Open Journal of Polymer Chemistry
Vol. 3 No. 1 (2013) , Article ID: 28474 , 7 pages DOI:10.4236/ojpchem.2013.31004
Nitroxide-Mediated Photo-Controlled/Living Radical Polymerization of Methacrylic Acid
Department of Environmental and Life Sciences, Toyohashi University of Technology, Toyohashi, Japan
Email: eyoshida@ens.tut.ac.jp
Received December 13, 2012; revised January 15, 2013; accepted January 25, 2013
Keywords: Photo-Controlled/Living Radical Polymerization; Nitroxide-Mediated Polymerization; Methacrylic Acid; 4-Methoxy-2,2,6,6-tetramethylpiperidine-1-oxyl; (4-Tert-butylphenyl)diphenylsulfonium Triflate; Molecular Weight Control
ABSTRACT
The photo-controlled/living radical polymerization of methacrylic acid (MAA) was performed at room temperature by irradiation with a high-pressure mercury lamp using azo initiators and 4-methoxy-2,2,6,6-tetramethylpiperidine-1-oxyl as the mediator in the presence of (4-tert-butylphenyl)diphenylsulfonium triflate (tBuS) as the accelerator. Whereas the bulk polymerization yielded polymers with a bimodal molecular weight distribution in both the absence and presence of tBuS, the solution polymerization in methanol produced unimodal polymers with the molecular weight distribution of 2.0 - 2.3 in the presence of tBuS. The molecular weight distribution of the resulting poly (MAA) decreased with an increase in tBuS. The dilution of the monomer concentration also reduced the molecular weight distribution. The use of the initiator with a low 10-h half-life temperature also effectively controlled the molecular weight. The livingness of the polymerization was confirmed by obtaining linear increases in the first-order conversion versus time, the molecular weight versus the conversion, and the molecular weight versus the reciprocal of the initiator concentration.
1. Introduction
Poly (methacrylic acid) (PMAA), an important watersoluble polymer, has many industrial and medical applications, such as a dispersant for coatings [1], a hydrophilic modifier on a surface [2], and a component of drug carriers, like micelles, in drug delivery systems [3,4]. With the aim of precisely synthesizing hydrophilic architectures based on PMAA, the controlled radical polymerization of methacrylic acid (MAA) was exploited using various catalysts. Reversible addition-fragmentation chain transfer (RAFT) polymerization produced PMAA with a narrow molecular weight distribution (Mw/Mn < 1.2) both in water [5] and in organic solvents [6-10]. Photoiniferter polymerization mediated by diethyldithiocarbamate yielded PMAA with the molecular weight distribution of ca. 1.2 with less than a 50% conversion [11]. Atom transfer radical polymerization (ATRP) was not applicable to MAA because this acidic monomer poisoned the catalysts by coordinating to the transition metal, therefore, protection of the monomer is needed for this polymerization [12,13]. Nitroxide-mediated polymerization (NMP) has been regarded as difficult to be applied to MAA as well as methacrylate esters due to the side reaction of the disproportionation termination caused during the high temperature polymerization. Hence, there are only a few examples of the copolymerization of MAA with the comonomers of styrene [14] and sodium 4-styrenesulfonate [15].
Recently, the photo-induced NMP was reported to be successfully applied to the methacrylate esters of methyl [16-29], tert-butyl [30], glycidyl [31], and 2-(dimethylamino) ethyl [32] to produce polymers with comparatively narrow molecular weight distributions (Mw/Mn ≈ 1.4). This photo-NMP was attained using azoinitiators and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as the mediator in the presence of photo-acid generators as the accelerator by both bulk and solution polymerizations. The polymerization was also applicable to sodium methacrylate in water, although the molecular weight distribution was not very narrow (Mw/Mn ≈ 2) [33]. It was found that the photo-induced NMP of MAA proceeded in accordance with a living mechanism in an organic solvent to produce PMAA. This paper describes the photo-NMP of MAA in methanol using 4-methoxy-TEMPO (MTEMPO) as the mediator.
2. Experimental
2.1. Instrumentation
The photopolymerization was carried out using an Ushio optical modulex BA-H502, an illuminator OPM2-502H with a high-illumination lens UI-OP2SL, and a 500 W super high-pressure UV lamp (USH-500SC2, Ushio Co. Ltd.). 1H NMR measurements were conducted using a Jeol ECS 400 FT NMR spectrometer. Gel permeation chromatography (GPC) was performed using a Tosoh GPC-8020 instrument equipped with a DP-8020 dual pump, a CO-8020 column oven, and a RI-8020 refractometer. Two gel columns, Tosoh TSK-GEL α-M were used with N,N-dimethylformamide (DMF) containing 30 mM LiBr and 60 mM H3PO4 as the eluent at 40˚C. The molecular weight and molecular weight distribution were estimated by GPC based on poly(methyl methacrylate) (PMMA) standards.
2.2. Materials
MTEMPO was prepared as reported previously [34]. (2RS,2’RS)-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (r-AMDV) was obtained by separation from a mixture of the racemic and meso forms of 2,2’-azobis (4-methoxy-2,4-dimethylvaleronitrile) [35]. Tetramethyl tetrazene (TMT) was prepared as reported previously [36]. Extrapure 2,2’-azobis[2-(2-imidazolin-2-yl)propane] (V- 61) and 2,2’-azobis[2-methyl-N-[1,1-bis (hydroxyl-methyl)-2-hydroxyethyl]propionamide] (V-80) were purchased from Wako Chemicals and used without further purification. tBuS was purchased from Sigma-Aldrich and used as received. MAA was purified by distillation under reduced pressure. Methanol was distilled over magnesium with a small amount of iodine.
2.3. Photopolymerization: General Procedure
MTEMPO (9.0 mg, 0.0483 mmol), V-61 (11.4 mg, 0.0455 mmol), tBuS (12.0 mg, 0.0256 mmol), MAA (1.015 g, 11.8 mmol), and methanol (2 mL) were placed in an ampoule. The contents in the ampoule were degassed several times using a freeze-pump-thaw cycle and were charged with nitrogen. The polymerization was carried out at room temperature for 7 h at 1.75 × 106-lux illuminance with irradiation by reflective light using a mirror with a 500 W high-pressure mercury lamp at 8.75 ampere. Methanol (8 mL) was added to the product to dissolve it. A small amount (ca. 0.1 mL) of the solution was withdrawn to determine the conversion by 1H NMR. The residue was poured into ether (500 mL). The precipitate was collected by filtration and dried in vacuo for several hours to obtain a polymer (824.3 mg).
3. Results and Discussion
The photoradical polymerization of MAA was performed using r-AMDV as the initiator and MTEMPO as the mediator at room temperature at 1.75 × 106-lux illuminance by irradiation with a high-pressure mercury lamp. The monomer conversions were estimated by 1H NMR based on the signal intensity of the methyl and methylene protons at 0.9 - 2.2 ppm for the resulting polymer and the methyl protons at 1.90 ppm for the remaining unreacted monomer (Figure 1). The signals at 5.58 ppm and 6.06 ppm were attributed to the vinyl protons of the unreacted monomer. The results are shown in Table 1. An orange-colored solution containing the monomer, r-AMDV, and MTEMPO turned into a white suspension within 1 h during the bulk polymerization. The change into the heterogeneous state during the bulk polymerization indicated that the resulting PMAA was insoluble in the monomer. The polymerization slowly proceeded in the absence of tBuS to produce a polymer at a 26% conversion after 6 h. It was revealed that the resulting polymer showed a bimodal GPC consisting of the number-average molecular weights of Mn = 71,400 and 3950, and the area ratio was Mn (71,400/3950) = 0.58/0.42. The polymerization was accelerated in the presence of tBuS, however, the resulting polymer still showed a bimodal GPC, although the proportion of the lower molecular weight polymer was reduced. In order to prevent the formation of the heterogeneous state during the polymerization, the solution polymerization was carried out in methanol. The polymerization homogeneously proceeded, while it was decelerated in methanol. The solution polymerization was accelerated by tBuS as well as the bulk polymerization. Figure 2 shows the GPC profiles of the resulting polymers obtained at the different tBuS/MTEMPO ratios.
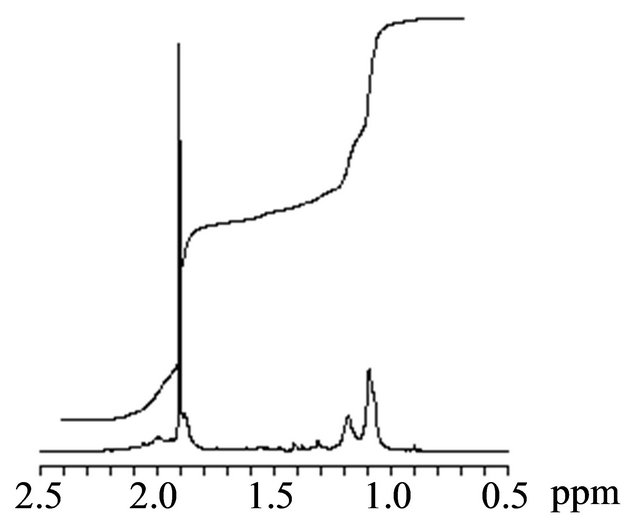
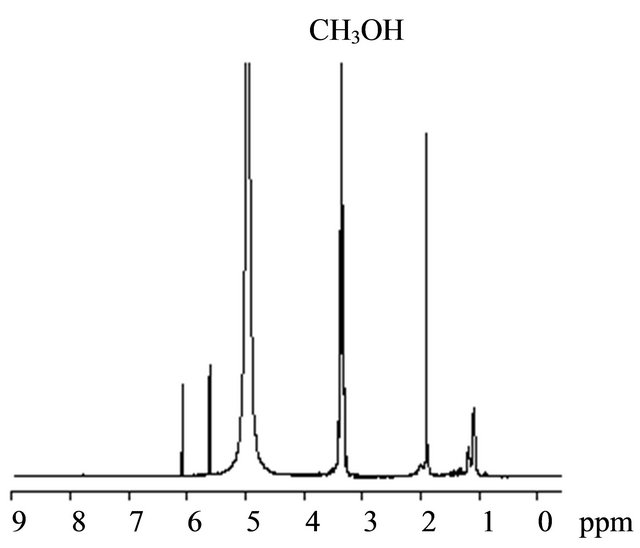
Figure 1. The 1H NMR spectra of the product obtained by the MTEMPO-mediated photoradical polymerization of MAA. [MAA]0 = 11.8 M, MTEMPO/r-AMDV = 1.0, tBuS/ MTEMPO = 0.265. Conversion = 68%. Solvent: CD3OD.
Table 1. The MTEMPO-mediated photoradical polymerization of MAA.a
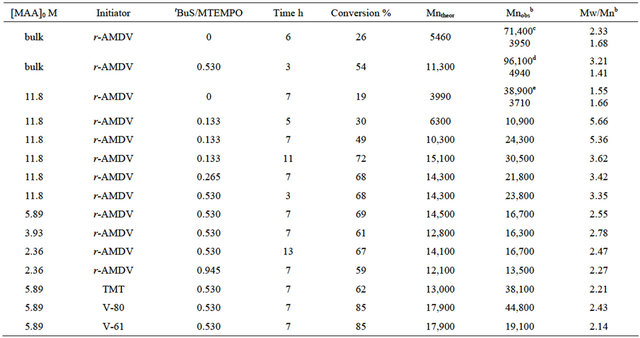
aMTEMPO/the initiator = 1.0; bEstimated by GPC based on PMMA standards; cArea ratio: Mn (71,400/3950) = 0.58/0.42; dArea ratio: Mn (96,100/4940) = 0.83/0.17; eArea ratio: Mn (38,900/3710) = 0.50/0.50.
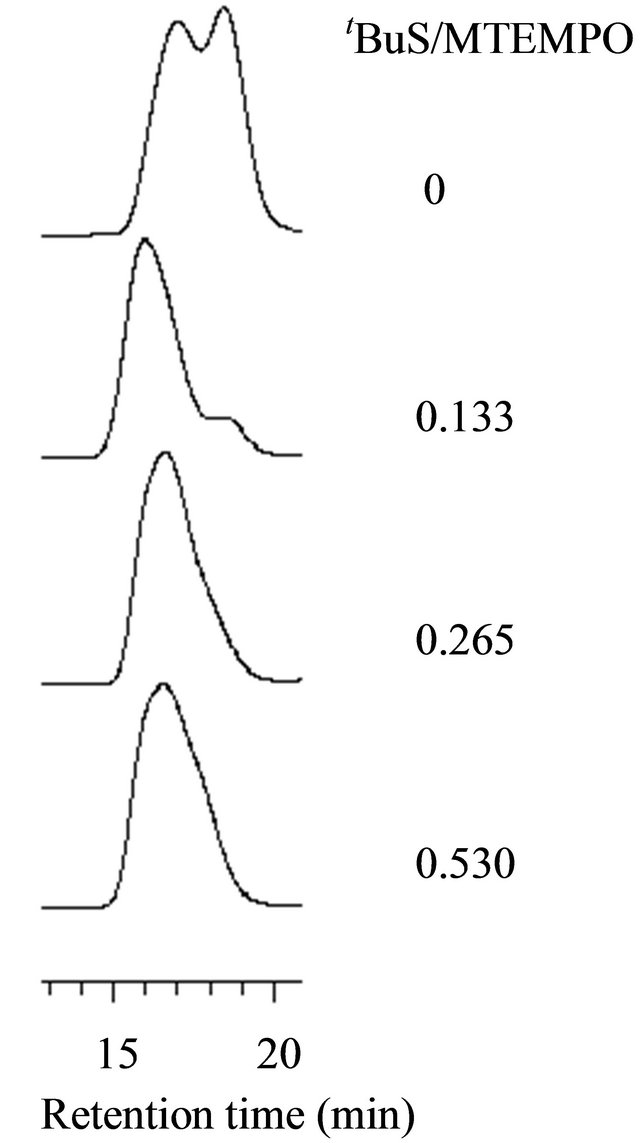
Figure 2. The GPC profiles of the PMAA by the MTEMPOmediated photopolymerization at the different tBuS/ MTEMPO ratios. [MAA]0 = 11.8 M, Initiator: r-AMDV.
An increase in tBuS reduced the proportion of the lower molecular weight polymer, resulting in a decrease in the molecular weight distribution. It is considered that tBuS promoted the cleavage of the C-ON bond at the propagating chain end to prevent the side reactions, such as the deactivation of the propagating end by the disproportionation termination. The dilution of the monomer concentration also decreased the molecular weight distribution. The experimental molecular weights (Mnobs) observed in the GPC of the polymers were close to the theoretical values (Mntheor) at the concentrations below 5.9 M. The theoretical molecular weight was calculated using the concentrations of MTEMPO and the monomer consumed on the basis of the previous result on the methyl methacrylate polymerization for which the molecular weight was determined by the ratio of the monomer to MTEMPO [17,28]. It was found that the properties of the initiators also affected the molecular weight distribution.
The structures of the initiators are shown in Scheme 1. TMT produced a single radical species of the N-center radical by the decomposition [37], different from the cyano-containing azoinitiators, like r-AMDV, that provided the two species of the C-center and N-center radicals with different initiation rate constants [38-41]. The single radical species from TMT decreased the molecular weight distribution, whereas the experimental molecular weight was much higher than the calculated value, implying its low initiator efficiency (Figure 3). V-80 and V-61 are more hydrophilic than r-AMDV and easily dissolved in methanol. Their high hydrophilicity also decreased the molecular weight distribution. In particular, V-61 more effectively reduced the molecular weight distribution and produced a polymer with a molecular weight close to the calculated value. This more effectiveness of V-61 regarding the molecular weight control as compared to V-80 should be due to its lower 10-h
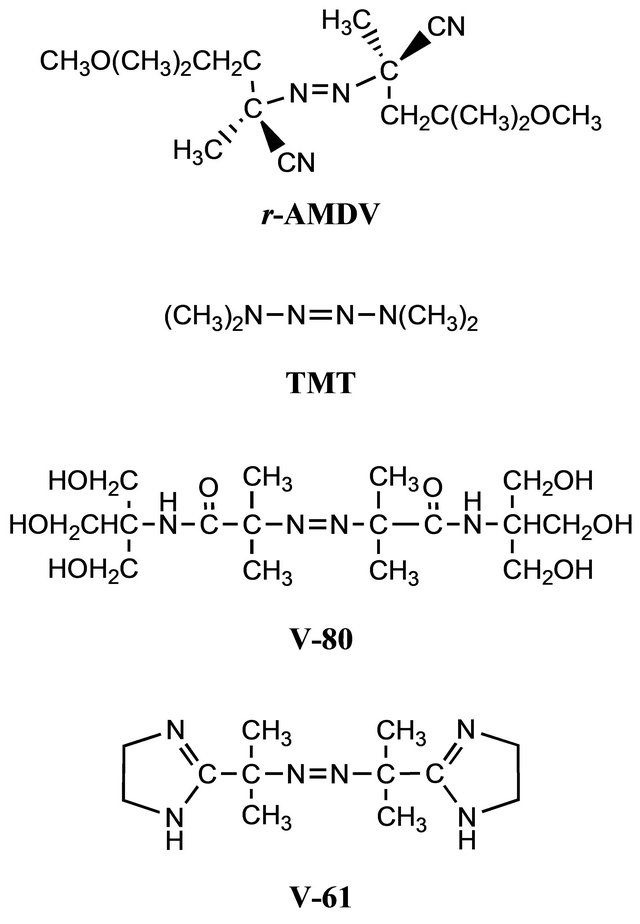
Scheme 1. The azoinitiators used for the MTEMPO-mediated photoradical polymerization.
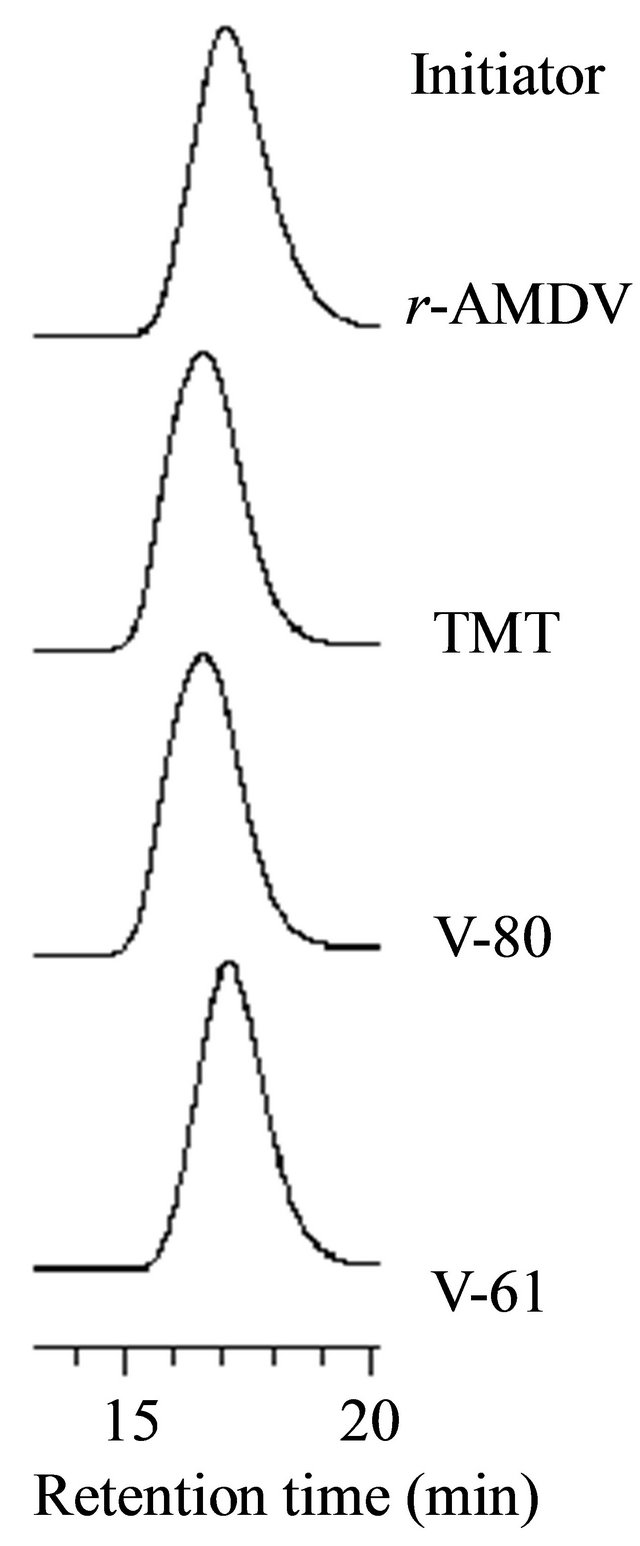
Figure 3. The GPC profiles of the PMAA prepared by the MTEMPO-mediated photopolymerization with the different initiators. [MAA]0 = 5.89 M, MTEMPO/the initiator = 1.0, tBuS/MTEMPO = 0.530. The polymerization time: 7 h.
half-life period temperature; 61˚C for V-61 and 80˚C for V-80. The half-life period temperature of V-61 is still lower under an acidic condition [35,42,43].
The living nature of the polymerization was explored using V-61 in the presence of tBuS in methanol at a 5.89-M monomer concentration, 1.0 MTEMPO/V-61, and 0.530 tBuS/MTEMPO. Figure 4 shows the timeconversion and its first-order time-conversion plots, ln([M]0/[M]t), for the polymerization. [M] denotes the monomer concentration. The conversion reached over 80% at around 7 h. The ln([M]0/[M]t) plots exhibited a linear increase, suggesting that the number of polymer chains was constant throughout the course of the polymerization. The variation in the GPC curve with the conversion is shown in Figure 5. The curves of the resulting polymers were shifted to the higher molecular weight side with an increase in the conversion, implying the livingness of the polymerization. Figure 6 shows the conversion-molecular weight plots for the polymerization. The linear increase in the molecular weight with the increasing conversion confirmed the living mechanism of the polymerization. The proposed mechanism of the polymerization is shown in Scheme 2 based on the previous results of the methyl methacrylate polymerization; tBuS only served as the accelerator of the polymerization and was not inserted into the resulting polymer structure [26, 28]. The line for the conversion-molecular weight plots did not pass through zero. This phenomenon can be accounted due to the existence of a non-steady state during
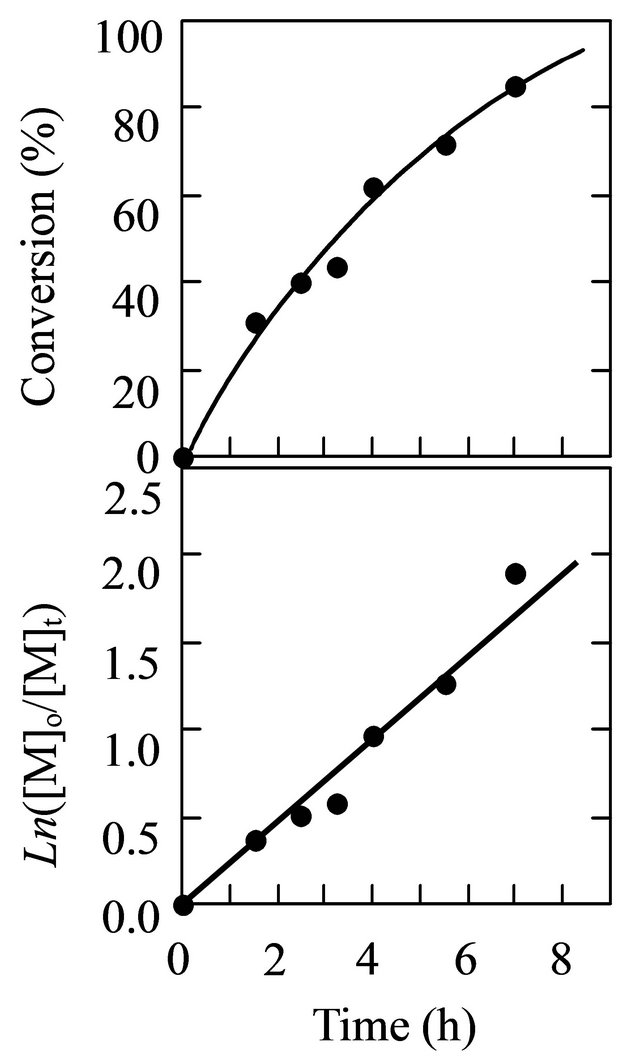
Figure 4. The time-conversion and its first order plots for the MAA polymerization. [MAA]0 = 5.89 M, MTEMPO/ V-61 = 1.0, tBuS/MTEMPO = 0.530.
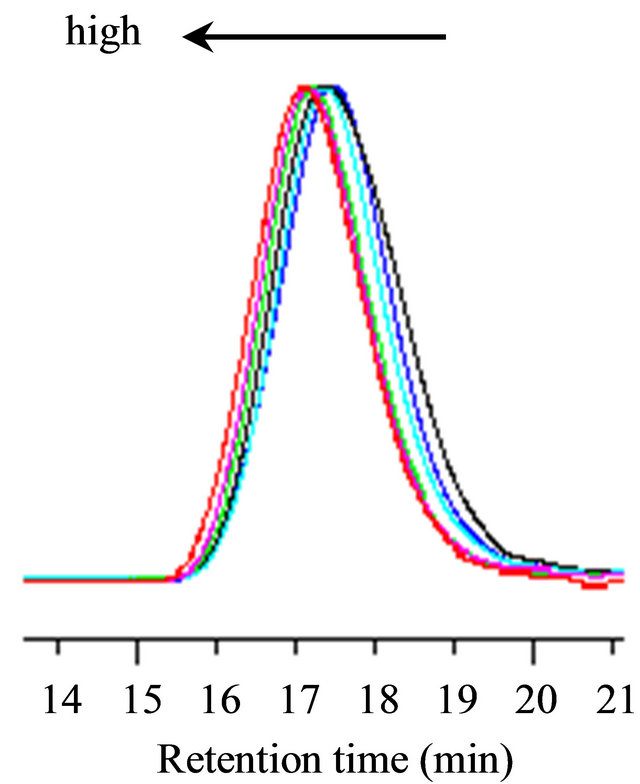
Figure 5. The variation in the GPC curves of the PMAA versus the conversion: 31% (1.5 h, ¾); 40% (2.5 h, ¾); 44% (3.25 h, ¾); 62% (4 h, ¾); 72% (5.5 h, ¾); and 85% (7 h, ¾). [MAA]0 = 5.89 M, MTEMPO/V-61 = 1.0, tBuS/ MTEMPO = 0.530.
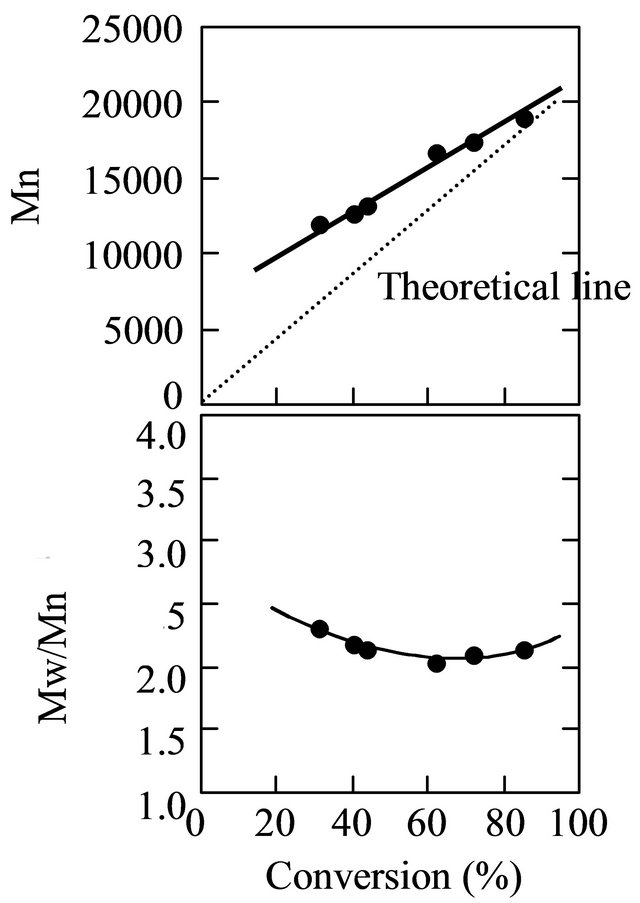
Figure 6. The plots of the molecular weight and its distribution versus the conversion for the MAA polymerization. [MAA]0 = 5.89 M, MTEMPO/V-61 = 1.0, tBuS/MTEMPO = 0.530.
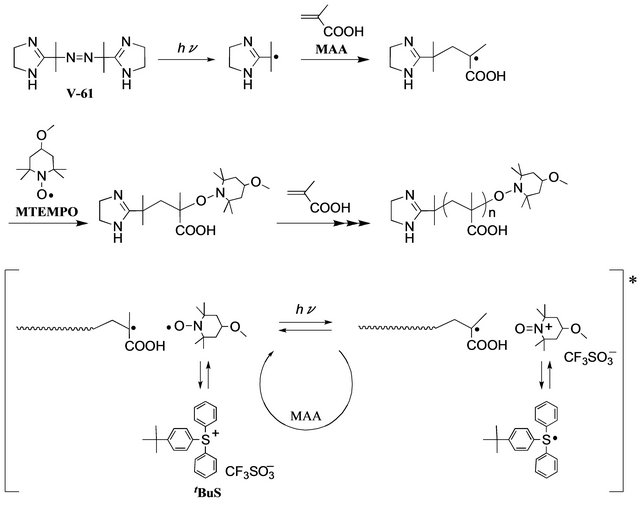
Scheme 2. The mechanism of the MTEMPO-mediated photocontrolled/living radical polymerization of MAA.
the very early stage of the polymerization [21,26]. Hence, there was a difference in the molecular weight between the observed and theoretical values at the low conversion. The molecular weight distribution was in the range of 2.0 - 2.3 throughout the polymerization.
In order to ensure the livingness of the polymerization, the relationship between the molecular weight of the resulting polymer and the initiator concentration was investigated. The results for the polymerization carried out at different V-61 concentrations are shown in Table 2. As the V-61 concentration increased, the molecular weight and its distribution decreased. The experimental molecular weights were in good agreement with the theoretical values. By plotting the molecular weight versus the reciprocal of the initiator concentration, a linear correlation was obtained (Figure 7). This led to the con-
Table 2. The MTEMPO-mediated photopolymerizaton of MAA with different initiator concentrationsa.
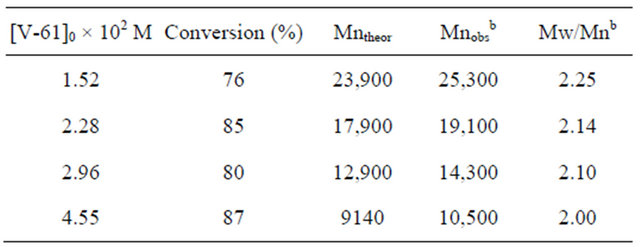
a[MAA]0 = 5.89 M, MTEMPO/V-61 = 1.0, tBuS/MTEMPO = 0.530. Polymerization time: 7 h; bEstimated by GPC based on PMMA standards.
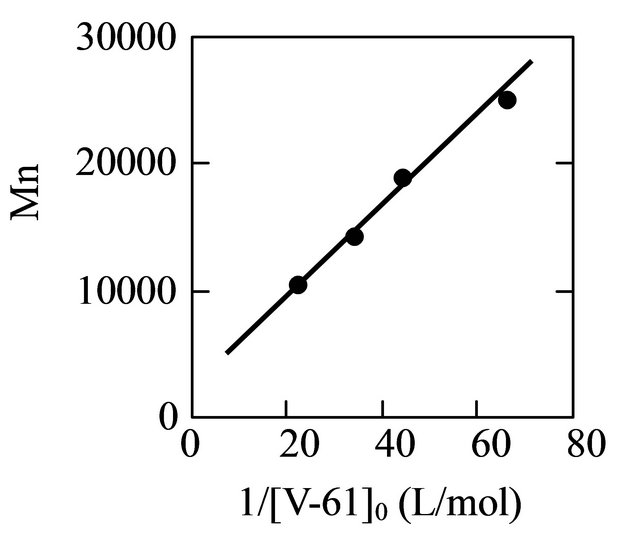
Figure 7. The plots of the molecular weight versus the reciprocal of the V-61 concentration for the MAA polymerization. [M]0 = 5.89 M, MTEMPO/V-61 = 1.0, tBuS/ MTEMPO = 0.530. The polymerization time: 7 h.
clusion that the MTEMPO-mediated photoradical polymerization of MAA proceeded in accordance with a living mechanism in methanol by V-61 in the presence of tBuS.
4. Conclusion
The photo-controlled/living radical polymerization of MAA was attained in methanol at room temperature using the MTEMPO mediator and the azoinitiators in the presence of tBuS. While tBuS accelerated the polymerization, it tended to decrease the molecular weight distribution. The dilution of the monomer concentration also reduced the molecular weight distribution. Furthermore, the initiators that generate a single radical species by decomposition and have a low 10-h half-life temperature also effectively controlled the molecular weight. The livingness of the polymerization was ensured with V-61 based on linear increases in the first-order conversion with time, the molecular weight versus the conversion, and the molecular weight versus the reciprocal of the initiator concentration. The experimental molecular weights of the resulting PMAA were in good agreement with the theoretical values at the high conversions.
5. Acknowledgements
The author is thankful for a Shiseido Female Researcher Science Grant.
REFERENCES
- R. Thomas, “ Dispersant Theory and Polyacrylate Dispersants,” Journal of Water Borne Coatings, Vol. 12, No. 1-2, 1989, pp. 8-10.
- R. Heeb, R. M. Bielecki, S. Lee and N. D. Spencer, “Room-Temperature, Aqueous-Phase Fabrication of Poly (methacrylic acid) Brushes by UV-LED-Induced, Controlled Radical Polymerization with High Selectivity for Surface-Bound Species,” Macromolecules, Vol. 42, No. 22, 2009, pp. 9124-9132. doi:10.1021/ma901607w
- T. Cao, P. Munk, C. Ramireddy, Z. Tuzar and S. E. Webber, “Fluorescence Studies of Amphiphilic Poly(methacrylic acid)-block-polystyrene-block-poly(methacrylic acid) Micelles,” Macromolecules, Vol. 24, No. 23, 1991, pp. 6300-6305. doi:10.1021/ma00023a036
- J. Gohy, S. K. Varshney and R. Jerome, “Water-Soluble Complexes Formed by Poly(2-vinylpyridinium)-blockpoly(ethylene oxide) and Poly(sodium methacrylate)- block-poly(ethylene oxide) Copolymers,” Macromolecules, Vol. 34, No. 10, 2001, pp. 3361-3366. doi:10.1021/ma0020483
- I. Chaduc, M. Lansalot, F. D’Agosto and B. Charleux, “RAFT Polymerization of Methacrylic Acid in Water,” Macromolecules, Vol. 45, No. 3, 2012, pp. 1241-1247. doi:10.1021/ma2023815
- J. M. Pelet and D. Putnam, “High Molecular Weight Poly(methacrylic acid) with Narrow Polydispersity by RAFT Polymerization,” Macromolecules, Vol. 42, No. 5, 2009, pp. 1494-1499. doi:10.1021/ma801433g
- B. Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, “A More Versatile Route to Block Copolymers and Other Polymers of Complex Architecture by Living Radical Polymerization: The RAFT Process,” Macromolecules, Vol. 32, No. 6, 1999, pp. 2071-2074. doi:10.1021/ma981472p
- E. Sprong, D. D. Wet-Roos, M. P. Tonge and R. D. Sanderson, “Characterization and Rheological Properties of Model Alkali-Soluble Rheology Modifiers Synthesized by Reversible Addition-Fragmentation Chain-Transfer Polymerization,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 41, No. 2, 2003, pp. 223-235. doi:10.1002/pola.10503
- C. Yang and Y. Cheng, “RAFT Synthesis of Poly(N-isopropylacrylamide) and Poly(methacrylic acid) Homopolymers and Block Copolymers: Kinetics and Characterization,” Journal of Applied Polymer Science, Vol. 102, No. 2, 2006, pp. 1191-1201. doi:10.1002/app.24415
- E. H. Nejad, P. Castignolles, R. G. Gilbert and Y. Guillaneuf, “Synthesis of Methacrylate Derivatives Oligomers by Dithiobenzoate-RAFT-Mediated Polymerization,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 46, No. 6, 2008, pp. 2277-2289. doi:10.1002/pola.22563
- K. Ishizu, H. Katsuhara and K. Itoya, “Controlled Radical Polymerization of Mathacrylic Acid Initiated by Diethyldithio-carbamate-Mediated Iniferter,” Journal of Polymer Science Part A: Polymer Chemistry, Vol. 43, No. 1, 2005, pp. 230-233. doi:10.1002/pola.20395
- K. Matyjaszewski and J. Xia, “Atom Transfer Radical Polymerization,” Chemical Reviews, Vol. 101, No. 9, 2001, pp. 2921-2990. doi:10.1021/cr940534g
- J. Yuan, Y. Shi, Z. Fu and W. Yang, “Synthesis of Amphiphilic Poly(methyl methacrylate)-block-poly(methacrylic acid) Diblock Copolymers by Atom Transfer Radical Polymerization,” Polymer International, Vol. 55, No. 3, 2006, pp. 360-364. doi:10.1002/pi.1977
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, “Nitroxide-Mediated Controlled/Living Free-Radical Surfactant-Free Emulsion Polymerization of Methyl Methacrylate Using a Poly(methacrylic acid)-Based Macroalkoxyamine Initiator,” Macromolecules, Vol. 42, No. 1, 2009, pp. 95-103. doi:10.1021/ma802083g
- S. Brusseau, F. D’Agosto, S. Magnet, L. Couvreur, C. Chamignon and B. Charleux, “Nitroxide-Mediated Copolymerization of Methacrylic Acid and Sodium 4-Styrenesulfonate in Water Solution and One-Pot Synthesis of Amphiphilic Block Copolymer Nanoparticles,” Macromolecules, Vol. 44, No. 14, 2011, pp. 5590-5598. doi:10.1021/ma2008282
- E. Yoshida, “Effects of Illuminance and Heat Rays on Photo-Controlld/Living Radical Polymerization Mediated by 4-Methoxy-2,2,6,6-tetramethylpiperidine-1-oxyl,” International Scholarly Research Network ISRN Polymer Science, 2012, Article ID 102186. http://www.hindawi.com/isrn/ps/2012/102186/ doi:10.5402/2012/102186
- E. Yoshida, “Photo-Controlled/Living Radical Polymerization Mediated by 2,2,6,6-Tetramethylpiperidine-1-oxyl in Inert Atmospheres,” Colloid and Polymer Science, Vol. 290, No. 11, 2012, pp. 1087-1091. doi:10.1007/s00396-012-2668-0
- E. Yoshida, “Nitroxide-Mediated Photo-Controlled/Living Radical Dispersion Polymerization of Methyl Methacrylate,” Colloid and Polymer Science, Vol. 289, No. 14, 2011, pp. 1625-1630. doi:10.1007/s00396-011-2487-8
- E. Yoshida, “Graft Copolymerization of Methyl Methacrylate on Polystyrene Backbone through Nitroxide-Mediated Photo-Living Radical Polymerization,” Colloid and Polymer Science, Vol. 289, No. 7, 2011, pp. 837-841. doi:10.1007/s00396-011-2385-0
- E. Yoshida, “Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate in the Presence of (h6-Benzene)(h5-cyclopentadienyl)FeII Hexafluorophosphate,” Colloid and Polymer Science, Vol. 288, No. 18, 2010, pp. 1745-1749.
- E. Yoshida, “Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate in Solution,” Colloid and Polymer Science, Vol. 288, No. 16-17, 2010, pp. 1639-1643. doi:10.1007/s00396-010-2287-6
- E. Yoshida, “Stability of Growing Polymer Chain Ends for Nitroxide-Mediated Photo-Living Radical Polymerization,” Colloid and Polymer Science, Vol. 288, No. 9, 2010, pp. 1027-1030. doi:10.1007/s00396-010-2230-x
- E. Yoshida, “Effects of Initiators and Photo-Acid Generators on Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate,” Colloid and Polymer Science, Vol. 288, No. 8, 2010, pp. 901-905. doi:10.1007/s00396-010-2220-z
- E. Yoshida, “Effect of Azoinitiators on Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate,” Colloid and Polymer Science, Vol. 288, No. 3, 2010, pp. 341-345. doi:10.1007/s00396-009-2163-4
- E. Yoshida, “Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate Using (4-TertButylphenyl)diphenylsulfonium Triflate as a Photo-Acid Generator,” Colloid and Polymer Science, Vol. 288, No. 2, 2010, pp. 239-243. doi:10.1007/s00396-009-2161-6
- E. Yoshida, “Photo-Living Radical Polymerization of Methyl Methacrylate Using Alkoxyamine as an Initiator,” Colloid and Polymer Science, Vol. 288, No. 1, 2010, pp. 7-13. doi:10.1007/s00396-009-2113-1
- E. Yoshida, “Synthesis of Poly(methyl methacrylate)- block-poly(tetrahydrofuran) by Photo-Living Radical Polymerization Using a 2,2,6,6-Tetramethylpiperidine-1- oxyl,” Colloid and Polymer Science, Vol. 287, No. 12, 2009, pp. 1417-1424. doi:10.1007/s00396-009-2105-1
- E. Yoshida, “Photo-Living Radical Polymerization of Methyl Methacrylate by 2,2,6,6-Tetramethylpiperidine-1- oxyl in the Presence of a Photo-Acid Generator,” Colloid and Polymer Science, Vol. 287, No. 7, 2009, pp. 767-772. doi:10.1007/s00396-009-2023-2
- E. Yoshida, “Photo-Living Radical Polymerization of Methyl Methacrylate by a Nitroxide Mediator,” Colloid and Polymer Science, Vol. 286, No. 14-15, 2008, pp. 1663-1666. doi:10.1007/s00396-008-1930-y
- E. Yoshida, “Photo-Controlled/Living Radical Polymerization of Tert-Butyl Methacrylate in the Presence of a Photo-Acid Generator Using a Nitroxide Mediator,” Colloid and Polymer Science, Vol. 290, No. 7, 2012, pp. 661-665. doi:10.1007/s00396-012-2605-2
- E. Yoshida, “Selective Controlled/Living Photoradical Polymerization of Glycidyl Methacrylate, Using a Nitroxide Mediator in the Presence of a Photosensitive Triarylsulfonium Salt,” Polymers, Vol. 4, No. 3, 2012, pp. 1580-1589. doi:10.3390/polym4031580
- E. Yoshida, “Photo-Controlled/Living Radical Polymerization of 2-(Dimethylamino)ethyl Methacrylate Using 4-Methoxy-2,2,6,6-tetramethylpiperidine-1-oxyl as a Mediator,” Colloid and Polymer Science, Vol. 290, No. 10, 2012, pp. 965-969. doi:10.1007/s00396-012-2641-y
- E. Yoshida, “Aqueous Photo-Living Radical Polymerization of Sodium Methacrylate Using a Water-Soluble Nitroxide Mediator,” International Scholarly Research Network ISRN Polymer Science, 2012, Article ID 630478. doi:10.5402/2012/630478 http://www.hindawi.com/isrn/ps/2012/630478/
- T. Miyazawa, T. Endo, S. Shiihashi and M. Ogawara, “Selective Oxidation of Alcohols by Oxoaminium Salts (R2N=O+ X−),” Journal of Organic Chemistry, Vol. 50, No. 8, 1985, pp. 1332-1334. doi:10.1021/jo00208a047
- Y. Kita, K. Gotanda, K. Murata, M. Suemura, A. Sano, T. Yamaguchi, M. Oka and M. Matsugi, “Practical Radical Additions under Mild Conditions Using 2,2’-Azobis(2,4- dimethyl-4-methoxyvaleronitrile) [V-70] as an Initiator,” Organic Process Research & Development, Vol. 2, No. 4, 1998, pp. 250-254. doi:10.1021/op970059z
- W. E. Bull, J. A. Seaton and L. F. Audrieth, “Some Properties of Tetraalkyl-2-tetrazenes,” Journal of American Chemical Society, Vol. 80, No. 10, 1958, pp. 2516-2518. doi:10.1021/ja01543a038
- K. Sugiyama, T. Nakaya and M. Imoto, “Vinyl Polymerization. 275. Polymerization of Vinyl Monomers Photosensitized by Tetramethyltetrazene,” Journal of Polymer Science Part A-1, Vol. 10, No. 1, 1972, pp. 205-215. doi:10.1002/pol.1972.150100119
- P. Smith and A. M. Rosenberg, “The Kinetics of the Photolysis of 2,2’-Azo-bis-isobutyronitrile,” Journal of American Chemical Society, Vol. 81, No. 9, 1959, pp. 2037- 2043. doi:10.1021/ja01518a002
- M. Talat-Erben and S. Bywater, “The Thermal Decomposition of 2,2’-Azo-bis-isobutyronitrile. Part 1. Products of the Reaction,” Journal of American Chemical Society, Vol. 77, No. 14, 1955, pp. 3710-3711. doi:10.1021/ja01619a010
- G. S. Hammond, O. D. Trapp, R. T. Keys and D. L. Neff, “Isolation and Study of the Intermediate [Dimethyl-N-(2- cyano-2-propyl)-ketenimine] Formed in the Decomposition of α,α’-Azoisobutyronitrile,” Journal of American Chemical Society, Vol. 81, No. 18, 1959, pp. 4878-4882. doi:10.1021/ja01527a030
- G. S. Hammond, C. S. Wu, O. D. Trapp, J. Warkentin, R. T. Keys and D. L. Neff, “The Mechanism of Decomposition of Azo Compounds. II. Cage Effects in the Decomposition of α,α’-Azoisobutyronitrile and Related Compounds,” Journal of American Chemical Society, Vol. 82, No. 20, 1960, pp. 5394-5399. doi:10.1021/ja01505a026
- G. S. Hammond and R. C. Neuman Jr., “The Mechanism of Decomposition of Azo Compounds. III. Cage Effects with Positively Charged Geminate Radical Pairs,” Journal of the American Chemical Society, Vol. 85, No. 10, 1963, pp. 1501-1508. doi:10.1021/ja00893a026
- “Wako Chemicals Information.” http://www.wako-chem.co.jp/kaseihin/waterazo/VA-061.htm