Open Journal of Physical Chemistry
Vol.3 No.1(2013), Article ID:27884,10 pages DOI:10.4236/ojpc.2013.31004
A Molecular Dynamics Simulation Study of Two Dipeptide Based Molecular Micelles: Effect of Amino Acid Order
1Department of Chemistry, Carthage College, Kenosha, USA
2Department of Physical and Environmental Sciences, Texas A&M University-Corpus Christi, Corpus Christi, USA
3Office of Naval Research, Arlington, USA
4Department of Biochemistry and Molecular Biology, Howard University College of Medicine, Howard University, Washington, USA
Email: *yfang@howard.edu
Received August 4, 2012; revised September 10, 2012; accepted October 15, 2012
Keywords: Chiral Recognition; Polymeric Surfactant; Molecular Modeling
ABSTRACT
Molecular dynamics (MD) simulations were used to compare the structures of the chiral molecular micelles (MM) poly-(sodium undecyl-(L,L)-leucine-valine) (poly(SULV)) and poly-(sodium undecyl-(L,L)-valine-leucine) (poly- (SUVL)). Both MM contained polymerized surfactant monomers terminated by chiral dipeptide headgroups. The study was undertaken to investigate why poly(SULV) is generally a better chiral selector compared to poly(SUVL) in electrokinetic chromatography separations. When comparing poly(SULV) to poly(SUVL), poly(SULV) had the more conformational flexible dipeptide headgroup and hydrogen bond analyses revealed that the poly(SULV) headgroup conformation allowed a larger number of intramolecular hydrogen bonds to form between monomer chains. In addition, a larger number of water molecules surrounded the chiral centers of the poly(SULV) molecular micelle. Poly(SULV) was also found to have a larger solvent accessible surface area (SASA) than poly(SUVL) and fluctuations in the poly(SULV) SASA during the MD simulation allowed dynamic monomer chain motions expected to be important in chiral recognition to be identified. Finally, approximately 50% of the Na+ counterions were found in the first three solvation shells surrounding both MM, with the remainder located in the bulk. Overall the MD simulations point to both greater headgroup flexibility and solvent and analyte access to the chiral centers of the dipeptide headgroup as factors contributing to the enhanced chiral selectivity observed with poly(SULV).
1. Introduction
The enantiomers of chiral drugs often have different pharmacological potencies and toxicities. As a result, FDA guidelines issued in 1992 required the activities of drug enantiomers to be investigated separately and for only the therapeutic enantiomer to be brought to market [1]. These guidelines have led to chiral separation methods playing an important role in modern drug discovery [2]. Examples of chiral separation techniques developed to date include chiral electrokinetic chromatography (EKC), thin layer chromatography, gas chomatography, supercritical fluid chromatography, and high performance liquid chromatography [2,3].
In EKC-based chiral separations, enantiomeric resolution results from differential analyte interactions with a chiral selector as both are pulled through a capillary by an electric field [4]. Chiral micelles, polysaccharides, polymers, crown ethers, ligand-exchange complexes, and cyclodextrins have all been used as EKC chiral selectors. Recent developments in the field have been reviewed by Sánchez-Hernández, et al. [5]. This investigation focused on amino acid-based molecular micelles (MM) which were first employed as EKC chiral selectors by Wang and Warner [6]. MM consist of polymerized surfactant monomers with covalent attachments at the end of each monomer’s hydrocarbon chain. In the MM investigated here, each monomer was terminated with a chiral dipeptide headgroup.
The roles played by MM headgroup amino acid order [7-9], steric factors [7,8,10-12], hydrogen bonding [12, 13], electrostatic interactions [15,16], and the number and position of the headgroup chiral centers [8,15] in governing chiral selectivity in EKC separations have been reported. Research has also addressed how chiral selectivity is affected by the concentration at which the surfactant monomers were polymerized [16] and the depth of analyte penetration into the hydrophobic core [17]. NMR experiments have been used as well to investigate the thermodynamics of chiral analyte: MM interactions and the structures of chiral analyte:MM intermolecular complexes [4,14,18,19].
This project is part of an ongoing effort to characterize more fully dipeptide terminated MM structure and the analyte:MM intermolecular interactions that lead to chiral recognition in EKC separations. Here we have used molecular dynamics (MD) simulations to investigate the structures of two dipeptide terminated MM. The rationale for choosing these MM is based upon the experimental observation that chiral selectivity is generally higher when the larger of the two amino acids is in the N-terminal position [8,20]. For example, the MM poly- (sodium undecyl-(L,L)-leucine-valine) (poly-SULV) is a relatively effective chiral selector [21], while chiral selectivity in separations with poly-(sodium undecyl-(L,L) -valine-leucine) (poly-SUVL) is generally relatively poor [8]. The structures of poly(SULV) and poly(SUVL) are shown in Figures 1(a) and (b). The MD simulations reported here were carried out to investigate the structures of these two MM in aqueous solution in an effort to rationalize the difference in chiral selectivity brought about by changing the amino acid order in the MM dipeptide headgroups.
A previous MD simulation and NMR study of poly- (SULV) and poly(SUVL) provided insights into differences between the two MM structures and offered clues as to why poly(SULV) is the better chiral selector [22]. For example, poly(SULV) was found to have an overall more open structure with correspondingly more potential pockets or spaces into which chiral analytes could insert. The structure of poly(SUVL), in contrast, was more closed and compact. Water was also shown to penetrate to a greater degree into the core and headgroup regions of poly(SULV), while poly(SUVL) excluded more water molecules from these regions. This result was found to be consistent with the experimental observation that polar binaphthyl analytes bound more strongly to poly- (SULV), while non-polar binaphthyl analytes interacted more strongly with poly(SUVL) [22]. In the study reported here, additional MD simulation analyses were used to delve deeper into the differences between the two MM structures and to gain further insight into the molecular features that make poly(SULV) the better chiral selector. The first MM structural property investigated was the distribution of dipeptide headgroup dihedral angles. By quantifying how these dihedral angles changed during the MD simulations, we were able to compare the conformational flexibilities of the poly(SULV) and poly- (SUVL) headgroups. MD simulations were also used to
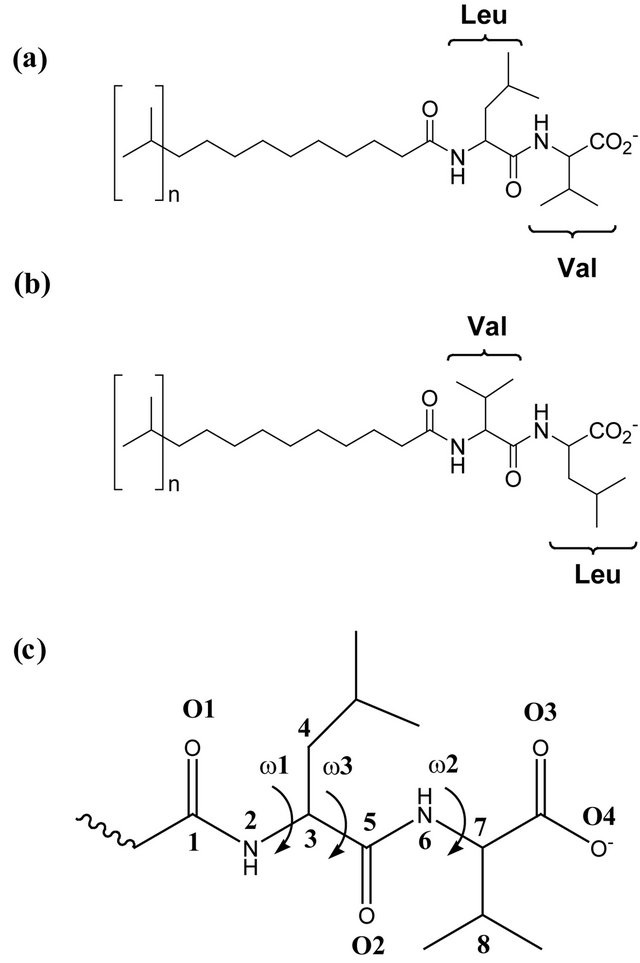
Figure 1. (a) Structure of poly(SULV); (b) Structure of poly(SUVL); (c) Headgroup atom labels for dihedral angle and hydrogen bond analyses.
investigate intramolecular hydrogen bonding between the MM monomer chains and intermolecular hydrogen bonding between the MM molecules and water. Hydrogen bonding would be expected to affect both the local structures and overall shapes of the MM. Also, in an effort to probe the access that poly(SULV) and poly(SUVL) provide to the solvent, and thus by extension to potential chiral analytes, MD simulations were used to compare the solvent accessible surface areas (SASA) of the two MM. The surface area analyses also allowed a number of monomer chain motions that open and close chiral grooves or pockets near the MM headgroups to be identified. The final two MM properties investigated with the MD simulations were the number of water molecules surrounding the two headgroup chiral centers and the distribution of Na+ counterions around the MM carboxylate groups.
2. Experimental Details
The details of the poly(SULV) and poly(SUVL) MD simulations have been previously reported [22]. Briefly, the methods utilized are summarized as follows. Each MM contained twenty surfactant monomers. This value was chosen based upon fluorescence quenching measurements of each micelle’s aggregation number [23]. In both MM, monomers were connected by covalent bonds at the end of each surfactant’s hydrocarbon chain. MD simulations were performed using AMBER 10 [24] and the parm 99 [25] force field. Each MD simulation contained the respective MM, twenty Na+ counterions, and 8360 or 8689 water residues for poly(SULV) and poly- (SUVL), respectively. The TIP3P water model was used in both MD simulations. The MM systems were first optimized and then 20 ps MD simulations were carried out to heat to 300 K. Next, a 1 ns simulation was done to equilibrate the system to 1 atm and 300 K, followed by a 10.0 ns production run to collect statistical data. The time step in the MD simulations was 2.0 fs and structures were saved every 0.2 ps. All analyses of the MD simulation trajectories were done with the ptraj utility in AMBER 10 [24].
3. Results and Discussion
3.1. Headgroup Conformation and Dihedral Angles
Distributions of selected headgroup dihedral angles were used to assess the conformational flexibilities of the poly(SULV) and poly(SUVL) headgroups. Figure 1(c) shows atom labels used to define the three dihedral angles monitored during the MD simulations. The angle ω1 was C1-N2-C3-C4, ω2 was C5-N6-C7-C8, and ω3 was C4-C3-C5-N6. Figure 2 shows a histogram of the ω1, ω2, and ω3 values during the poly(SULV) MD simulation. Note that ω2 takes on a relatively narrow range of values centered around 134˚. The dihedral angle ω3 has a bimodal distribution with maxima at 171˚ and 233˚, corresponding to trans and anti-(-) conformations, respectively. The populations of all other ω3 dihedral angle
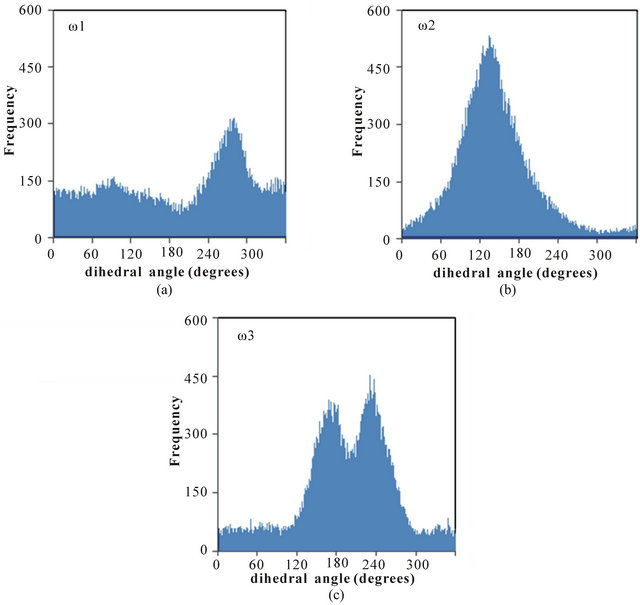
Figure 2. Distribution of the dihedral angles (a) ω1, (b) ω2, and (c) ω3 for poly(SULV).
values are relatively small. In contrast, angle ω1 is very flexible with smaller maxima at 94˚ and 274˚ but with significant dihedral angle population ranging from 0 to 360˚. This flexibility may contribute to the high chiral selectivity observed for poly(SULV). For example, rotation of the headgroup about angle ω1 would allow the poly(SULV) headgroup to open and close thus giving chiral molecules an opportunity to insert into chiral pockets near the dipeptide headgroup and experience stereo selective interactions with both the leucine and valine chiral centers. Access to the N-terminal leucine may be important because NMR and EKC studies of analyte association with poly(SULV) have shown that binaphthyl analytes such as 1,1’-binaphthyl-2,2’-diyl hydrogen phosphate and 1,1-’bi-2-naphthol interact predominately with the leucine chiral center [4,18].
The distributions of dihedral angles ω1, ω2, and ω3 in poly(SUVL) are shown in Figure 3. The MD simulations show that all three dihedral angles take on relatively well-defined values centered at 132˚, 141˚, and 142˚, for ω1, ω2, and ω3, respectively. None of the poly(SUVL) dihedral angles show the wide-ranging variability observed for ω1 in poly(SULV). The smaller variability of the poly(SUVL) dihedral angles suggests that the headgroup adopts a more fixed, less dynamic conformation when compared to poly(SULV). This lack of flexibility may be especially effective in denying chiral analyte access to the valine chiral center. Fewer analyte interactions with this chiral center likely contribute to the low chiral selectivity in separations with poly(SUVL).
3.2. Hydrogen Bonding
Analyses of the intraand intermolecular hydrogen bonds
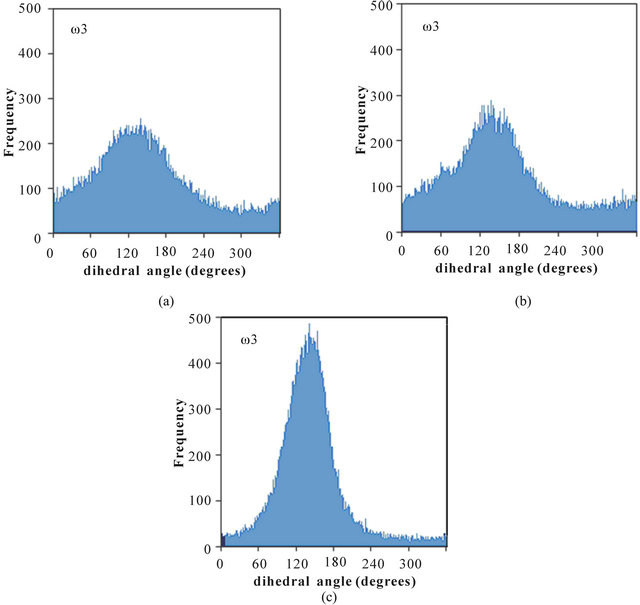
Figure 3. Distribution of the dihedral angles (a) ω1, (b) ω2, and (c) ω3 for poly(SUVL).
formed during the MD simulations give further insight into the relative abilities of the two MM investigated to act as chiral selectors. The intra-molecular hydrogen bonds formed by poly(SULV) and poly(SUVL) are shown in Table 1. The atom labels used to index the hydrogen bonds are shown in Figure 1(c). Eight intra-molecular poly(SULV) hydrogen bonds with percent occupancies greater than 10% formed during the MD simulation. Note, though, that in all but two of these hydrogen bonds the H-bond acceptor atom was the carbonyl oxygen separating the monomer hydrocarbon chain from the dipeptide headgroup (atom O1 in Figure 1). This result is consistent with poly(SULV) monomers adopting predominately open wing-like conformations that allow potential H-bond donor atoms to access the oxygen at the end of the hydrocarbon chain [22]. Also, in all but one of the poly(SULV) hydrogen bonds listed in Table 1, the N-terminal leucine amide hydrogen was the H-bond donor atom. Overall, the poly(SULV) H-bond results show that most of the intra-molecular H-bonds formed in the MD simulation involved oxygen atom O1 and NH atoms on the leucine amino acid. Figure 4(a) shows an intramolecular hydrogen bond with a percent occupancy of 76.82% formed between poly(SULV) chains 12 and 15. In the hydrogen-bonded structure, the open wing-like headgroup conformation of both mono-
Table 1. Intramolecular hydrogen bonds with occupancies greater than 10% from the poly(SULV) and poly(SUVL) MD simulations.
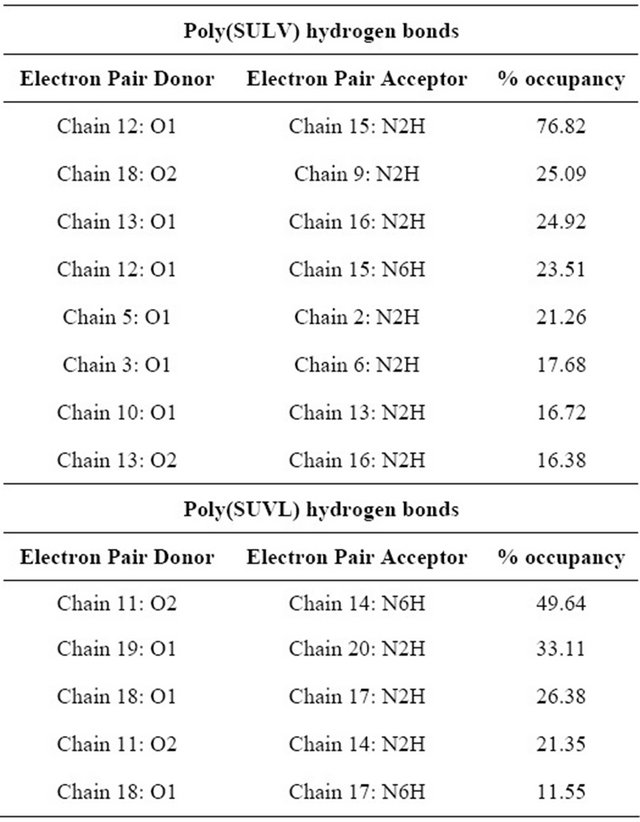
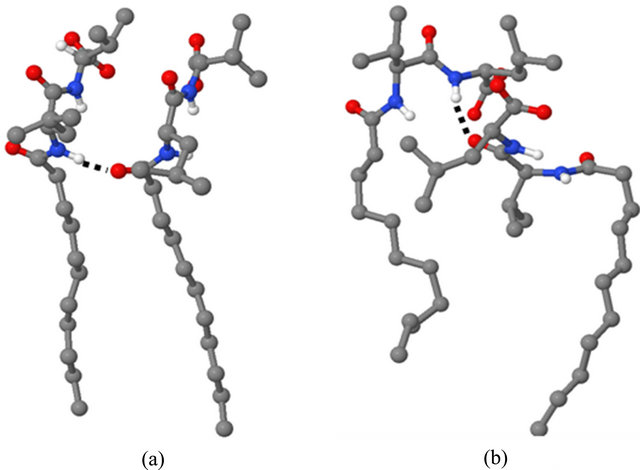
Figure 4. Hydrogen bonded structures for (a) poly(SULV) and (b) poly(SUVL).
mer chains facilitates H-bond formation between the donor and acceptor atoms.
Results from the poly(SUVL) hydrogen bond analysis are also shown in Table 1. For this MM, five intra-molecular H-bonds had occupancies greater than 10%. Table 1 reveals, though, that three of these hydrogen bonds formed between monomers at the end of the MM polymer chain. For example, a H-bond formed between chains 19 and 20 and two H-bonds with occupancies of 26.38% and 11.55% formed between chains 17 and 18. It would be expected that monomer chains at either end of the MM would have greater motional freedom, making it easier for atoms to come close enough to form H-bonds. The two remaining poly(SUVL) hydrogen bonds in Table 1 formed between atoms on monomer chains 11 and 14. Overall, the results in Table I indicate that relatively fewer intra-molecular H-bonds formed in poly(SUVL) than in poly(SULV). In addition, in poly(SUVL) the H-bonds were more evenly distributed between atoms on the leucine and valine amino acids, with three of the five H-bond acceptor atoms coming from the N-terminal valine and two from the C-terminal leucine. A similar distribution is seen for the donor atoms. Therefore, unlike poly(SULV), there is no clear preference in poly- (SUVL) for formation of intra-molecular H-bonds with the N-terminal amino acid.
The observations that in poly(SUVL) there are fewer overall intra-molecular H-bonds and relatively fewer intra-molecular H-bonds with the N-terminal amino acid are consistent with a large fraction of the poly(SUVL) headgroups forming folded conformations that point the valine amino acid side chain toward the hydrocarbon core. This conformation shields potential donor-acceptor atoms on adjacent chains from one another and reduces the potential for inter-chain hydrogen bond formation. Figure 4(b) shows chains 11 and 14 of poly(SUVL) forming an intra-molecular hydrogen bond. Note that chain 11 adopts the folded conformation discussed above and that the donor and acceptor atoms are respectively, O2 and the leucine amide hydrogen. The H-bond depicted in Figure 4 (b) had an occupancy of 49.64%. The occupancy of a H-bond between O1 and the valine amide hydrogen of the same monomer chains was 21.35%. Overall, the poly(SUVL) intra-molecular hydrogen bond results suggest that the same steric factors that restrict H-bond formation between poly(SUVL) headgroup atoms on different monomer chains likely also prevent chiral analyte molecules from accessing and forming hydrogen bonds with these headgroup atoms. The result is fewer stereo selective analyte:MM hydrogen bonding interactions in poly(SUVL) and thus a relatively low chiral selectivity.
The number of intermolecular hydrogen bonds between water molecules and the poly(SULV) and poly- (SUVL) headgroup atoms were also analyzed. It has been previously reported that of the two MM investigated here, poly(SULV) had more water molecules in the micelle headgroup region [22]. This result was shown to be consistent with the relative magnitudes of association constants for polar and non-polar analytes binding to each MM [22]. During the MD simulations, the total number of intermolecular hydrogen bonds formed by the two MM were very similar, with poly(SULV) and poly- (SUVL) forming, respectively 200,594 and 199,502 intermolecular H-bonds with water. While the total number of intermolecular H-bonds differed by less than 1% for the two MM, differences were observed in the distribution of these hydrogen bonds within the dipeptide headgroup. We first consider intermolecular hydrogen bonds between the water oxygen and MM headgroup NH atoms. In poly(SULV), the donor atom in 57.9% of these Hbonds was the C-terminal valine NH atom. The remaining 42.1% of the hydrogen bonds were formed by the Nterminal leucine amide hydrogen. In poly(SUVL), there was a greater preference for hydrogen bonding with the C-terminal amino acid. Here 67.4% of the intermolecular hydrogen bonds to water oxygen atoms were formed by the C-terminal leucine NH, while 32.6% formed with the N-terminal valine amide hydrogen.
A similar trend was observed for intermolecular hydrogen bonds between water hydrogens and MM headgroup oxygen atoms. In poly(SULV), 56.4% of these intermolecular H-bonds formed between water and the C-terminal carboxylate oxygens. The remaining 43.6% formed with the oxygen atomsO1 and O2 in Figure 1. In poly(SUVL), there was again a slightly greater preference for intermolecular H-bond formation with the C-terminal amino acid. Here 60.9% of the intermolecular H-bonds with water hydrogens formed with the C-terminal leucine carboxylate oxygens, while 39.1% formed with the other oxygen atoms in the dipeptide headgroup. These results are consistent with poly(SULV) forming a more open headgroup structure that allows greater access by water molecules to both amino acids of the dipeptide headgroup. Poly(SUVL) in contrast has been shown to have fewer water molecules in both the core and headgroup regions and the hydrogen bond analysis presented here shows relatively fewer H-bonds forming between water and the N-terminal valine amino acid. If water is less able to access and H-bond to the poly(SUVL) Nterminal amino acid atoms, chiral analyte access to these atoms and to the valine chiral center is likely restricted as well. This idea is explored in greater detail below where the number of water molecules surrounding each chiral center is compared.
Finally, hydrogen bond analyses showed that while the total number of H-bonds formed by poly(SULV) and poly(SUVL) were nearly identical, poly(SUVL) had a larger number of intermolecular hydrogen bonds with percent occupancies greater than 10%. In poly(SUVL), five H-bonds formed between water and the headgroup amide hydrogens with occupancies ranging from 16.13% to 10.30%. Three H-bonds with occupancies of 11.94%, 11.85%, and 10.90% were formed between water and the poly(SUVL) headgroup oxygen atoms. In contrast, only three H-bonds with occupancies greater than 10% were found to form between water and the poly(SULV) amide hydrogens and all H-bonds between water and the poly(SULV) oxygen atoms had occupancies less than 10%. These results suggest that the intermolecular hydrogen bonds formed between water and the poly(SULV) headgroup atoms are shorter-lived or that the H-bonding environment around the poly(SULV) headgroup is relatively dynamic. This environment could provide more opportunities for chiral analytes to access and interact with the poly(SULV) headgroup atoms. In poly(SUVL), though, the H-bond analysis suggested that there is a more fixed, less dynamic solvation shell around the poly- (SUVL) headgroups as evidenced by the larger number of hydrogen bonds with high percent occupancies.
3.3. Surface Area
A plot of the MM solvent accessible surface area (SASA) versus simulation time for both poly(SULV) and poly- (SUVL) is shown in Figure 5. The SASA for both MM were ~10000 Å2 at the beginning of the MD simulations. The values decreased steadily and plateaued around 2.0 ns. Therefore, the changes in SASA observed from 2.0 ns to the end of the MD simulation are shown in Figure 5. The SASA results show that throughout the MD simulations, the poly(SULV) SASA was consistently greater than that of poly(SUVL). The mean SASA for poly- (SULV) in the 2.0 ns to 11.0 ns range was also 8179 Å2, while the poly(SUVL) mean was 7608 Å2. As discussed above, previously reported MD simulation results for
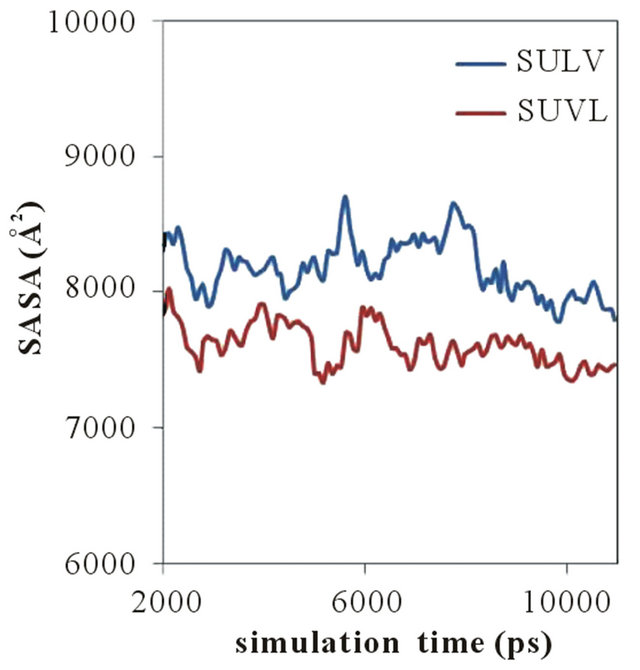
Figure 5. Solvent assessable surface area versus simulation time plots for poly(SULV) and poly(SUVL).
these MM showed that poly(SULV) had a more open structure than poly(SUVL) that allowed greater water penetration into the hydrocarbon core and didpeptide headgroup regions [22]. These results are consistent with poly(SULV) also having the greater SASA.
Insight into differences between the MM investigated here and conventional micelles formed from surfactants such as sodium dodecyl sulfate (SDS) can be gained by calculating the SASA per monomer chain. Dividing the average SASA for each MM by the twenty chains making up each macromolecule yields values of 409 Å2 and 380 Å2 per monomer chain for poly(SULV) and poly- (SUVL), respectively. In contrast, MD simulations by Bruce, et al. yielded a SASA per monomer residue of only 176 Å2 for SDS micelles [26]. This comparison suggests that in both the poly(SULV) and poly(SUVL) MM structures, more of the surfactant chain area is accessible to water molecules than in a corresponding SDS micelle. It is also interesting to note that NMR diffusion studies have shown that small non-polar probe molecules solubilize in the interior of SDS and unpolymerized amino acid-based surfactant micelles to a much greater degree than into the corresponding MM [27]. For example, the mole fraction of toluene molecules solubilzed into unpolymerized micelles formed by the amino acidbased surfactants sodium undecyl-(L,L)-valine and sodium undecyl-(L,L)-valine-valine were found to be 0.88 and 0.90 respectively. In contrast, the mole fraction of toluene solubilzed into the core of polymerized forms of these surfactants were 0.15 for poly-(sodium undecyl- (L,L)-valine) and 0.05 for poly-(sodium undecyl-(L,L)- valine-valine) [27]. It would be expected that water penetration into the MM cores would decrease their ability to solubilize or bind small nonpolar molecules such as toluene. Therefore, both MD simulation and NMR solubilization studies suggest that these MM exclude water from the micelle structure to a lesser extent than unpolymerized surfactant micelles.
The results plotted in Figure 5 also show that in the 2.0 to 11.0 ns range of the MD simulation, the SASA of both MM fluctuated over a relatively wide range. For example, the poly(SULV) SASA fluctuated between 8704 and 7786 Å2 while the poly(SUVL) values varied between 8023 and 7338 Å2. This result suggests that both MM experience relatively large scale, dynamic monomer chain motions during the MD simulations. These motions are likely important for analyte binding and chiral recognition because they could allow space or pockets to form between the monomer chains. These pockets would in turn allow chiral analytes to access or penetrate into the MM and interact stereo selectively with the dipeptide headgroups.
In order to test this hypothesis and to identify the monomer chain motions associated with the observed changes in the SASA, MM structures with high and low SASA values were identified. These structures were then overlaid to show the structural changes leading to the fluctuations in the SASA observed in Figure 5. Figures 6(a)-(d) show super positions of poly(SULV) monomer chains 9, 13, 11 and 16, respectively for structures extracted at 4.420 ns (low SASA) and 7.735 ns (high SASA) of the MD simulation. The structures in Figure 6(a) show the two hydrocarbon chains lying almost on top of one another, but with the headgroup atoms in different positions. This superposition suggests that one motion
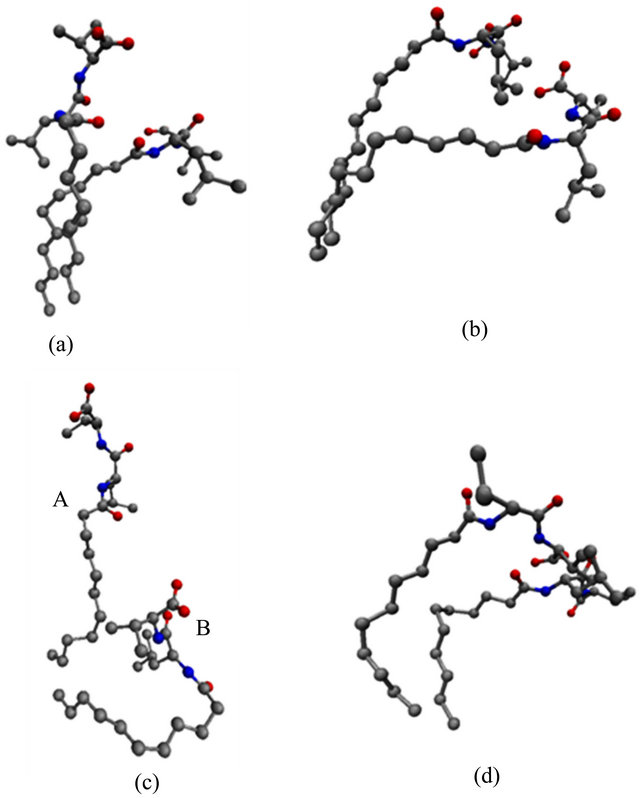
Figure 6. Overlaid structures extracted at 4.420 and 7.735 ns of the MD simulation for (a) chain 9, (b) chain 13, (c) chain 11, and (d) chain 16 of poly(SULV).
leading to the changes observed in the SASA is an opening and closing of cavities or pockets near the monomer headgroups. This motion could allow movement of chiral analytes into the cavities created between the monomer headgroups where they could then bind to the MM in a stereo selective fashion. The motion depicted in Figure 6(a) is thus expected to be important in chiral recognition. Figure 6(b) depicts another cavity-opening motion observed during the MD simulation. Here both of the headgroups and five carbon atoms along the hydrocarbon chain move apart to create a potential analyte binding pocket. Finally, Figure 6(c) shows a poly(SULV) monomer chain changing its shape from a folded to a more open wing-like conformation. This change increases the SASA and the open conformation labeled A in Figure 6(c) likely gives both water and chiral analytes greater access to the MM chiral centers.
Other chain motions, though, identified by the superimpositions discussed above produced changes in the SASA that were likely not important in chiral recognition. For example, the superimposed structures in Figure 6(d) show the two MM headgroups overlapped and the hydrocarbon chain atoms in different positions. These structures suggest that a motion that could produce changes in the SASA involves gaps or cavities opening between the hydrocarbon chains. These motions would give water molecules access to the core region of the micelle, but would not be expected to be important in governing chiral selectivity. Finally, very similar chain motions to those shown in Figure 6 were observed when analogous super positions of poly(SUVL) structures with high (5.95 ns) and low (10.12 ns) SASA values were carried out.
As discussed above, poly(SULV) has been found experimentally to be a relatively effective chiral selector, while chiral selectivity with poly(SUVL) is generally poor [8,21]. Therefore, it seems reasonable to expect that the motions identified above leading to fluctuations in the SASA should occur to a greater extent in poly(SULV) than in poly(SUVL). This should especially be true for those motions opening pockets near the dipeptide headgroup that give chiral analytes access to the MM chiral centers. To test this hypothesis, the variability in the positions of selected poly(SULV) and poly(SUVL)atoms during the MD simulations were compared using the following method. Four carbon atoms were chosen that spanned the midpoint of the monomer hydrocarbon chain to the end of the dipeptide headgroup. Moving up toward the headgroup, these atoms are labeled C6, C10, N-terminal Ca, and C-terminal Ca in Figure 7(a). The root mean squared deviation (rmsd) in the positions of these four atoms with respect to their positions in the average MD simulation structures were then calculated. Changes in these rmsd values in the 2.50 to 11.00 ps range of the MD simulations are plotted in Figures 7(b)-(e) for car
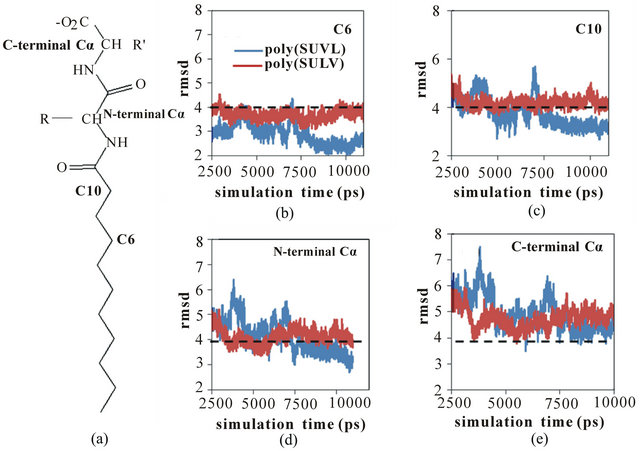
Figure 7. (a) MM structure identifying carbon atoms C6, C10, and the Nand C-terminal acarbon atoms. RMDS versus simulation time plots for poly(SULV) (blue) and poly(SUVL) (red) atoms; (b) C6; (c) C10; (d) N-terminal Cα; (e) C-terminal Cα.
bon atoms C6, C10, and the Nand C-terminal Ca atoms, respectively. The dashed line at 4.0 Å is included to facilitate comparison between the four graphs.
In Figure 7(b), the rmsd values for C6 are consistently less than 4 Å and both the poly(SULV) and poly(SUVL) values show relatively little variability. However, moving up to atom C10 (Figure 7(c)), the rmsd values for both MM increase and the poly(SULV) plot begins to show greater fluctuations in the atoms’ positions when compared to poly(SUVL). The rmsd values continue to increase moving up the monomer chain to the Nand C-terminal Ca carbons. This observation suggests than in the MD simulations of both MM, there was greater variability in the positions of the dipeptide headgroup atoms than for the carbon atoms along the hydrocarbon chain. The results plotted in Figures 7(d) and (e) also show more variability in the poly(SULV) rmsd values suggesting that the poly(SULV) headgroup atoms undergo larger fluctuations in their positions than corresponding atoms in poly(SUVL). Therefore, it can be concluded that chain motions leading to changes in the SASA, and likely opening and closing chiral analyte binding pockets, occur to a greater extent in poly(SULV) than in poly (SUVL). If chiral analytes are provided with more potential binding pockets, we would then expect poly(SULV) to have the higher chiral selectivity, as is observed experimentally.
3.4. Water Shell Results
For chiral recognition to take place chiral pockets must open as described above, but analytes must also have access to the chiral centers of the dipeptide headgroup where stereo selective interactions take place. Thereforeto compare the access that each MM provides to the dipeptide headgroup chiral centers, the number of water molecules surrounding the chiral carbons in both MM was investigated. The rationale for this comparison was that greater water access to the chiral centers should also allow greater access for chiral analytes. Figure 8(a) shows comparisons of the number of water molecules within 5.0 Å of the N-terminal Ca chiral centers of poly(SULV) and poly(SUVL). Note that more water molecules surround the poly(SULV)chiral center, therefore, chiral analytes likely have greater access to this chiral center as well. A similar result was obtained when the number of water molecules surrounding the C-terminal chiral centers in each MM were compared. These results are shown in Figure 8(b), again probing the number of water molecules within 5.0 Å of the selected chiral atom. As expected, overall more water molecules were found to surround the C-terminal Ca atom than surrounded the N-terminal Ca in both MM. In addition, as with the N-terminal Ca analysis, poly(SULV) has more water molecules surrounding the C-terminal Ca atom when compared to poly(SUVL). Therefore, it can be concluded that the more open structure of poly(SULV) gives water molecules and thus by extension chiral solutes greater access to the chiral atoms in the dipeptide headgroup.
3.5. Sodium Ion Distribution
The final MM property investigated with the MD simulations was the distribution of sodium counterions around each macromolecule. The counterion distribution was studied because the charge and the distribution of counterions around each monomer headgroup would be expected to influence the electrostatic repulsion between monomer chains and thus the overall size and shape of the macromolecule. The first step of this analysis was to calculate the radial distribution functions (RDF) between sodium ions and the carboxylate oxygen atoms. The RDF plot for poly(SULV) is shown in Figure 9(a). These data indicate that some of sodium ions are relatively close to
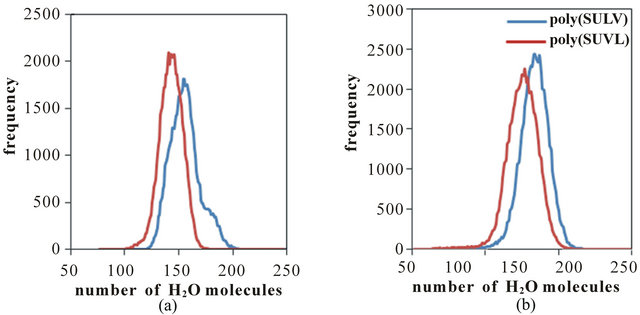
Figure 8. Water shells surrounding the poly(SULV) and poly(SUVL) chiral centers. (a) N-terminal amino acid chiral center; (b) C-terminal amino acid chiral center.
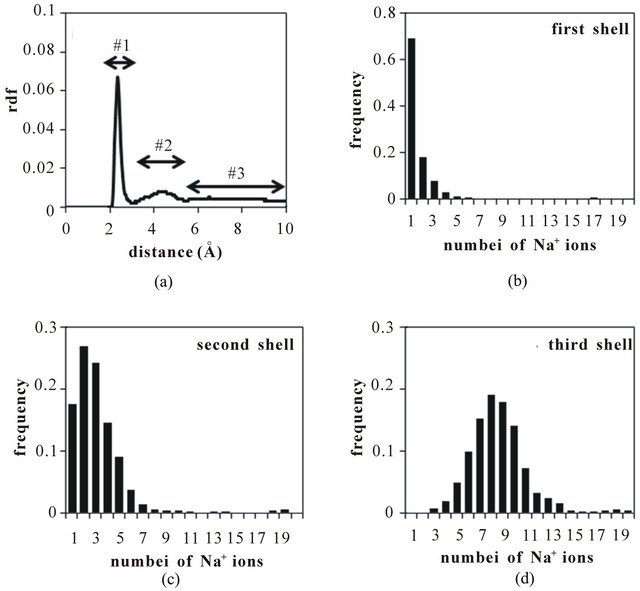
Figure 9. (a) Radial distribution function for Na+ ions identifying counterion shells #1, #2, and #3. Number is Na+ ions occupying the (b) first; (c) second; (d) third shells.
the MM carboxylate anions, thus producing the first peak in the RDF in the 2.0 - 3.3 Å range. Other sodium ions were found to have moved farther from the carboxylate headgroups yielding the second peak in the RDF between 3.3 and 5.2 Å. Finally, the remaining sodium ions are located farther than 5.2 Å from the MM carboxylate headgroups.
To better understand the distributions of sodium ions around the MM, the system was divided into three Na+ shells extending, respectively, 3.3 Å, 5.2 Å, and 10.0 Å from the carboxylate oxygens. The populations of sodium ions in these shells for LV are plotted in Figures 9(b)-(d). Figures 9(b) and (c) show that relatively few sodium occupy the first two shells closest to the carboxylate oxygens. The distribution in Figure (d) indicates that on average about 9 sodium ions occupy the third shell, with the remaining ions dissociated from the MM and found in the bulk aqueous phase. Overall, the distributions shown in Figures 9(b)-(d) suggest that about 50% of sodium ions are within 10.0 Å of carboxylate anions while the remaining 50% of the sodium ions diffuse farther from the micelle during the MD simulation. The RDF plot and sodium ion distribution plots for poly(SUVL) were found to be very similar to the poly- (SULV) plots shown in Figure 9.
4. Conclusion
MD simulations were used to investigate the chiral MM poly(SULV) and poly(SUVL). The more effective chiral selector poly(SULV) was found to have a more conformationally flexible headgroup and a larger SASA. Poly- (SULV) also allowed more intraand intermolecular H-bonds to form with atoms on the N-terminal amino acid of the dipeptide headgroup. In addition, the MD simulations suggested that large-scale monomer chain motions occur in both MM. These motions may open cavities or pockets that could be used by chiral analytes to access the chiral centers of the MM dipeptide headgroups. Finally, water molecules were found to have greater access to the poly(SULV) chiral centers and an analysis of the distribution of sodium ions showed that approximately 50% of the sodium counterions are found within 10.0 Å of the MM carboxylate groups.
5. Acknowledgements
This work was supported by grant # 8G12 MD007597 from NIMHD, NIH to the RCMI program at Howard University, a NSF CAREER grant to Dr. Eugene Billiot (No. 0449742), and a Robert A. Welch Chemistry Departmental Grant to the Chemistry Program at Texas A&M University-Corpus Christi, a NSF grant to Drs Kevin F. Morris and Fereshteh H. Billiot (No. CHE- 1213532), Howard University College of Medicine Bridge Funds and Pilot Study Awards program (BFPSAP) to Dr. Yayin Fang (No. U400040). We also acknowledge the Donors of the American Chemical Society Petroleum Research Fund (46707-B4) and the generosity of the Ralph E. Klingenmeyer family.
REFERENCES
- FDA Policy Statement for the Development of New Stereoisomeric Drugs, US Food and Drug Administration, Center for Drug Evaluation and Research, Department of Health and Human Services, 1992.
- G. Subramanian, “Chiral Separation Techniques,” 3rd Edition, Wiley-VCH, New York, 2007.
- T. J. Ward and K. D. Ward, “Chiral Separations: Fundamental Review,” Analytical Chemistry, Vol. 82, No. 12, 2010, pp. 4712-4722. doi:10.1021/ac1010926
- B. C. Valle, K. F. Morris, K. A. Fletcher, V. Fernand, D. M. Sword, S. Eldridge, C. K. Larive and I. M. Warner, “Understanding Chiral Molecular Micellar Separations Using Steady-State Fluorescence Anisotropy, Capillary Electrophoresis, and NMR,” Langmuir, Vol. 23, No. 2, 2007, pp. 425-435. doi:10.1021/la0612623
- L. Sanchez-Hernandez, M. Castro-Puyana, M. L. Marina and A. L. Crego, “Recent Approaches in Sensitive Enantioseparations by CE,” Electrophoresis, Vol. 33, No. 1, 2012, pp. 228-242. doi:10.1002/elps.201100404
- J. Wang and I. M. Warner, “Chiral Separations Using Micellar Electrokinetic Capillary Chromatography and a Polymerized Chiral Micelle,” Analytical Chemistry, Vol. 66, No. 21, 1994, pp. 3773-3776. doi:10.1021/ac00093a037
- S. J. Thibodeaux, E. J. Billiot and I. M. Warner, “Enantiomeric Separations Using Poly(L-Valine) and Poly(LLeucine) Surfactants: Investigation of Steric Factors Near the Chiral Center,” Journal of Chromatography A, Vol. 966, No. 1-2, 2002, pp. 179-186. doi:10.1016/S0021-9673(02)00747-1
- E. J. Billiot and I. M. Warner, “Examination of Structural Changes of Polymeric Amino Acid-Based Surfactants on Enantioselectivity: Effect of Amino Acid Order, Steric Factors, and Number and Position of Chiral Centers,” Analytical Chemistry, Vol. 72, No. 8, 2000, pp. 1740-1748. doi:10.1021/ac9908804
- E. J. Billiot, R. A. Agbaria, S. A. Shamsi and I. M. Warner, “Amino Acid Order in Polymeric Dipeptide Surfactants: Effect on Physical Properties and Enantioselectivity,” Analytical Chemistry, Vol. 71, No. 7, 1999, pp. 1252-1256. doi:10.1021/ac980461r
- F. H. Haddadian, E. J. Billiot, S. A. Shamsi and I. M. Warner, “Chiral Separations using Polymeric Dipeptide Surfactants: Effect of Number of Chiral Centers and Steric Factors,” Journal of Chromatography A, Vol. 858, 1999, pp. 219-227. doi:10.1016/S0021-9673(99)00810-9
- S. J. Thibodeaux, E. J. Billiot and I. M. Warner, “Enantiomeric Separations Using Polymeric L-Glutamate Surfactant Derivatives: Effect of Increasing Steric Factors,” Electrophoresis, Vol. 24, No. 6, 2003, pp. 1077-1082. doi:10.1002/elps.200390125
- F. Haddadian-Billiot, M. E. McCarroll, E. J. Billiot, J. K. Rugutt, K. F. Morris and I. M. Warner, “Comparison of the Aggregation Behavior of 15 Polymeric and Monomeric Dipeptide Surfactants in Aqueous Solution,” Langmuir, Vol. 18, No. 8, 2002, pp. 2993-2997. doi:10.1021/la0110592
- J. L. Haynes III, E. J. Billiot, H. H. Yarabe, S. A. Shamsi and I. M. Warner, “Chiral Separation with Dipeptide-Terminated Polymeric Surfactants: The Effect of an Extra Heteroatom on the Polar Head Group,” Electrophoresis, Vol. 21, No. 8, pp. 1597-1605. doi:10.1002/(SICI)1522-2683(20000501)21:8<1597::AID-ELPS1597>3.0.CO;2-L
- J. K. Rugutt, E. J. Billiot and I. M. Warner, “NMR Study of the Interaction of Monomeric and Polymeric Chiral Surfactants with (R)- and (S)-1,1’-Binaphthyl-2,2’-diyl Hydrogen Phosphate,” Langmuir, Vol. 16, No. 7, 2000, pp. 3022-3029. doi:10.1021/la990539e
- E. J. Billiot, J. Macossay, S. J. Thibodeaux, S. A. Shamsi and I. M Warner, “Chiral Separations Using Dipeptide Polymerized Surfactants: Effect of Amino Acid Order,” Analytical Chemistry, Vol. 70, No. 7, 1998, pp. 1375-1381. doi:10.1021/ac9709561
- C. W. Harrell, K. F. Morris, M. E. McCarroll, E. J. Billiot and I. M. Warner, “Fluorescence and Nuclear Magnetic Resonance Spectroscopic Studies of the Effect of the Polymerization Concentration on the Properties of an Amino Acid-Based Polymeric Surfactant,” Langmuir, Vol. 19, No. 26, 2003, pp. 10684-10691. doi:10.1021/la0348362
- F. Haddadian-Billiot, E. J. Billiot and I. M. Warner, “Depth of Penetration of Binaphthyl Derivatives into the Micellar Core of Sodium Undecenoyl Leucyl-Leucinate Surfactants,” Journal of Chromatography A, Vol. 950, No. 1-2, 2002, pp. 233-239. doi:10.1016/S1570-0232(02)00008-9
- K. F. Morris, B. A. Becker, B. C. Valle, I. M. Warner and C. K. Larive, “Use of NMR Binding Interaction Mapping Techniques to Examine Interactions of Chiral Molecules with Molecular Micelles,” Journal of Physical Chemistry B, Vol. 110, No. 35, 2006, pp. 17359-17369. doi:10.1021/jp0627224
- S. A. Kingsbury, C. J. Ducommun, B. M. Zahakaylo, E. H. Dickinson and K. F. Morris, “NMR Characterization of 1,1’-Binaphthyl-2,2’-Diyl Hydrogen Phosphate Binding to Chiral Molecular Micelles,” Magnetic Resonance in Chemistry, Vol. 48, No. 3, 2010, pp. 184-191.
- H. H. Yarabe, S. A. Shamsi and I. M Warner, “Characterization and Thermodynamic Studies of the Interactions of Two Chiral Polymeric Surfactants with Model Substances: Phenylthiohydantoin Amino Acids,” Analytical Chemistry, Vol. 71, No. 18, 1999, pp. 3992-3999. doi:10.1021/ac990212i
- S. A. Shamsi, B. C. Valle, F. H. Billiot and I. M. Warner, “Polysodium N-Undecanoyl-l-Leucylvalinate: A Versatile Chiral Selector for MicellarElectrokinetic Chromatography,” Analytical Chemistry, Vol. 75, No. 3, 2003, pp. 379- 387. doi:10.1021/ac020386r
- K. F. Morris, E. J. Billiot, F. H. Billiot, K. B. Lipkowitz, W. H. Southerland and Y. Fang, “Investigation of Chiral Molecular Micelles by NMR Spectroscopy and Molecular Dynamics Simulation,” Open Journal of Physical Chemistry, Vol. 2, No. 4, 2012, pp. 240-251.
- E. J. Billiot, “Chiral Recognition with Polymerized Dipeptide Surfactants in Capillary Electrophoresis,” Ph.D. Dissertation, Louisiana State University, Baton Rouge, 1998.
- D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P.A. Kollman, AMBER 10, University of California, San Francisco, 2008.
- J. Wang, P. Cieplak and P. A. Kollman, “How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules?” Journal of Computational Chemistry, Vol. 21, No. 12, 2000, pp. 1049-1074. doi:10.1002/1096-987X(200009)21:12<1049::AID-JCC3>3.0.CO;2-F
- C. D. Bruce, M. L. Berkowitz, L. Perera and M. D. E. Forbes, “Molecular Dynamics Simulation of Sodium Dodecyl Sulfate Micelle in Water: Micellar Structural Characteristics and Counterion Distribution,” Journal of Physical Chemistry B, Vol. 106, No. 15, 2002, pp. 3788-3793. doi:10.1021/jp013616z
- R. S. Hickok, S. A. Wedge, A. L. Hansen, K. F. Morris, F. H. Billiot and I. M. Warner, “Pulsed Field Gradient NMR Investigation of Solubilization Equilibria in Amino Acid and Dipeptide Terminated Micellar and Polymeric Surfactant Solutions,” Magnetic Resonance in Chemistry, Vol. 40, No. 12, 2002, pp. 755-761. doi:10.1002/mrc.1099
NOTES
*Corresponding author.