American Journal of Analytical Chemistry
Vol. 3 No. 2 (2012) , Article ID: 17283 , 5 pages DOI:10.4236/ajac.2012.32016
Validation of a HPLC Method for Determination of Glutamine in Food Additives Using Post-Column Derivatization
1Departamento de Fármacos e Medicamentos, Faculdade de Ciências Farmacêuticas de Araraquara, Universidade Estadual Paulista (UNESP), Araraquara, Brazil
2Faculdade de Americana, Americana, Brazil
3Programa de Pós-Graduação em Ciências Farmacêuticas, Faculdade de Ciências Farmacêuticas de Araraquara, Universidade Estadual Paulista (UNESP), Araraquara, Brazil
Email: *lucmsil@yahoo.com.br
Received November 1, 2011; revised December 4, 2011; accepted December 15, 2011
Keywords: Glutamine; Food Additive Product; Liquid Chromatography
ABSTRACT
An ion-exchange chromatography (IEC) followed by post-column derivatization method was validated for the determination of glutamine in food additive products. The chromatographic method was carried out on a sodium cation-exchange column and using detection at 570 nm. The mobile phase consisted of sodium eluant, pH 3.15, with 5% sulfolane, sodium eluant, pH 7.40 and sodium column regenerant. The method was linear in the range of 4 - 50 µg/mL (r2 = 0.9999). The accuracy was 99.78%. Moreover, method validation demonstrated acceptable results for precision and robustness. The method was found to be able to use for routine analysis of the glutamine.
1. Introduction
Glutamine is a multifaceted amino acid that plays key roles in many metabolic pathways and also fulfils essential signaling functions. Although classified as non-essential, recent evidence suggests that glutamine is a conditionally essential amino acid in several physiological situations [1]. Glutamine might be a good nitrogen source in the mammals diet, since higher organisms are not capable to synthesize nitrogen-containing organic compounds from inorganic salts [2]. It has multiple physiological roles and functions: a precursor of nucleic acids, amino sugars, and proteins, an important nitrogen transporter, and a carrier of ammonia. It serves as a primary fuel source for rapidly dividing cells such as enterocytes and lymphocytes and as a prime source or energy for small intestine mucosa cells [3]. The chemical structure of glutamine is represented in Figure 1.
This amino acid may become essential under conditions of catabolic disease states such as sepsis, trauma, major surgery, burns, and uncontrolled diabetes [4]. Thus, during events of stress, the requirement for glutamine may be greater than the ability of the body to produce it, and exogenous supplementation may be required. These observations serve as the rationale for inclusion of glutamine in nutritional products formulated for patients under conditions of metabolic stress or malabsorptive disorders [3].
The literature describe HPLC method for the quantitative analysis of glutamine, glucose, lactate and alanine on suspended mammalian cell reactors, using isocratic elution of a mobile phase constituted of 5 mM Ca(NO3)2 (pH 5.5) [5]. A reversed-phase liquid chromatography method (RP-LC) was described for the determination of glutamine and N-acetylglutamine in aqueous solution and in a liquid nutritional product, using isocratic elution at a flow-rate of 1 mL/min [3]. Moreover, HPLC method
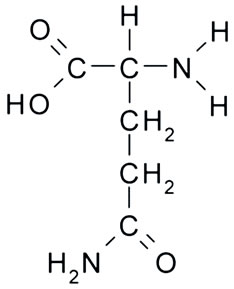
Figure 1. Chemical structure of glutamine.
was validated with electrochemical detection for glutamate, glutamine and GABA determination in brain homogenates. The protocol was based on a precolumn derivatization of amino acids with o-phthalaldehyde and sodium sulfite, a separation through a C18 column and an isocratic elution [6].
Although there are recently developed methods of amino acid analyses, such as gas chromato-graphy, which are faster and less expensive, ion exchange method of amino acid analysis is the best despite its being expensive, and will continue to be used for this purpose for years to come because of its reproducibility, recently achieved better speed, and complete automation [7].
The ion exchange takes place on resin, consisting of small spherical beads of polystyrene, reacted with divinylbenzene to achieve the required degrees of cross linkage between the two polymerised chains of styrene, and sulphonated to provide an electrical charge. The chromatographic column is filled with resins of negative charge, and the amino acids are put on the column at a low pH value (about 2.0), hence all of them bear a positive charge. In these conditions all of the amino acids will link to the resin, no chromatographic division will occur, and the amino acids are waiting at the beginning of the column for a change in conditions. If the pH and the ionic strength of the elution buffers increase, the isoelectric point of the amino acids will be reached, and the attraction of the ions towards the resin diminishes and so the amino acids will be eluted from the column. The conditions of the separation of the amino acids can be modifed in a way that the isoelectric points, for all amino acids, are to be reached at various times [8].
There is a limitation in amino acids analysis concerning sensitive and/or selective detection after chromatographic run. These substances often lack from a suitable chromophore, fluorophore or electrophore and have to be detected by the UV-absorption within the wavelength range from 205 to 230 nm. This range, however, is very unspecific because a lot of substances show UV-absorption therein [9,10]. This detection problem could be solved by derivatization procedures which allow transforming amino acids onto detectable forms. Many publications have reported the use of derivatization methods for improving a protein’s detectability [11].
Ion-exchange chromatography followed by post-column derivatization has been a method of choice for amino acid analysis for many years. In the post-column derivatization, the analytes are reacted with derivatizing reagent after their HPLC separation and before they reach the detector [9]. The advantage of this process is the previous separation of reaction interfering analytes. This constitutes an advantage when dealing with unknown, complex mixtures [12]. Furthermore, the amino acids are directly injected into the chromatographic column, which avoids the decomposition of derivates before injection or inside the chromatographic system. In this approach, the resolution of the derivates and quantitativity of the reactions is not needed [13]. Ninhydrin is the most frequently applied reagent for residue derivatization in the postcolumn approach. It is still considered the standard method of the technology. The advantages of the method are that it is robust, reliable, and reproducible and it can detect a large number of amino acids and homologs [12]. Ninhydrin reacts to decarboxylate the amino acids, and yield an intensively colored purple blue product having absorption maximum at 570 nm. The reaction of ninhydrin with amino acid is very sensitive and is represented in Figure 2 [7].
Validated procedures to evaluation of glutamine in food additive products were not found in the literature. Thus, the aim of this study was to validate IEC method for analysis of this important amino acid. The method uses a mobile phase without organic solvents, UV-visible detection and does not require complicated sample preparation. The availability of this rapid and selective method will be very useful for determination of glutamine. The method was validated in accordance with the specifications of the International Conference on Harmonization [14].
2. Material and Methods
2.1. Chemicals and Reagents
The glutamine reference substance and glutamic acid reference substance were supplied by Sigma-Aldrich (St Louis, Missouri, USA). Batches of glutamine in food additive products were obtained from production process of Ajinomoto (São Paulo, Brazil). Analytical grade sodium eluant, pH 3.15, with 5% sulfolane, sodium eluant, pH 7.40, sodium column regenerant and ninhydrin reagent were acquired from Pickering Laboratories (Mountain View, California, USA). All chemicals used were special analytical grade. For all of the analyses, ultrapure water was purified using an Elix 3 coupled to a Milli-Q Gradient A10 system (Millipore, Bedford, MA, USA).
2.2. Apparatus and Chromatographic Conditions
An Agilent 1100 LC system (Agilent Technologies, Waldbronn, Germany) equipped with quaternary pump, column oven, refrigerated autosampler and UV-Vis detector was used. The LC system was coupled with Pickering Laboratories PCX 5200 post-column derivatization system (Mountain View, California, USA).
2.3. Ion-Exchange Chromatography (IEC)
The experiments were performed on a high efficiency sodium cation-exchange Pickering Laboratories column (4.0 × 150 mm). A security guard holder was used to
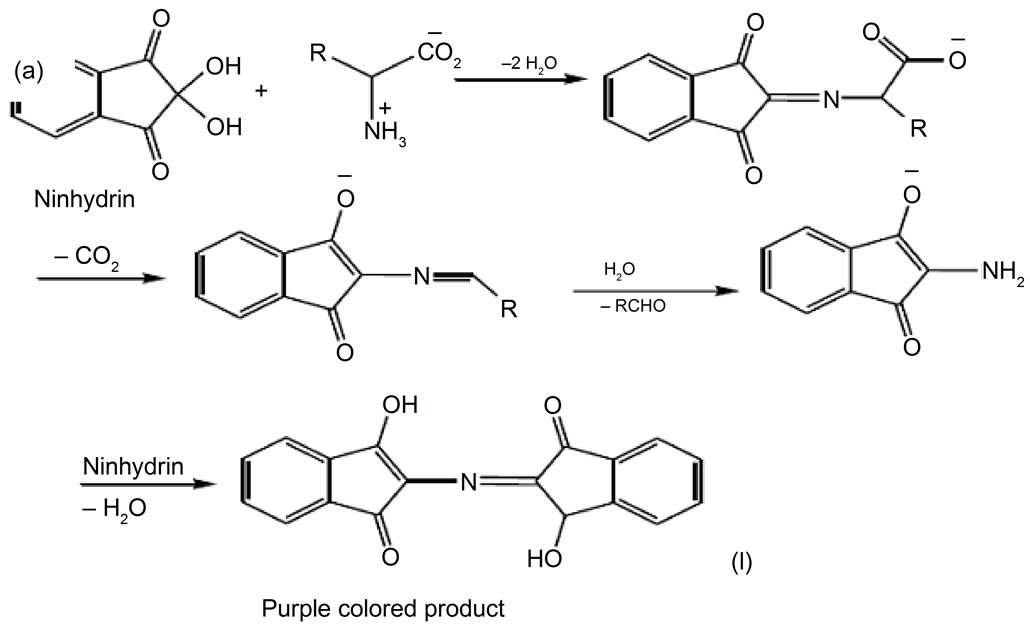
Figure 2. Reactions of ninhydrin with amino acid (Khan and Faiz, 2008).
protect the analytical column. The Agilent LC system was operated using a mobile phase consisted sodium eluant, pH 3.15, with 5% sulfolane, sodium eluant, pH 7.40, sodium column regenerant and using UV-Vis detection at 570 nm. The gradient program was implemented as follows: time = 0 min, A:B:C (100:0:0); time = 12 min, A:B:C (100:0:0); time = 34 min, A:B:C (0:100:0); time = 53 min, A:B:C (0:100:0); time = 53.1 min, A:B:C (0:0:100); time = 55 min, A:B:C (0:0:100); time = 55.1 min, A:B:C (100:0:0), maintaing this proportion until time = 67 min. The flow-rate of the mobile phase was 0.4 mL/min, and the flow-rate of the derivatizating reagent was 0.3 mL/min. The column temperature was set at 48˚C and the post column reaction equipment was kept at 130˚C temperature. The temperature of the auto-sampler was kept at 5˚C and the injection volume was 10 µL for both standard and samples.
2.4. Preparation of Samples and Standard Solutions
Working standard and sample solutions of glutamine were prepared daily by diluting the standard for glutamine and the samples of food additive products in ultrapure water, to a final concentration of 25 µg/mL.
3. Experimental
3.1. Validation of the IEC Method
Once the chromatographic and the experimental conditions were optimized, the method was validated by the determination of the following parameters: specificity, linearity, precision, accuracy, limit of detection and limit of quantitation following the International Conference on Harmonisation guidelines [14].
3.1.1. Specificity
Specificity of the method towards the amino acid was established through the determination of the resolution between peaks of glutamine and glutamic acid reference substances. The solution of two amino acids was prepared by mixture of equal volumes of the glutamine (25 µg/mL) and glutamic acid (25 µg/mL) solutions.
3.1.2. Linearity
Linearity was determined by constructing an analytical curve with five reference substance concentrations of the glutamine in the range of 4 - 50 µg/mL prepared in ultrapure water. Before injection of the solutions, the column was equilibrated for at least 20 min with the mobile phase flowing through the system. Three replicate of 10 μL injections of the reference solutions were made to verify the repeatability of the detector response. The peak areas of the chromatograms were plotted against the respective concentrations of glutamine reference substance to obtain the analytical curve. The results were subjected to regression analysis by the least squares method to calculate calibration equation and determination coefficient.
3.1.3. Precision
The precision of the method was determined by repeatability (intra-day) and intermediate precision (inter-day). Repeatability was examined by three evaluations of the three concentrations of glutamine standard, on the same day, under the same experimental conditions. The intermediate precision of the method was assessed by carrying out the analysis on three different days (inter-days). Intra-and inter-day precision were expressed as relative standard deviation (RSD).
3.1.4. Accuracy
To confirm the accuracy of the method, recovery was determined at three concentrations, by adding known amounts of the reference substance at the samples of production process, to obtain solutions at concentrations of 4, 25 and 50 µg/mL, equivalent to 16%, 100% and 200% of the nominal analytical concentration, respectively. The accuracy was calculated as the percentage of the reference substance recovered from the samples.
3.1.5. Limits of Detection and Quantitation
The limits were defined by the determination of the signalto-noise ratio by comparison of measured signals from samples with known low concentrations of analyte with those of blank samples and by establishing the minimum concentration at which the analyte was reliably detected, for the limit of detection (signal-to-noise ratio is 3:1), and quantified with acceptable accuracy and precision, for the limit of quantitation (signal-to-noise ratio is 10:1).
4. Results and Discussion
4.1. Optimization of Chromatographic Conditions
To obtain the best chromatographic conditions, the mobile phases were optimized to provide appropriate selectivity and sensitivity. The use of a gradient elution resulted in better sensitivity, improving the peak symmetry with the retention time also suitable for the separation of glutamic acid and glutamine. The optimized conditions of the LC method were validated for the analysis of glutamine in food additive products.
4.2. Method Validation
The results of the method validation are shown in Table 1.
4.2.1. Specificity
The analysis of the glutamine standard showed the specificity of the analytical method. The resolution of glutamic acid and glutamine peaks was 5.31 and the tailing factor of glutamine peak was 0.99. The symmetrical peak corresponding to glutamine, with the retention time of 10.1 min were shown in Figure 3, demonstrating also that the proposed method is able to separate glutamic acid and glutamine.
4.2.2. Linearity
The analytical curve constructed for glutamine was found to be linear in the 4 - 50 µg/mL range. The value of the
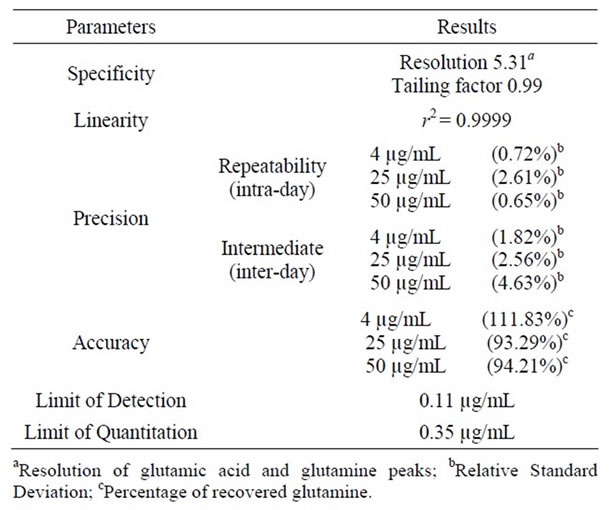
Table 1. Validation results of IEC for glutamine in food additive products.
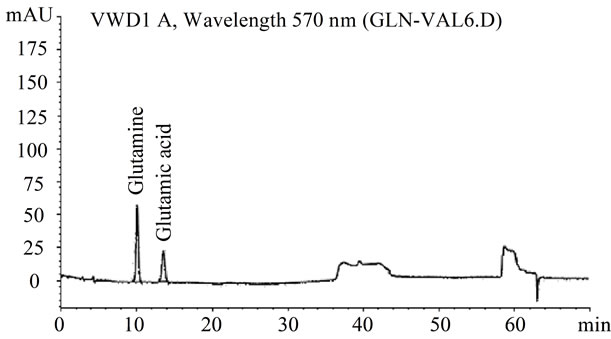
Figure 3. Representative IEC chromatograms of glutamic acid and glutamine.
determination coefficient calculated (r2 = 0.9999, y = 72.97350x – 16.22441, where, x is concentration and y is the peak absolute area) indicated the linearity of the analytical curve for the method.
4.2.3. Precision
The precision evaluated as the repeatability of the method was studied by calculating the relative standard deviation (RSD) for three determinations of the concentrations 4, 25 and 50 µg/mL performed on the same day and under the same experimental conditions. The RSD values obtained were 0.72%, 2.61% and 0.65%, respectively.
The intermediate precision was assessed by calculating the relative standard deviation (RSD) for determinations of the three levels of concentration on three different days (inter-day). The RSD values obtained between days were 1.82%, 2.56% and 4.63%, for samples 4, 25 and 50 µg/mL, respectively.
4.2.4. Accuracy
The accuracy was calculated as the percentage of the drug recovered from three replicate determinations of three different solutions containing 4, 25 and 50 µg/mL, constituted of glutamine sample and standard (50:50). The absolute means obtained of recovered glutamine were 111.83%, 93.29% and 94.21%, respectively, demonstrating that the method is accurate within the desired range.
4.2.5. Limits of Detection and Quantitation
The limit of detection and limit of quantitation determined were 0.11 µg/mL and 0.35 µg/mL, respectively.
Based on the findings of the present work, it can be concluded that analysis via ion-exchange chromatography (IEC) followed by post-column derivatization method, is indeed a viable alternative for determining glutamine quality. This method can be useful in quality control during and after production of glutamine food additive products.
5. Conclusion
The results of the validation studies show that the IEC method is sensitive with a limit of quantitation of 0.35 µg/mL, accurate with a mean value of 99.78%, and possesses significant linearity (r2 = 0.9999). The separation was achieved with the retention time of 10.1 min, and the method has been successfully used for analysis of glutamine. Moreover, the validated physicochemical technique employed, offered a high degree of resolving power and selectivity and is suggested as an analytical alternative to improve the quality control of food additive products containing glutamine.
6. Acknowledgements
This work was supported by CNPq-Brazil program and PACD-FCF-UNESP.
REFERENCES
- J. M Matés, J. A. Segura, J. A. Campos-Sandoval, C. Lobo, L. Alonso, F. J. Alonso and J. Márquez, “Glutamine Homeostasis and Mitochondrial Dynamics,” The International Journal of Biochemistry & Cell Biology, Vol. 41, No. 10, 2009, pp. 2051-2061. doi:10.1016/j.biocel.2009.03.003
- U. Hellmann, I. Lüderwald, M. Neuhaüser and Z. Fresen, “Determination of N-acetyl-L-glutamine in Urine by HPLC,” Fresenius’ Journal of Analytical Chemistry, Vol. 325, No. 3, 1986, pp. 290-292. doi:10.1007/BF00498176
- M. K. Snowden, J. H. Baxter, M. Mamula Bergana, I. Reyzer and V. Pound, “Stability of N-Acetylglutamine and Glutamine in Aqueous Solution and in a Liquid Nutritional Product by an Improved HPLC Method,” Journal of Food Science, Vol. 67, No. 1, 2002, pp. 384-389. doi:10.1111/j.1365-2621.2002.tb11415.x
- J. M. Lacey and D. W. Wilmore, “Is Glutamine Conditionally Essential Amino Acid?” Nutrition Reviews, Vol. 48, No. 8, 1990, pp. 297-309. doi:10.1111/j.1753-4887.1990.tb02967.x
- E. Frave, P. Pugeaud and P. Péringer, “Automated HPLC Monitoring of Glucose, Glutamine, Lactate and Alanine on Suspended Mammalian Cell Reactors,” Biotechnology Techniques, Vol. 4, No. 5, 1990, pp. 315-320.
- A. A. Monge-Acuña and J. Fornaguera-Trías, “A High Performance Liquid Chromatography Method with Electrochemical Detection of Gamma-Aminobutyric Acid, Glutamate and Glutamine in Rat Brain Homogenates,” Journal of Neuroscience Methods, Vol. 183, No. 2, 2009, pp. 176-181. doi:10.1016/j.jneumeth.2009.06.042
- A. S. Khan and F. Faiz, “Amino Acid Analysis Using Ion Exchange Resins,” Journal of Natural Sciences and Mathematics, Vol. 48, No. 1-2, 2008, pp. 1-17.
- J. Csapó, Cs. Albert, K. Lóki and Zs. Csapó-Kiss, “Separation and Determination of the Amino Acids by Ion Exchange Column Chromatography Applying Postcolumn Derivatization,” Acta Universitatis Sapientiae Alimentaria, Vol. 1, 2008, pp. 5-29.
- M. Koller and H. Eckert, “Derivatization of Peptides for Their Determination by Chromatographic Methods,” Analytica Chimica Acta, Vol. 352, No. 1-3, 1997, pp. 31- 59. doi:10.1016/S0003-2670(97)00321-8
- K. M. De Antonis, P. R. Brown and S. A. Cohen, “High-Performance Liquid Chromatographic Analysis of Synthetic Peptides Using Derivatization with 6-Aminoquinolyl-N-Hydroxysuccinimidyl Carbamate,” Analytical Biochemistry, Vol. 223, No. 2, 1994, pp. 191-197. doi:10.1006/abio.1994.1572
- C. Sun, J. Yang, L. Li, X. Wu, Y. Liu and S. Liu, “Advances in the Study of Luminescence Probes for Proteins,” Journal of Chromatography B, Vol. 803, No. 2, 2004, pp. 173-190. doi:10.1016/j.jchromb.2003.12.039
- M. Fountoulakis and H. Lahm, “Hydrolysis and Amino Acid Composition Analysis of Proteins,” Journal of Chromatography A, Vol. 826, No. 2, 1998, pp. 109-134. doi:10.1016/S0021-9673(98)00721-3
- S. López-Grío, J. R. Torres-Lapasió, J. J Baeza-Baeza and M. C. García-Alvarez-Coque, “Micellar Liquid Chromatographic Separation of Amino Acids Using Preand PostColumn O-phthalaldehyde/N-acetylcysteine Derivatization,” Analytica Chimica Acta, Vol. 418, No. 2, 2000, pp. 153-165. doi:10.1016/S0003-2670(00)00965-X
- ICH Q2(R1), “Validation of Analytical Procedures: Text and Methodology,” International Conference on Harmonization of Technical Requirements for the Registration of Pharmaceutical for Human Use, Geneva, Switzerland, 2005.
NOTES
*Corresponding author.