocoumarol in plasma samples of 28 healthy subjects who participated in a pharmacokinetics study.
Keywords:
Stereoselective, Chromatography, Mass Spectrometry, LC-MSMS, Acenocoumarol, Pharmacokinetics

1. Introduction
Acenocoumarol, 4-hydroxy-3-[1-(4-nitrophenyl)-3-oxobutyl]-2H-1-benzopyran-2-one (Figure 1), is a vitamin K antagonists that belongs to the group of most frequently used drugs worldwide [1] -[3] . Acenocoumarol is indicated for the long-term prevention of thromboembolic. It is characterized by a narrow therapeutic index and large inter and intraindividual variations of their dose-response relationship [4] . Acenocoumarol has a single, chiral centre that gives rise to two different enantiomeric forms, of which the S-form is approximately 2- to 5- fold more potent than its R-counterpart [1] - [3] .
Several analytical techniques have been published for the analysis of acenocoumarol in different matrices, e.g., racemic determination by high performance liquid chromatography (HPLC-UV) in plasma [5] or plasma and urine [6] . Thijssen et al. reported in 2001 [7] the use of HPLC-UV stereoselective quantification of the R and S- acenocoumarol in human plasma after derivatization with N-carbobenzoyl-Lproline. The authors also reported the determination of 6- and 7-hydroxylated metabolites in urine by HPLC-UV. On the other hand, a two-dimen- sional-enantioselective LC-MSMS technique was reported for the determination of warfarin, phenprocoumon and acenocoumarol [8] . Kollroser and Schober described in 2002 achairal LC-MSMS method for the determination of coumarin-type anticoagulants in human plasma by using ion trap detector [9] . The mentioned methods either suffered drawbacks since either were not validated [5] - [7] or implicated derivatization [7] , or were tedious and somehow complicated [8] or were achairal [9] .
Surprisingly, though of its major importance, to date, acenocoumarol detailed pharmacokinetics in human is still not well documented in the literature. One of the reasons is due to the absence of well-documented specific and sensitive validated chiral method for the determination of R- and S-acenocoumarol after oral administration of the drug in a well controlled clinical/pharmacokinetics study. Therefore, the main aim of our work was to develop and validate a highly specific and sensitive stereoselective liquid chromatographic-mass spectrometric method for the determination of R- and S-acenocoumarol in human plasma, as well, its application for the enantioselective pharmacokinetics evaluation of both enantiomers after oral dosing in human healthy subjects.
2. Experimental
2.1. Reagents
R- and S-acenocoumarol were purchased from Cayman (Ann Arbor, MI, USA), while and acenocoumarol-D5 (Internal Standard, IS) was purchased from BDG (Lower Hutt, NZ). Analytical grade for HPLC acetonitrile, methanol, formic acid, ammonium acetate, hydrochloric acid were supplied by Merck (Darmstadt, Germany). Water was purified using a Milli-Q® (Millipore, France). Six different batches of plasma were obtained from healthy blood donors who were proved to be HIV, hepatitis B & C negative.
2.2. Preparation of Stock and Working Solutions
Stock solution of R- and S-acenocoumarol were prepared using dissolving appropriate amount in methanol to

Figure 1. Structures of R- and S-acenocoumarol and the internal standard acenocoumarol-D5.
produce a concentration of 100 μg/ml of either analytes. Stock solutions were stored at −70˚C. Working solu- tions of R- and S-acenocoumarol were prepared by diluting the above mentioned stocks in methanol/dionized water to a final concentration of 8 and 4 μg/ml of R- and S-acenocoumarol, respectively.
The internal standard (acenocoumarol-D5) stock Standard solution was prepared in methanol with a final con- centration of 0.10 mg/ml. Working solution was prepared by dilution in methanol/dionized water to a final concentration of 20.0 ng/ml.
2.3. Calibration Curves
Calibration curve standards were prepared by spiking human blank plasma with serial dilutions of the working standards to produce the calibration curve standards of (0.40/0.20), (1.00/0.50), (2.00/1.00), (4.00/2.00), (10.00/ 5.00), (20.00/10.00), (30.00/15.00) and (40.00/20.00) of R-acenocoumarol/S-acenocoumarol, respectively. Each calibration curve standards contained also 20.0 ng/ml of the internal standard acenocoumarol-D5.
A double blank plasma sample (no IS) and a single blank plasma (containing 20.0 ng/ml of the internal standard) were used as part of each run. Neither the double blank sample nor the single blank was used to construct the calibration function. Calibration curves were run daily together with quality control samples.
2.4. Quality Control Samples
Quality control samples were prepared and labeled as: QC-2, QC-3, QC-4 and QC-5 with final concentration of (1.20/0.60), (8.00/4.00), (16.00/8.00), and (28.00/14.00), R-acenocoumarol/S-acenocoumarol, respectively. Each quality control sample contained also 20.0 ng/ml of the internal standard acenocoumarol-D5.
2.5. Extraction
All QC, calibration curve standards, single blank and double blank samples were extracted using a solid phase extraction technique. Two hundred µl of the unknown plasma samples were spiked with 100 µl of acenocoumarol- D5 of 20 ng/ml and vortexed for 10 s. Then after, two hundred µl of 2% formic acid were added to each sample and vortexed for 30 s. Samples then were centrifuged for 5 min at 16,000 g. Samples were applied to solid phase extraction cartridges that were conditioned previously with water and methanol. The cartridges were then washed with 1 ml 0.1% HCl. Samples elution was done by using 1 ml of methanol. Immediately after, samples were evaporated under a flow of nitrogen gas. Samples were reconstituted in 300 µl of mobile phase then vortexed for 30 s and transferred to 250 ml glass insert tubes. Only 25 µl were injected into the LC-MSMS system.
2.6. Chromatographic Conditions
After several attempts using different columns and mobile phase combinations, the separation and determination of the R- and S-acenocoumarol and IS were carried out using Astec chirobiotc V chiral column, 5 µm × 4.6 mm × 100 mm (Supelco, Bellefonte, PE, USA) thermostated at 35˚C. The mobile phase consisted of AcN-MeOH-5 mM ammonium acetate with formic acid (15:15:70). The flow rate was fixed at 0.6 ml/min. Samples were kept in the autosampler at 5˚C.
2.7. Mass Spectrometric Conditions
R- and S-acenocoumarol monitoring and quantitation were achieved using Waters Micromass® Quattro Premier mass spectrometer (Milford, MA, USA) set at unit resolution in the multiple reaction monitoring (MRM) mode using turbo ion spray with negative mode ionization. The analyses were run by Waters MassLynx® software. The mass spectrometric conditions were optimized to obtain maximum sensitivity for R- and S-acenocoumarol and (IS). To achieve this, 100 ng/ml solution from each one of these solutions were prepared separately in mobile phase and infused into mass detector using syringe pump in the infusion mode. The best mass detector specific parameters were found to be: capillary (KV): 0.60; cone (V): 43; extractor (V): 4.00; RF lens: 0.90; source temperature: 120; desolvation temperature: 400; cone gas flow (L/Hr): 250; desolvation gas flow (L/Hr): 900; collision energy for R- and S-acenocoumarol: 30; collision energy for acenocoumarol-D5: 30; multiplier (V): 650.
Monitoring and quantitation were conducted at m/z 352.10 > 265.00 (R- and S-acenocoumarol), while IS (acen- ocoumarol-D5) monitoring and quantitation was done at m/z 357.10 > 270.00.
The detection in MS/MS technique is highly specific and sensitive, nevertheless, endogenous substances can exist in much higher concentration than the analytes of interest and may coelute with those affecting the ionization of the analytes leading to high imprecision and loss of sensitivity [10] . In order to determine ion suppression matrix effect profiles, analytes were infused into the mobile phase through a T-connection between the column and the interface while injecting the extracted blank plasma samples. The purpose of this post- column infusion with the analytes is to raise the background level so the suppression matrix will appear as negative peaks.
2.8. Data Treatment
The linearity of this method of analysis was tested for the range of concentrations of 0.40 - 40.00 ng/ml for R- acenocoumarol and 0.20 - 20.00 ng/ml for the S-acenocoumarol. Calibration curves were prepared by determining the best-fit of peak area ratios (peak area analyte/peak area internal standard) vs. concentration, and fitted to the equation y = bx + a by weighted least-squares regression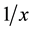 .
.
2.9. Application
The method was applied to analyze the samples of healthy male adult subjects who participated in a pharmacokinetics study. Healthy Jordanian male subjects gave written informed consent to participate in the study before screening. The study was approved by the Institutional Review Board at IPRC and was performed in accordance with the Good Clinical Practice Guideline and the Declaration of Helsinki [11] [12] . The twenty eight subjects who participated in the study were within 15% of their ideal body weight and were judged to be healthy based on medical history, physical examination, complete blood count, serum chemistry, and lack of hepatic, renal, respiratory, cardiac, or gastrointestinal conditions. Subjects were negative for HIV, hepatitis B and C viruses, and the presence of illegal drugs. The subjects did not participate in any other clinical study, donate blood, or require hospitalization within 3 months before initiation of both studies.
After 10-hour overnight fasting, a single dose of 1 mg racemic acenocoumarol tablets was administered orally followed by 240 ml of water. Blood samples (7 ml) were collected in citrate tubes from an indwelling catheter in the antecubital vein of the forearm before dosing and at 0.00 (pre-dose), 0.33, 0.66, 1.00, 1.33, 1.66, 2.00, 2.50, 3.00, 3.50, 4.00, 4.50, 5.00, 6.00, 8.00, 12.00, 24.00 and 48.00 hours (post-dose). Blood samples were centrifuged for 5 minutes at 1789 g and stored at −70˚C until analysis.
Subjects were under continuous medical observation by the investigators and attending physicians during the study. Follow-up medical testing (vital signs, physical examination, ECG, biochemistry, hematology, and urinalysis) was conducted on all volunteers at discharge from the study.
3. Results and Discussion
3.1. Separation and Specificity
ESI is a “gentle” ionization technique that produces high mass-to-charge [M-]-precursor ions with minimal frag- mentation of the analyte. R- and S-acenocoumarol and internal standards gave protonated precursor [M-1]-in the MS mode. The major ions observed were m/z 352.10 for R- and S-acenocoumarol and 357.10 for the internal standard acenocoumarol-D5. The most intense product ions observed in the MSMS spectra were m/z 265.00 for R- and S-acenocoumarol and 270.00 for the internal standard.
The combination of HPLC (under the isocratic conditions described) with ESI-MSMS leads to relatively short retention times and high selectivity and sensitivity. The SRM chromatograms obtained from extracted double blank plasma samples are depicted in Figure 2, indicating no endogenous peaks at the retention times (tR) of analytes or internal standard. No interferences of the analytes were observed due high selectivity of the MSMS technique.
As observed, Figure 3 shows the chromatogram of blank plasma samples where no ion suppression effects were observed.
3.2. Method Validation
The LC-MSMS method has been validated according to currently accepted US Food and Drug Administration


Figure 2. LC-MSMS chromatograms of R-acenocoumarol, S-acenocoumarol double blank plasma sample obtained from one of the volunteers listed top to bottom.
(FDA) bioanalytical method validation guidance [13] . The following parameters were considered.
To test the specificity, six double blank and six blank samples were prepared using six different batches of human plasma. Samples were tested for interference using the proposed extraction procedure and chromato- graphic/mass spectrometric conditions and were compared with those at LLOQ. No significant interference at the retention time of the drug or internal standard were observed as illustrated in the chromatograms presented in Figure 4.
The method was validated over a linear range of 0.40 - 40.00 ng/ml for R-acenocoumarol with back calculated accuracy of 98.33 - 101.83 (CV% 1.23 - 5.00), while the range was validated as 0.20 - 20.00 ng/ml for the S-acenocoumarol with backcalculated accuracy of 96.67% - 102.82% (CV% 1.33 - 7.07). In all analytical runs, the coefficients of determination were better than 0.98.


Figure 3. LC-MSMS chromatograms of R-acenocoumarol, S-acenocoumarol blank plasma sample obtained from one of the volunteers listed top to bottom.
The intra-day accuracy results obtained by analyzing six plasma samples at LLOQ, low, mid and high QC for R-acenocoumarol ranged between 98.75% - 103.33% (CV% 1.39 - 2.78); while for S-acenocoumarol ranged 90.00% - 103.29% (CV% 1.57 - 6.09).
The inter-day precision and accuracy results obtained by analyzing six spiked samples of R-acenocoumarol and S-acenocoumarol, low, mid and high QC over three consecutive days are depicted in Table 1. The limits of detection were 0.4 ng/ml for R-acenocoumarol and 0.2 ng/ml for S-acenocoumarol. The lower limits of quantitation chromatograms of the two analytes are depicted in Figure 4. Results are presented in Table 1.
The obtained results were within the acceptance criteria of not more than 20% deviation at LLOQ and not more than 15% deviation for standards above this point (LLOQ).
The matrix suppression was determined by injecting pure authentic standard solution of R-acenocoumarol, S-acenocoumarol or internal standard at low, mid and high QC and compared to the same solution reconstituted


Figure 4. LC-MSMS chromatograms showing plasma samples spiked with of R-acenocoumarol, S-acenocoumarol at LLOQ, chromatograms are listed top to bottom.
in extracted blank human plasma from 6 different batches. Matrix mean factor was found to be 5.28% (4.31 CV%) for R-acenocoumarol and 4.24% (5.31 CV%) for the S-acenocoumarol.
Table 2 shows the data representing the stability of R- and S-acenocoumarol in plasma samples over four cycles of freeze (−70˚C) and thawing (room temperature). The tests indicated that the analytes were stable in human plasma for four cycles of freeze and thaw when stored at −70˚C and thawed to room temperature.
The post-preparative stability of QC samples kept in the autosampler for 39 hrs was also assessed. The mean stability at low and high QC was 102.02% - 103.89% for R-acenocoumarol; while for S-acenocoumarol it was 101.08% - 101.57%. The precision ranged between 0.89% - 3.95%. The results indicated that analytes can remain at the autosampler at 5˚C for at least 39 hr, without showing significant loss in the quantified values, and that samples should be processed within this period of time.

Table 1. Interday accuracy and precision.
N: Number of QC samples included in the calculations of mean accuracy, SD & CV%.
Table 2. Freeze and thaw stability after 4 cycles at −70˚C.
For short-term stability determination, stored plasma aliquots were thawed and kept at room temperature for a period of time exceeding that expected to be encountered during the routine sample preparation. Samples were extracted and analyzed as abovementioned. Results are given below in Table 3. Short-term stability indicated reliable stability behavior under the experimental conditions of the regular runs.
Table 4 summarizes the long-term stability data of the two analytes in human plasma samples stored for a period of more than 130 days at −70˚C. The stability study of R- and S-acenocoumarol in human plasma showed reliable stability behavior as the mean of the results of the tested samples were within the acceptance criteria of ±15% of the initial values. These findings indicated that storage of R- and S-acenocoumarol plasma samples at −70˚C is adequate, and no stability-related problems would be expected during the samples routine analysis for pharmacokinetics or clinical studies.
The recoveries were evaluated for R- and S-acenocoumarol by comparing peak areas of the extracted samples with the unextracted pure authentic standard solutions peak areas at two low and high QC. Results are presented in Table 5.
The stability of the stock solution of R-acenocoumarol was tested and established for 45 days at −70˚C and was found to be 99.620% (CV% 0.69), while kept at room temperature was stable for at least for 6 hr with 98.75% recovery (1.27 CV%). S-acenocoumarol stock solution stability was evaluated to be at −70˚C equal to 100.09% (CV% 1.44), while at room temperature was 97.85% (CV% 0.71). Working solution of R-acenocoumarol was stable at −70˚C 100.21% (CV% 1.82) and at room temperature 98.56% (CV% 2.40). The S-acenocoumarol showed similar behavior where stability at −70˚C was 101.79% (2.01 CV%) and 97.62% (2.56 CV%) at room temperature.
The stability of the stock solution of the internal standard was tested for 16 days at −70˚C and was found to be 100.37% (CV% 0.69), while at room temperature was stable for at least 6 hrs with 101.27% recovery (1.28 CV%). Its working solution was stable for 10 days at −70˚C (100.80%) and for six hours (100.26%) at room temperature. CV% were 1.21 and 2.13, respectively.
Table 3. Short-term stability at room temperature.
Table 4. Long-term stability at −70˚C.
Table 5. Analytical recovery*.
N: Number of QC samples included in the calculations of mean accuracy, SD & CV%. *Analytical recovery calculated by comparing peak area of (QC spiked plasma samples) with standard solution spiked in extracted blank plasma samples.
4. Application
This specific and precise method was applied to analyze plasma samples obtained after the administration of a single dose of 1 mg racemic acenocoumarol to 28 healthy subjects in a controlled pharmacokinetic study. Figure 5 shows chromatograms of subjects’ extracted samples.
The mean pharmacokinetics parameters are depicted in Table 6, while the mean plasma concentration-time profiles of R- and S-acenocoumarol of all participants are represented in Figure 6.


Figure 5. Representative chromatograms of a plasma sample obtained from a volunteer after oral administration of 1 mg of racemic acenocoumarol. Backcalculated concentrations were 11.069 ng/ml of R-acenocoumarol, and 13.736 ng/ml of S-acenocoumarol. Chromatograms are listed top to bottom.
Table 6. The mean pharmacokinetics parameters in twenty-eight healthy subjects after a single oral dose of 1 mg racemic acenocoumarol tablets.

Figure 6. Mean plasma concentration-time profiles of R-acenocoumarol, S- acenocoumarol in 28 healthy subjects after a single-dose oral administration of 1 mg tablet containing racemic acenocoumarol.
5. Conclusion
The proposed method of analysis provided a sensitive and specific assay for R- and S-acenocoumarol determination in human plasma. It was shown that this method is suitable for the analysis of both enantiomers in human plasma samples collected for pharmacokinetics or clinical studies in humans.
Conflict of Interest
The authors confirm that this article content has no conflict of interest.
Acknowledgements
The authors wish to thank the staff (physicians, pharmacists, chemists, nurses and technicians) at IPRC for their help and cooperation.
References
- Hirsh, J., Dalen, J.E., Anderson, D.R., Poller, L., Bussey, H., Ansell, J., Deykin, D. and Brandt, J.T. (1998) Oral Anticoagulants: Mechanism of Action, Clinical Effectiveness, and Optimal Therapeutic Range. Chest, 114, 445S-469S. http://dx.doi.org/10.1378/chest.114.5_Supplement.445S
- Hirsh, J., O’Donnell, M. and Weitz, J.I. (2005) New Anticoagulants. Blood, 105, 453-463. http://dx.doi.org/10.1182/blood-2003-12-4195
- Weitz, J.I. (2004) New Anticoagulants for Treatment of Venous Thrombo-Embolism. Circulation, 110, I19-I26. http://dx.doi.org/10.1161/01.CIR.0000140901.04538.ae
- Saraeva, R.B., Paskaleva, I.D., Doncheva, E., Eap, C.B. and Ganev, V.S. (2007) Pharmacogenetics of Acenocoumarol: CYP2C9, CYP2C19, CYP1A2, CYP3A4, CYP3A5 and ABCB1 Gene Polymorphisms and Dose Requirements. Journal of Clinical Pharmacy and Therapeutics, 32, 641-649. http://dx.doi.org/10.1111/j.1365-2710.2007.00870.x
- Thijssen, H.H., Baars, L.G. and Reijnders, M.J. (1983) Analysis of Acenocoumarin and Its Amino and Acetamido Metabolites in Body Fluids by High-Performance Liquid Chromatography. Journal of Chromatography B, 274, 231-238. http://dx.doi.org/10.1016/S0378-4347(00)84426-6
- Thijssen, H.H., Janssen, G.M. and Baars, L.G. (1986) Lack of Effect of Cimetidine on Pharmacodynamics and Kinetics of Single Oral Doses of R- and S-Acenocoumarol. European Journal of Clinical Pharmacology, 30, 619-623. http://dx.doi.org/10.1007/BF00542424
- Thijssen, H.H., Drittij, M.J., Vervoort, L.M. and de Vries-Hanje, J.C. (2001) Altered Pharmacokinetics of R- and S-Aceno- coumarol in a Subject Heterozygous for CYP2C9*3. Clinical Pharmacology & Therapeutics, 70, 292-298. http://dx.doi.org/10.1067/mcp.2001.117936
- Vecchione, G., Casetta, B., Tomaiuolo, M., Grandone, E. and Margaglione, M. (2007) A Rapid Method for the Quantification of the Enantiomers of Warfarin, Phenprocoumon and Acenocoumarol by Two-Dimensional-Enantioselective Liquid Chromatography/Electrospray Tandem Mass Spectrometry. Journal of Chromatography B, 850, 507-514. http://dx.doi.org/10.1016/j.jchromb.2006.12.050
- Kollroser, M. and Schober, C. (2002) Determination of Coumarin-Type Anticoagulants in Human Plasma by HPLC- Electrospray Ionization Tandem Mass Spectrometry with an Ion Trap Detector. Clinical Chemistry, 48, 84-91.
- Willoughby, R., Sheehan, E. and Mitrovich, S. (2002) A Global View of LC/MS. 2nd Edition, Global View Publishing, Pittsburgh.
- The European Medicines Agency (EMA) (2014) Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95). www.ema.europa.eu/
- World Medical Association (2000) Declaration of Helsinki. Amended by the 52nd World Medical Assembly (WMA), Edinburgh. (Note Added in Tokyo, 2004 and Updated in Seoul, 2008)
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) (2001) Guidance for Industry, Bioanalytical Method Validation.
NOTES

*Corresponding author.