American Journal of Molecular Biology
Vol.2 No.2(2012), Article ID:18946,11 pages DOI:10.4236/ajmb.2012.22013
Modulation of gastric mucosal inflammatory responses to Helicobacter pylori by ghrelin: Role of cNOS-dependent IKK-β S-nitrosylation in the regulation of COX-2 activation
![]()
Research Center, University of Medicine and Dentistry of New Jersey, Newark, USA
Email: slomiabr@umdnj.edu
Received 25 January 2012; revised 15 February 2012; accepted 27 February 2012
Keywords: H. pylori; Gastric Mucosa; iNOS Induction; COX-2 Activation; Ghrelin; cNOS Phosphorylation; IKK-β; S-Nitrosylation
ABSTRACT
Disturbances in nitric oxide synthase (NOS) and cyclooxygenase (COX) isozyme systems, manifested by the excessive NO and prostaglandin (PGE2) generation, are well-recognized features of gastric mucosal inflammatory responses to H. pylori infection. In this study, we report that H. pylori LPS-induced enhancement in gastric mucosal inducible (i) iNOS expression and COX-2 activation was accompanied by the impairment in constitutive (c) cNOS phosphorylation, up-regulation in the inhibitory κB kinase-β (IKKβ) activation and the increase in the transcriptional factor, NF-κB, nuclear translocation. Further, we show that abrogation of cNOS control over NF-κB activation has lead to induction of iNOS expression and COX-2 activation through S-nitrosylation. Moreover, we demonstrate that the modulatory effect of peptide hormone, ghrelin, on the LPS-induced changes was reflected in the increase in Src/Akt-dependent cNOS activation through phosphorylation and the suppression of IKK-β activity through cNOS-mediated IKK-β protein S-nitrosylation. As a result, ghrelin exerted the inhibitory effect on NF-κB nuclear translocation, thus causing the repression of iNOS gene induction and the inhibition in COX-2 activation through iNOSdependent S-nitrosylation. Our findings point to cNOS activation as a pivotal element in the signaling cascade by which ghrelin exerts modulatory control over proinflammatory events triggered in gastric mucosa by H. pylori infection.
1. INTRODUCTION
Infection with Helicobacter pylori is a primary factor in the etiology of gastric disease, and the excessive nitric oxide (NO) and prostaglandin (PGE2) generation along with up-regulation in proinflammatory cytokine production, are well-documented features of H. pylori-induced gastritis as well as characterize gastric mucosal inflammatory responses to H. pylori lipopolysaccharide (LPS) [1-4]. While NO and PGE2 generated by the constitutive (c) nitric oxide synthase (cNOS) and cyclooxygenase-1 (COX-1) enzymes are responsible for maintaining normal physiological functions, and are recognized as essential elements of gastric mucosal defense mechanism, the overexpression of inducible (i) iNOS and cyclooxygenase-2 (COX-2) has been intimately implicated as the promoting event in H. pylori-associated chronic gastritis and gastric cancer [2,4-6].
A growing body of evidence, moreover, supports the existence of a functional and signaling relationship between NO generated by NOS isozyme system, and the formation of PGE2 synthesized from arachidonic acid by the action of COX systems [7-10]. The cross-talk between the products of NOS and COX pathways is supported by the studies indicating that stimulation of NO production through iNOS induction leads to COX enzymes activation and the increase in PGE2 generation, whereas NOS gene deletion or inhibition of NOS enzymes with pharmacological agents results in a decrease in PGE2 formation [9-12]. The role of cNOS in the iNOS-dependent COX-2 activation has also been suggested [10,13], and we have reported that the disturbances in NO and PGE2 generation elicited by H. pylori LPS are reflected in the massive up-regulation of iNOS and COX-2 activity, and the suppression in Src/Aktdependent cNOS activation [14-16].
Investigations into LPS-induced signaling events underlying the expression of proinflammatory mediators indicate that H. pylori LPS, like LPS of other Gram-negative bacteria, is known to trigger the activation of tolllike receptor-4 (TLR-4), which through downstream effectors leads to activation of transcriptional factors involved in iNOS and COX-2 gene induction [10,12,17,18]. While the induction of iNOS gene expression in response to LPS involves activation of transcriptional factor NF- κB [8,10,19], the role of NF-κB in the transcriptional control of COX-2 expression is less apparent and remains controversial [17,18,20]. Depending on the cell type, the regulation of COX-2 expression has been attributed to transcriptional factors, NF-κB, activator protein-1 (AP-1), cAMP response element biding protein (CREB), and CCATT/enhancer binding protein (C/EBP), as well as kinases of MAPK and PKC family [10,12,18, 20,21]. Moreover, up-regulation in COX-2 activation has been linked to posttranslational modification of the enzyme protein through S-nitrosylation via LPS-elicited induction in iNOS expression [9,10]. Indeed, as demonstrated recently, the induction in iNOS expression by H. pylori LPS leads to COX-2 S-nitrosylation that results in an excessive PGE2 generation [16].
In this study, we investigated further the role of cNOS in the signaling cascade of H. pylori LPS-induced expression of iNOS and COX-2 in gastric mucosal cells. Our results demonstrate that the LPS-induced abrogation of cNOS control over NF-κB activation results in the induction of iNOS expression and leads to COX-2 activation through S-nitrosylation. Moreover, our results show that peptide hormone, ghrelin, recognized for its modulatory control over NOS and COX enzyme systems [16,22-25], suppresses these untoward consequences of the LPS through up-regulation in cNOS activation that interferes with NF-κB nuclear translocation, thus causing the repression of iNOS gene induction and the inhibition of COX-2 activation through iNOS-dependent S-nitrosylation.
2. MATERIALS AND METHODS
2.1. Gastric Mucosal Cell Incubation
The cells were collected form the mucosa of freshly dissected rat stomachs with a blunt spatula, and suspended in five volumes of ice-cold Dulbecco’s modified (Gibco) Eagle’s minimal essential medium (DMEM), supplemented with fungizone (50 µg/ml), penicillin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf serum. The cells were then gently dispersed by trituration with a syringe, settled by centrifugation, and following rinsing resuspended in the medium to a concentration of 2 × 107 cell/ml [14]. Cell aliquots (1 ml) were then transferred to DMEM in culture dishes and incubated under 95% O2 - 5% CO2 atmosphere at 37˚C for up to16h in the presence of 0 - 100 ng/ml of H. pylori LPS [15]. H. pylori used for LPS preparation was cultured from clinical isolates obtained from ATCC No. 4350 [3]. In the experiments evaluating the effect of ghrelin (rat, Sigma), cNOS inhibitor, L-NAME, iNOS inhibitor, 1400W, Akt inhibitor, SH-5, Src inhibitor, PP2, NF-κB inhibitors, Bay11-7082 and PPM-18 (Calbiochem), and ascorbate (Sigma), the cells were first preincubated for 30 min with the indicated dose of the agent or vehicle before the addition of the LPS. The viability of cell preparations before and during the experimentation, assessed by Trypan blue dye exclusion assay [15], was greater than 97%.
2.2. cNOS and iNOS Activity Assay
Nitric oxide synthase activities of cNOS and iNOS enzymes in the gastric mucosal cells were measured by monitoring the conversion of L-[3H] arginine to L-[3H] citrulline using NOS-detect kit (Stratagene). The cells from the control and experimental treatments were homogenized in a sample buffer containing either 10 mM EDTA (for Ca2+-independent iNOS) or 6 mM CaCl2 (for Ca2+- depenedent cNOS), and centrifuged. The aliquots of the resulting supernatant were incubated for 30 min at 25˚C in the presence of 50 µCi/ml of L-[3H] arginine, 10 mM NAPDH, 5 µM tetrahydrobiopterin, and 50 mM Tis-HCl buffer, pH 7.4, in a final volume of 250 µl. Following addition of stop buffer and Dowex-50 W (Na+) resin, the mixtures were transferred to spin cups, centrifuged and the formed L-[3H] citrulline contained in the flow through was quantified by scintillation counting [15].
2.3. COX-1 and COX-2 Activity Assay
The cyclooxygenase activity of COX-1 and COX-2 isoforms in gastric mucosal cells was measured with the COX Activity Assay Kit (Cayman) by monitoring the appearance of oxidized TMPD at 590 nm [16]. The cells from the control and various experimental treatments were settled by centrifugation, rinsed with phosphatebuffered saline, and homogenized in 0.3 ml of cold sample buffer containing 0.1 M TRIS-HCl, pH 7.8, and 1 mM EDTA, centrifuged at 12,000 × g for 10 min at 4˚C, and the supernatant collected. The COX-1 activity in 40 µl sample aliquots was measured in the absence and the presence of COX-2 inhibition (DuP-697), while the COX-2 activity assays were conducted in the absence and the presence of COX-1 inhibition (SC-560), according to manufacturer’s instruction.
2.4. Nuclear Protein Extraction
The aliquots of gastric mucosal cell suspension from the control and various experimental conditions were settled by centrifugation at 1500 × g for 5 min, rinsed with phosphate-β uffered saline, and lysed by incubation for 10 min on ice in the lysis buffer, containing 10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5% Nonidet P-40, 1 mM dithiothreitol, and 0.5 mM PMSF [26]. Following centrifugation at 12,000 × g for 10 min at 4˚C, the supernatant was subjected to centrifugation at 100,000 × g for 1 h and the obtained soluble fraction was used as source of cytosolic extract [27]. The pellets, from 12,000 × g centrifugation, containing crude nuclei were suspended for 20 min at 4˚C in the extraction buffer, containing 20 mM HEPES, pH 7.9, 25% glycerol, 400 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM PMSF. The samples were centrifuged at 15,000 × g for 10 min at 4˚C, and the supernatants containing nuclear extracts were collected and stored at –70˚C until use.
2.5. IκB Kinase Activity Assay To measure the IKK-β activity we utilized the ELISAbased detection kit, K-LISATM (Calbiochem). The GSTIκB-α 50-amino acid peptide that includes the Ser32 and Ser36 of IκB-α phosphorylation sites was used as a substrate [27]. The gastric mucosal cell cytosolic extracts were incubated a glutathione-coated 96-well plate with GST-tagged IκB-α at room temperature for 30 min, and the phosphorylated GST-IκB-α substrate was detected using anti-phospho IκB-α (Ser32/Ser36) as first antibody, followed by horseradish peroxidase-conjugated secondary antibody. Following washing the retained complex was probed TMB reagent for spectrophotometric quantification at 450 nm.
2.6. IKK-β and COX-2 Protein S-Nitrosylation Assay
A biotin switch procedure was employed to assess IKK-β and COX-2 protein S-nitrosylation [28,29]. The gastric mucosal cells were treated with ghrelin (0.5 µg/ml), or Akt inhibitor, SH-5 (20 µM) + ghrelin (0.5 µg/ml), and incubated in the presence of 100 ng/ml of H. pylori LPS. Following centrifugation at 500 × g for 5 min, the recovered cells were lysed in 0.2 ml of HEN lysis buffer (250 mM HEPES, 1 mM EDTA, 0.1 mM neocuprin, pH 7.7), and the unnitrosylated thiol groups were blocked with S-methyl methanethiosulfonate reagent at 50˚C for 20 min [29]. The proteins were precipitated with acetone, resuspended in 0.2 ml of HEN buffer containing 1% SDS, and subjected to targeted nitrothiol group reduction with sodium ascorbate (100 mM). The free thiols were then labeled with biotin and the biotinylated proteins were recovered on streptavidin beads. The formed streptavidin bead-protein complex was washed with neutralization buffer, and the bound proteins were dissociated from streptavidin beads with 50 µl of elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, pH 7.7) containing 1% 2-mercaptoethanol [29]. The obtained proteins were then analyzed by Western blotting.
2.7. Immunoblot Analysis
The mucosal cells from the control and experimental treatments were collected by centrifugation and resuspended for 30 min in ice-cold lysis buffer (20 mM TrisHCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X- 100, 2 mM EDTA, 1 mM sodium orthovanadate, 4 mM sodium pyrophosphate, 1 mM PMSF, and 1 mM NaF), containing 1 µg/ml leupeptin and 1 µg/ml pepstatin [16]. Following brief sonication, the lysates were centrifuged at 12,000 × g for 10 min, and the supernatants were collected and analyzed for protein concentration using BCA protein assay kit (Pierce). The samples, including those subjected to biotin switch procedure, were then normalized with respect to protein content to ensure equal protein loading on SDS-PAGE. The proteins were suspended in loading buffer, boiled for 5 min, and subjected to SDS-PAGE using 40 µg protein/lane. The separated proteins were transferred onto nitrocellulose membranes, blocked for 1 h with 5% skim milk in Tris-buffered Tween (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and probed with specific polyclonal rabbit antibodies directed against IκB-α, COX-1, COX-2, iNOS, and cNOS (Calbiochem), NF-κB p65 and IKK-β (EMD Millipore). The phosphorylated cNOS (pcNOS) was analyzed using specific antibody (Calbiochem) directed against phospho-cNOS (mouse anti-eNOS, pSer1179).
2.8. Data Analysis
All experiments were carried out using duplicate sampling, and the results are expressed as means ± SD. Analysis of variance (ANOVA) and nonparametric KruskalWallis tests were used to determine significance. Any difference detected was evaluated by means of post hoc Bonferroni test, and the significance level was set at P < 0.05.
3. RESULTS
Infection of gastric mucosa by H. pylori in humans or stimulation of gastric mucosal cells with H. pylori LPS elicits a rapid proinflammatory reaction characterized by the excessive NO and PGE2 generation caused by the disturbances in NOS and COX isozyme systems [1-4,16]. Hence, to further ascertain the nature of the disturbances, we first assessed the time course of protein expression and the activity of NOS and COX isozyme systems in rat gastric mucosal cells exposed to H. pylori LPS. The results revealed that the LPS (100 ng/ml) elicited a significant induction in COX-2 (Figure 1(a)) and iNOS (Figure 2(a)) protein levels within 4 h of incubation, with further sustained increase for at least 16 h. This effect of the LPS, furthermore, was accompanied by a significant increase in the mucosal cell COX-2 (Figure 1(b)) and iNOS (Figure 2(b)) enzymatic activities. We have also observed that within the time frame of incubation, the LPS exerted no apparent effect on the mucosal cell ex-

Figure 1. Time course of COX-1 and COX-2 protein expression (a), and the activity of COX-1 and COX-2 (b) in rat gastric mucosal cells subjected to H. pylori LPS. The cells were treated with the LPS at 100 ng/ml and incubated for up to 16 h. At the indicated time points, the cell lysates were analyzed by Western blotting with anti-COX-1 and anti-COX-2 antibody for protein expression, and the relative densities are expressed as fold of control. The activities of COX-1 and COX-2 were measured as described under “Materials and Methods”. The data represent the means ± SD of four separate experiments. *P < 0.05 compared with that of control (time-0).
pression of COX-1 (Figure 1(a)) and cNOS (Figure 2(a)) proteins. However, while the COX-1activity was not discernibly affected by the LPS (Figure 1(b)), the activity of cNOS in the presence of the LPS decreased markedly with the incubation time (Figure 2(b)).
Moreover, we established that preincubation of gastric mucosal cells with peptide hormone, ghrelin, lead to a concentration-dependent suppression of the LPS-induced COX-2 (Figure 3(a)) and iNOS (Figure 3(b)) activities, and the reversal in the LPS inhibitory effect on the cNOS activity. Ghrelin, however, showed no discernible effect on the mucosal cell activity of COX-1 enzyme. Furthermore, examination of the influence of the LPS and ghrelin on the gastric mucosal cell expression of cNOS, iNOS, COX-1, and COX-2 proteins revealed that the LPS-induced suppression in cNOS activity was associated with the inhibition in the enzyme phosphorylation, while up-regulation in cNOS activity by ghrelin was reflected in a marked increase in the enzyme phosphoryla-
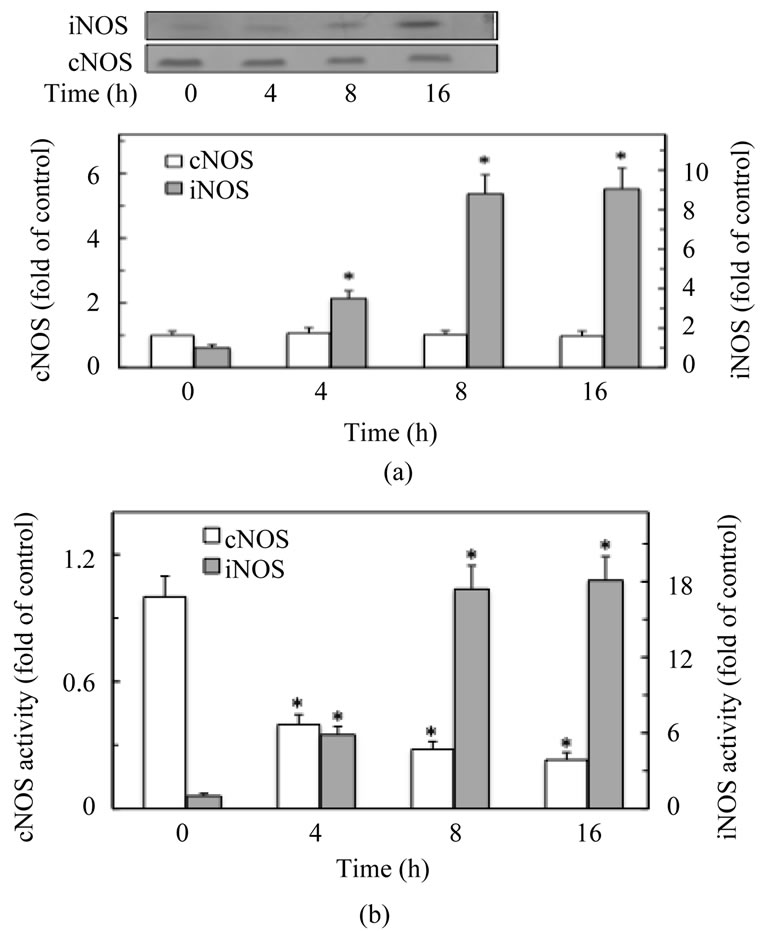
Figure 2. Time course of cNOS and iNOS protein expression (a), and the activity of cNOS and iNOS (b) in rat gastric mucosal cells subjected to H. pylori LPS treatment. The cells were treated with the LPS at 100 ng/ml and incubated for up to 16 h. At the indicated time points, the cell lysates were analyzed by Western blotting with anti-cNOS and anti-iNOS antibody for protein expression, and the relative densities are expressed as fold of control. The activities of cNOS and iNOS were measured as described under “Materials and Methods”. The data represent the means ± SD of four separate experiments. *P < 0.05 compared with that of control (time-0).
tion at Ser1179 (Figure 4). We also found that the countering effect of ghrelin on the LPS elicited induction in iNOS and COX-2 activity was manifested in a marked inhibition of the iNOS protein expression, but no apparent change in COX-2 protein expression. Moreover, neither the LPS nor ghrelin affected the expression of COX-1 protein. These results point to a role of iNOS in the LPS-induced up-regulation in COX-2 activation.
Hence, to provide further leads as to the requirements for iNOS-dependent up-regulation in COX-2 activation by H. pylori LPS, the mucosal cells prior to incubation with ghrelin were pretreated with the inhibitors of cNOS phosphorylation, an Akt inhibitor, SH-5 and Src inhibitor, PP2, and assayed for COX-2 and iNOS activity. As shown in Figure 5, the ghrelin-induced suppression in COX-2 and iNOS activation was susceptible to reversal by both inhibitors. Moreover, the LPS-induced up-regulation in COX-2 and iNOS activity displayed susceptibility to the inhibitors of NF-κB activation, PPM-18 and Bay11-7082. Further, we found that both these inhibitors

Figure 3. Effect of ghrelin on H. pylori LPS-induced expression of COX-1 and COX-2 (a), and cNOS and iNOS (b) activities in gastric mucosal cells. The cells, preincubated for 30 min with the indicated concentrations of ghrelin, were treated with the LPS at 100 ng/ml) and incubated for 8 h. Values are presented as fold of control, and represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone.
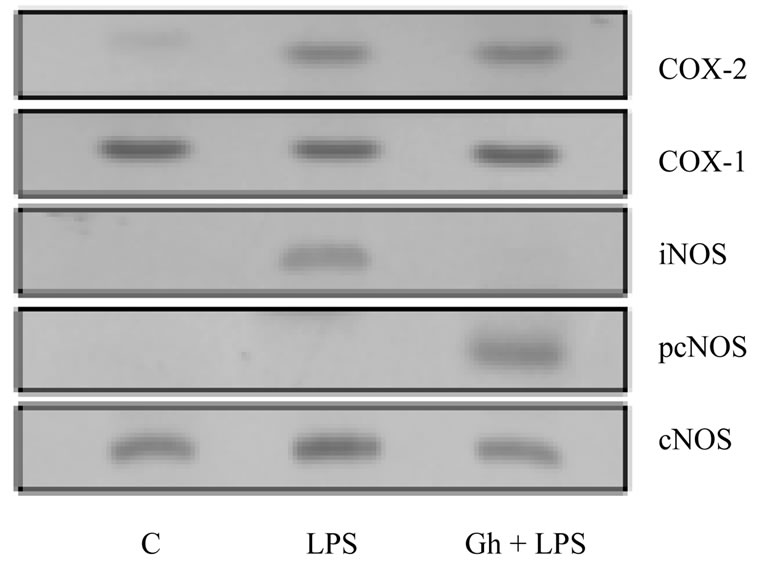
Figure 4. Effect of ghrelin (Gh) on H. pylori LPS-induced changes in expression of COX-1, COX-2, iNOS and cNOS proteins, and cNOS phosphorylation in gastric mucosal cells. The cells were treated with the LPS at 100 ng/ml or Gh at 0.5 µg/ml, and incubated for 8 h. Cell lysates were analyzed by Western blotting with anti-COX-1, anti-COX2, anti-iNOS, anticNOS, and phosphorylation specific cNOS (pcNOS) antibody. The immunoblots shown are representative of three experiments.
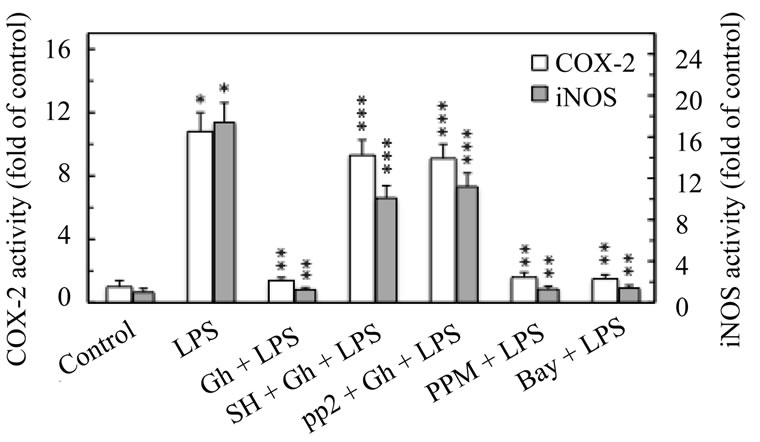
Figure 5. Effect of Akt inhibitor, SH-5, Src inhibitor, PP2, and NF-κB inhibitors, PPM-18 and Bay 11-7082, on the ghrelin (Gh)-induced changes in COX-2 and iNOS activities in gastric mucosal cells exposed to H. pylori LPS. The cells, preincubated with 20 µM SH-5 (SH), 30 µM PP2, 15 µM PPM-18 (PPM), or 20 µM Bay 11-7082 (Bay), were treated with Gh at 0.5 µg/ml and incubated for 8 h in the presence of 100 ng/ml of LPS. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. ***P < 0.05 compared with that of Gh + LPS.
of NF-κB activation, while showing no inhibitory effect on the LPS-induced expression of COX-2 protein, caused a marked inhibition in the LPS-induced expression of iNOS protein (Figure 6). Thus the countering effect of ghrelin on the LPS-induced COX-2 activation, like that of NF-κB inhibitors, is the consequence of iNOS suppression at the transcriptional level, and depends on cNOS activation through phosphorylation.
Therefore, to understand the mechanism underlying up-regulation in COX-2 activation, we examined the effect of ghrelin on the LPS-induced NF-κB activation. As NF-κB activation requires degradation of the inhibitory protein IκB-α that leads to translocation of p65 component of NF-κB from the cytoplasm to nucleus [30 -33], we exposed the gastric mucosal cells to H. pylori LPS in the absence or presence of ghrelin, and the cell lysates were analyzed for IκB-α level, while the nuclear extracts were assessed for p65 NF-κB. The results of immunoblots analysis revealed that, the LPS effect was manifested by a distinct reduction in IκB-α protein level and a marked increase of p65 in the nuclear extract (Figure 7). We also found that in the presence of ghrelin, the LPS-induced IκB-α degradation was significantly inhibited, while the extent of the LPS-induced nuclear translocation of p65 NF-κB showed a significant decrease.
Next, we analyzed the influence of ghrelin on H. pylori LPS-induced changes in the activity of IKK-β, a key enzyme of NF-κB activation pathway that controls the extent of IκB-α phosphorylation and its proteasomal degradation. The results revealed that ghrelin exerted a profound countering effect on the LPS-induced up-regulation in gastric mucosal cell IKK-β activity (Figure 8).

Figure 6. Effect of NF-κB inhibitors, Bay11-7082 (Bay) and PPM-18 (PPM), on H. pylori LPS-induced expression of COX-2 and iNOS proteins in gastric mucosal cells. The cells were treated with the LPS at 100 ng/ml, or Bay at 20 µM + LPS, or PPM at 15 µM + LPS, and incubated for 8 h. Cell lysates were resolved on SDS-PAGE, transferred to nitrocellulose, and probed with anti-COX-2 and anti-iNOS antibody. The immunoblots shown are representative of three experiments.

Figure 7. Effect of ghrelin on H. pylori LPS-induced IκB-α protein degradation and p65 NF-κB nuclear translocation in rat gastric mucosal cells. The cells were preincubated for 30 min with 0 or 0.5 µg/ml of ghrelin and incubated for 30 min with the LPS at 100 ng/ml. Cell lysates were analyzed for IκB-α by Western blotting with anti-IκB-α antibody, while the level of p65 NF-κB protein was assessed in the nuclear fraction with anti-p65 antibody (a). Relative densities are expressed as fold of control (b). The data represent the means ± SD of four experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS.
Moreover, the effect of ghrelin on LPS-induced upregulation in IKK-β activity was subject to suppression by the inhibitors of Src/Akt pathway, PP2 and SH-5. A significant decrease in the countering effect of ghrelin on the LPS-induced up-regulation in IKK-β activity was also attained in the presence of cNOS inhibitor, LNAME, while the inhibitor of NF-κB activation, PPM-18, and iNOS inhibitor, 1400 W, had no effect. This indicates that the countering effect of ghrelin on H. pylori LPSinduced up-regulation in gastric mucosal IKK-β activation, and the suppression of COX-2 and iNOS enzymes,

Figure 8. Effect of Src inhibitor, PP2, Akt inhibitor, SH-5, cNOS inhibitor, L-NAME, iNOS inhibitor, 1400W, and NF-κB inhibitor, PPM-18, on the ghrelin (Gh)-induced changes in IKK-β activity in gastric mucosal cells exposed to H. pylori LPS. The cells, preincubated with 30 µM PP2, 20 µM SH-5 (SH), 200 µM L-NAME (LN), 40 µM 1400W (14W), or 15 µM PPM-18 (PPM), were treated with Gh at 0.5 µg/ml and incubated for 30 min in the presence of 100 ng/ml of LPS. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. ***P < 0.05 compared with that of Gh + LPS.
occurs with the involvement of Src/Akt-mediated cNOS activation, and shows dependence on NO generated by the cNOS system.
Consequently, to reveal further the role of NO generated by cNOS isozyme system in the regulation of the LPS-induced IKK-β and COX-2 activation, we assessed the effect of nitrosothiols reducing agent, ascorbate. While preincubation with ascorbate produced no discernible effect on the extent of the LPS-induced IKK-β activation, a marked decrease was observed in the LPS-induced COX-2 activation (Figure 9). In addition, ascorbate elicited amplification in the effect of ghrelin on COX-2 activity, and produced a significant relieve in the inhibitory effect of ghrelin on the LPS-induced IKK-β activity. Moreover, examination of the effect of the LPS and ghrelin on the IKK-β and COX-2 S-nitrosylation patterns by the biotin switch method [28,29], revealed that gastric mucosal cells exposed to H. pylori LPS showed a marked increase in COX-2 S-nitrosylation, while the countering effect of ghrelin on the LPS-induced up-regulation in IKK-β activity was manifested by an increase in IKK-β S-nitrosylation and the substantial loss in COX-2 S-nitrosylation (Figure 10). Furthermore, the up-regulation in IKK-β S-nitrosylation by ghrelin, like that of IKK-β enzymatic activity, was susceptible to suppression by Akt inhibitor, SH-5. We have also observed that Akt inhibition with SH-5 caused the reversal in the inhibitory effect of ghrelin on the LPS-induced COX-2 protein S-nitrosylation. Thus, ghrelin-induced IKK-β S-nitrosylation causes the repression of iNOS
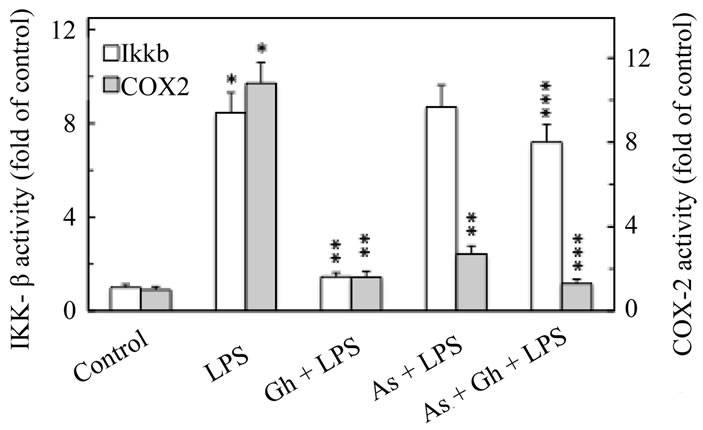
Figure 9. Effect of ascorbate on the ghrelin (Gh)-induced changes in the expression of IKK-β and COX-2 activities in gastric mucosal cells exposed to H. pylori LPS. The cells, preincubated with 300 µM ascorbate (As), were treated with Gh at 0.5 µg/ml and incubated for 30 min (in the case of IKKβ) and 8 h (in the case of COX-2) in the presence of 100 ng/ml LPS. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of LPS alone. ***P < 0.05 compared with that of Gh + LPS.

Figure 10. Effect of H. pylori LPS and ghrelin (Gh) on IKK-β and COX-2 protein S-nitrosylation in gastric mucosal cells exposed to Akt inhibitor, SH-5 (SH). The cells, were preincubated for 30 min with Gh at 0.5 µg/ml, or SH at (20 µM) + Gh, and incubated for 1 h (IKKβ) and 8 h (COX-2) in the presence of 100 ng/ml LPS. A portion of the cell lysates was processed by biotin switch procedure for protein S-nitrosylation and, along with the reminder of the lysates, resolved on SDS-PAGE, transferred to nitrocellulose probed with anti-IKKβ and anti-COX-2 antibody. The immunoblots shown are representative of three experiments.
induction and hence leads to the inhibition of COX-2 activation through iNOS-dependent S-nitrosylation.
4. DISCUSSION
Colonization of gastric mucosa by H. pylori in humans or stimulation of gastric mucosal cells with H. pylori LPS is known to elicit a cascade of inflammatory responses resulting in the release of proinflammatory mediators that increase the risk of gastric disease [2,4-6,14]. Primary among these mediators, is up-regulation in proinflammatory cytokine production and the excessive generation of NO and PGE2 [2,4,14,16]. While the NO and PGE2 generated by the constitutive cNOS and COX-1 enzyme systems are deemed essential for the maintenance of normal housekeeping functions, the overexpression of inducible iNOS and COX-2 isozymes, and consequent increase in NO and PGE2 production, are considered of major importance in defining the extent of gastric mucosal inflammatory involvement [4,5,16]. The signaling events underlying the induction of iNOS and COX-2 enzymes by bacterial LPS involves the stimulation of TLR-4, which then through a series of downstream effectors triggers the activation of transcriptional factors that exert control over iNOS and COX-2 gene expression [10,17,18,34,35]. Principal among the factors implicated in the regulation of inflammatory responses to H. pylori, is the nuclear transcriptional factor, NF-κB [34-36]. While the induction of iNOS gene by LPS has been convincingly linked to NF-κB activation [10,19, 26,30], the role of NF-κB in COX-2 gene expression remains less apparent [17,18,20]. Hence, in the present study we assessed the relationship between H. pylori LPS-induced NF-κB activation, iNOS gene induction, and COX-2 activation.
Our data, obtained with rat gastric mucosal cells, revealed that the LPS-elicited enhancement in the expression of iNOS COX-2 is accompanied by the impairment in cNOS phosphorylation, up-regulation in IKK-β activation, and the increase in NF-κB nuclear translocation. Further, we demonstrated that abrogation of cNOS control over NF-κB activation and its nuclear translocation leads to induction in iNOS expression, and COX-2 activation through S-nitrosylation. Moreover, we showed that ghrelin modulates the LPS-induced changes by exerting the inhibitory effect on NF-κB nuclear translocation, thus causing repression of iNOS gene and the inhibition of COX-2 activation through iNOS-dependent Snitrosylation. Indeed, examination of gastric mucosal cell expression of COX and NOS proteins and the activity of the individual isozymes demonstrated that H. pylori LPS-induced enhancement in iNOS and COX-2 enzymatic activities was associated with the induction in iNOS and COX-2 protein levels. The LPS, however, had no apparent effect on the expression of COX-1 and cNOS proteins, and COX-1 activity, while the activity of cNOS showed a marked decrease. Furthermore, assessment of the effect of peptide hormone, ghrelin, revealed that the induced increase in cNOS phosphorylation at Ser1179 was associated with a significant up-regulation in cNOS activity, inhibition of iNOS expression, and the suppression in COX-2 activity without affecting its protein expression. These findings are thus in concordance with the rapidly accumulating literature data attesting to a central role of ghrelin in modulation of gastric mucosal inflammatory responses to H. pylori colonization [14-16, 22-25], as well as point to cNOS activation through phosphorylation as a pivotal element of ghrelin signaling cascade.
The mechanism that underlies the regulation of NOS system by ghrelin involves the receptor (GHSR1a)- mediated activation of heterotrimeric G protein-dependent pathway that results in signal propagation through a multiple network of protein kinases, including that of Src/ Akt cascade that controls the process of cNOS activation [14,15,24,37,38]. Indeed, we found that in keeping with the documented involvement of Src/Akt in posttranslational cNOS activation through phosphorylation at Ser1179 [37,38], the countering effect of ghrelin on the LPS-induced up-regulation in COX-2 and iNOS activation was susceptible to suppression by Akt inhibitor, SH- 5, as well as Src inhibitor, PP2. Moreover, the LPS-induced increase in COX-2 and iNOS activity displayed susceptibility to PPM-18 and Bay 11-7082, the inhibitors of NF-κB activation with different mechanism of action [39,40]. However, while this inhibitory effect on iNOS activity was also reflected in a marked suppression in the expression of iNOS protein, neither PM-18 nor Bay 11-7082 affected the LPS-induced expression of COX-2 protein. Hence, considering the fact that PPM-18 is a potent blocker of NF-κB binding to its nuclear promoter response elements [39], while Bay 11-7082 inhibits the IκB kinase-β (IKK-β) enzymatic activity [18,40], we concluded that NF-κB does not appear to play a role in the regulation of H. pylori LPS-induced COX-2 protein expression and that the countering effect of ghrelin on the LPS-induced COX-2 activation, like that of NF-κB inhibitors, is the consequence of iNOS suppression at the transcriptional level, and depends on cNOS activation through phosphorylation.
Indeed, our studies confirmed an important role for ghrelin in the inhibition of NF-κB activation in response to H. pylori LPS-induced up-regulation in iNOS expression. NF-κB is a rapid transcriptional activator that is held in the cytoplasm of resting cells bound to a family of inhibitory IκB proteins. Upon stimulation by LPS or TNF-α, IκB undergoes phosphorylation at two critical serine residues by the an IκB kinase (IKK) complex, which targets IκB for degradation through the ubiquitinproteasomal pathway and leads to nuclear translocation of NF-κB, its biding to promoter response elements, and activation of the target gene transcription [30-34]. In concordance with this classical pathway of NF-κB activation [35,41], we have shown that the effect of H. pylori LPS was manifested by a distinct reduction in the inhibitory protein, IκB-α and a marked increase in p65 NF- κB nuclear translocation, whereas in the presence of ghrelin, the extent of the LPS-induced IκB-α degradation and the nuclear translocation of NF-κB decreased significantly. Moreover, we found that countering effect of ghrelin on the LPS-induced up-regulation in gastric mucosal cell IKK-b activity was subject to suppression by the inhibitors of Src/Akt pathway, PP2 and SH-5. A significant decrease in the effectiveness of ghrelin to counter the LPS-induced IKK-b activation was also observed in the presence of cNOS inhibitor, L-NAME, while the inhibitor of NF-κB activation, PPM-18, and iNOS inhibitor, 1400W, had no effect. From this, we inferred that the countering effect of ghrelin on H. pylori LPS-induced up-regulation in gastric mucosal IKK-b activation, as well as COX-2 and iNOS enzyme suppression, occurs with the involvement of Src/Akt-mediated cNOS activation, and shows dependence on NO generated by the cNOS. In this connection, it is pertinent to reiterate that signaling through Src/Akt pathway is known to occupy a central stage in the receptor (GHSR1a)-mediated responses to ghrelin stimulation [14,15, 24,38]. Deserving equally insightful consideration in furthering our understanding the signaling pathways of ghrelin are the reports indicating that the activity of IKK-β complex as well as that of COX-2 protein may also be regulated through S-nitrosylation [9,10,33,42- 44].
Indeed, as demonstrated recently, the induction in iNOS expression by LPS leads to COX-2 S-nitrosylation that result in an excessive PGE2 generation [9,10,16], and S-nitrosylation of a specific cysteine residue within the activation loop of IKK-β by endogenous NO donors eerts the inhibitory effect on the extent of IκB-α degradation, and hence affects the nuclear translocation of NF- κB [33,42,44]. Hence, to examine further the role of cNOS in the regulation of H. pylori LPS-induced IKK-β and COX-2 activation by ghrelin, we assessed the effect of nitrosothiol reducing agent, ascorbate. While preincubation with ascorbate produced no discernible effect on the extent of the LPS-induced IKK-β activation, a marked decrease was observed in the LPS-induced COX- 2 activity. Moreover, ascorbate elicited an amplification in the inhibitory effect of ghrelin on COX-2 activity and produced a significant relieve in the inhibitory effect of ghrelin on the LPS-induced IKK-β activity. Therefore, consistent with our findings, the regulation of IKK-β activity as well as the activation of COX-2 appears to be intimately linked to the events of S-nitrosylation by NO generated by the cNOS system. This contention is supported further by the results of biotin switch assay [28, 29]. Western blot analysis of COX-2 and IKK-β protein S-nitrosylation patterns revealed that gastric mucosal cells exposed to the LPS alone showed a marked increase in COX-2 S-nitrosylation, while the countering effect of ghrelin on the LPS-induced up-regulation in IKK-β activity was manifested by an increase in the kinase Snitrosylation as well as the substantial loss in COX-2
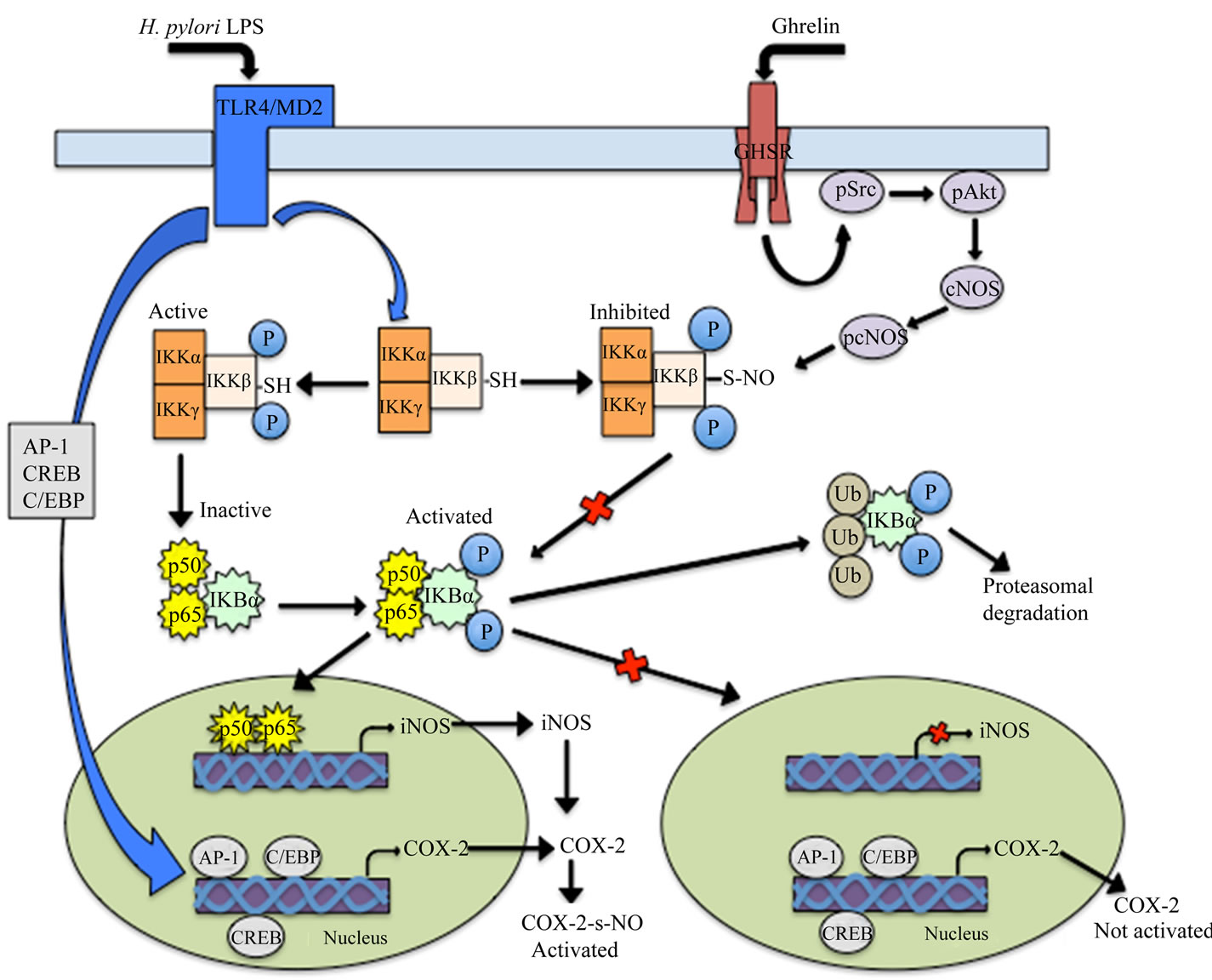
Figure 11. Proposed mechanism of ghrelin action in countering the gastric mucosal proinflammatory events triggered by H. pylori LPS. Binding of the LPS to Toll-like receptor 4 (TLR4)/MD2 triggers the activation and nuclear translocation of transcriptional factors involved in the induction of COX-2 (AP-1, CREB and C/EBP), and iNOS (NF-κB) genes transcription. The up-regulation in NO generated by iNOS leads to COX-2 protein S-nitrosylation that results in the induction of COX-2 enzymatic activity and the excessive PGE2 production. Binding of ghrelin to growth hormone secretagogue receptor (GHSR) triggers up-regulation in Src/Akt-dependent cNOS activation through phosphorylation that leads to the inhibition of the LPS-induced IKK-β ctivation by cNOS-mediated IKK-β S-nitrosylation, which interferes with IκB-α proteasomal degradation and NF-κB nuclear translocation, thus causing repression of iNOS gene induction and the inhibition of COX-2 activation through iNOS-dependent S-nitrosylation. AP-1, activator protein-1; CREB, cAMP response element binding protein; C/EBP, CCAAT/enhancer binding protein.
S-nitrosylation. Moreover, the induced by ghrelin increase in IKK-β S-nitrosylation was susceptible to suppression by Akt inhibitor, SH-5, which also caused the reversal of the countering effect of ghrelin on the LPSinduced COX-2 S-nitrosylation. Thus, our findings lend further support to increasingly apparent assertion that cNOS activation through Src/Akt-mediated phosphorylation is a pivotal element in the signaling cascade by which ghrelin exerts the modulatory control over proinflammatory events triggered in gastric mucosa by H. pylori infection [14-16,24,26,41].
Together, the data provided in our study demonstrate that H. pylori LPS-induced abrogation of cNOS control over NF-κB activation leads to the induction of iNOS expression that triggers up-regulation COX-2 activation through S-nitrosylation that results in an excessive PGE2 generation (Figure 11). We also show that peptide hormone, ghrelin, counters these untoward consequences of the LPS via up-regulation in Src/Akt-dependent cNOS activation that results in up-regulation in cNOS-mediated IKK-β S-nitrosylation which interferes with IκB-α proteasomal degradation and NF-κB nuclear translocation, thus causing repression of iNOS gene induction and hence the inhibition of COX-2 activation through iNOSdependent S-nitrosylation
REFERENCES
- Romano, M., Ricci, V., Memoli, A., et al. (1998) Helicobacter pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. Journal of Biological Chemistry, 273, 28560-28563. doi:10.1074/jbc.273.44.28560
- Fu, S., Ramanujam, K.S., Wong, A., et al. (1999) Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase-2 in Helicobacter pylori gastritis. Gastroenterology, 116, 1319-1329. doi:10.1016/S0016-5085(99)70496-8
- Slomiany, B.L. and Slomiany, A. (2002) Suppression of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide by peroxisome proliferator-activated receptor γ activation. IUBMB Life, 53, 303-308. doi:10.1080/15216540213459
- Reider, G., Hofmann, J.A., Hatz, R.A., Stolte, M. and Enders, G.A. (2003) Up-regulation of inducible nitric oxide synthase in Helicobacter pylori-associated gastritis may represent an increased risk factor to develop gastric carcinoma of the intestinal type. International Journal of Medical Microbiology, 293, 403-412. doi:10.1078/1438-4221-00280
- Wilson, K.T., Fu, S., Ramanujam, K.S. and Meltzer, S.J. (1998) Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinomas. Cancer Research, 58, 2929-2934.
- Wroblewski, L.A., Peek, R.M. and Wilson, K.T. (2010) Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clinical microbiology Review, 23, 713-739. doi:10.1128/CMR.00011-10
- Clancy, R., Varenika, B., Huang, W., et al. (2000) Nitric oxide synthase/COX cross-talk: Nitric oxide activates COX-1 but inhibits COX-2-derived prostaglandin production. Journal of Immunology, 165, 1582-1587.
- Cuzzocrea, S. and Salvemini, D. (2007) Molecular mechanisms involved in the reciprocal regulation of cyclooxygenase and nitric oxide synthase enzymes. Kidney International, 71, 290-297. doi:10.1038/sj.ki.5002058
- Kim, S.F., Huri, D.A. and Snyder, S.H. (2005) Inducible nitric oxide synthase binds, s-nitrosylates, and activates cyclooxygenase-2. Science, 310, 1966-1970. doi:10.1126/science.1119407
- Ye, Y., Martinez, J.D., Perez-Polo, R.J., Lin, Y., Uretsky, B.F. and Birnbaum, Y. (2008) The role of eNOS, iNOS, and NF-κB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastin. American Journal of Physiology Heart and Circulatory Physiology, 295, H343-H351. doi:10.1152/ajpheart.01350.2007
- Marnett, L.J., Wright, T.L., Crews, B.C., Tannenbaum, S.R. and Morrow, J.D. (2000) Regulation of prostaglandin biosynthesis by nitric oxide is revealed by targeted deletion of inducible nitric-oxide synthase. Journal of Biological Chemistry, 275, 13427-13430. doi:10.1074/jbc.275.18.13427
- Lamon, B.D., Upmacis, R.K., Deeb, R.S., Koyuncu, H. and Haijar, D. (2010) Inducible nitric oxide synthase gene deletion exaggerates MAPK-mediated cyclooxy-genase-2 induction by inflammatory stimuli. American Journal of Physiology Heart and Circulatory Physiology, 299, H613- H623. doi:10.1152/ajpheart.00144.2010
- Bell, R.M., Smith, C.C. and Yellon, D.M. (2002) Nitric oxide as a mediator of delayed pharmacological (A1 receptor triggered) preconditioning; is eNOS masquerading as iNOS? Cardiovascular Research, 53, 405-413. doi:10.1016/S0008-6363(01)00472-2
- Slomiany, B.L. and Slomiany, A. (2011) Helicobacter pylori induces disturbances in gastric mucosal Akt activetion through inducible nitric oxide synthase-dependent S-nitrosylation: Effect of ghrelin. ISRN Gastroenterology, 2011, 8 pages. doi:10.5402/2011/308727
- Slomiany, B.L. and Slomiany, A. (2011) Role of ghrelin-induced cSrc activation in modulation of gastric mucosal inflammatory responses to Helicobacter pylori. Inflammopharmacology, 19, 197-204.
- Slomiany, B.L. and Slomiany, A. (2011) Role of constitutive nitric oxide synthase in regulation of Helicobacter pylori-induced gastric mucosal cyclooxygenase-2 activation through S-nitrosylation: Mechanism of ghrelin action. Open Journal of Gastroenterology, 1, 13-22.
- Joo, M., Wright, J.G.., Hu, N.N., et al. (2007) Yin yang 1 enhances cyclooxygenase-2 gene expression in macrophages. American Journal of Physiology Lung and Cell Molecular Physiology, 292, L1219-L1226. doi:10.1152/ajplung.00474.2006
- Grishin, A.V., Wang, J., Potoka, D.A., et al. (2006) Lipopolysaccharide induces cyclooxygenase-2 in intestinal epithelium via a noncanonical p38 MAPK pathway. Journal of Immunology, 176, 580-588.
- Korhonen, R., Lahti, A., Kankaanrata, H. and Moilanen, E. (2005) Nitric oxide production and signaling in inflammation. Current Drug Targets: Inflammation and Allergy, 4, 471-479. doi:10.2174/1568010054526359
- Cho, I. and Kim, S.G. (2009) A novel mito-gen-activated protein kinase phosphatase-1 and gluco-corticoid receptor (GR) interacting protein-1-dependent combinatorial mechanism of gene transrepression by GR. Molecular Endocrinology, 23, 86-99. doi:10.1210/me.2008-0257
- Kang, Y.J., Wingerd, B.A., Arakawa, T. and Smith, W.L. (2006) Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. Journal of Immunology, 177, 8111-8122.
- Kojima, M., Hosoda, H., Date, Y., Nakazato, M. and Kangawa, K. (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature, 402, 656- 660. doi:10.1038/45230
- Sibilia, V., Pagani, F., Rindi, G., et al. (2008) Central ghrelin gastroprotection involves nitric oxide/prostaglandin cross-talk. British Journal of Pharmacology, 154, 688-697. doi:10.1038/bjp.2008.120
- Xu, X., Jhun, B.S., Ha, C.H. and Jin, Z.G. (2008) Molecular mechanisms of ghrelin-mediated endothelial nitricoxide synthase activation. Endocrinology, 149, 4183- 4192. doi:10.1210/en.2008-0255
- Chen, Y.T., Tsai, S.H., Sheu, S. Y. and Tsai, L.H. (2010) Ghrelin improves LPSinduced gastrointestinal mobility disturbances: Role of NO and prostaglandin E2. Shock, 33, 205-212. doi:10.1097/SHK.0b013e3181ae841b
- Kang, K.W., Choi, S.Y., Cho, M.K., Lee, C.C. and Kim, S.G. (2003) Thrombin induces nitric-oxide synthase via Ga12/13-coupled protein kinase C-dependent I-κBa phosphorylation and JNK-mediated I-κBa degradation. Journal of Biological Chemistry, 278, 17368-17378. doi:10.1074/jbc.M300471200
- Noha, S.M., Atanasov, A.G., Schuster, D., et al. (2011) Discovery of a novel IKK-β inhibitor by ligand-based virtual screening techniques. Bioorganic and Medicinal Chemistry Letters, 21, 577-583. doi:10.1016/j.bmcl.2010.10.051
- Jaffrey, S.R., Erdjument-Bromage, H., Ferris, D., Tempst, P. and Snyder, S.H. (2001) Protein S-nitrosylation: A physicological signal for neuronal nitric acid. Nature Cell Biology, 3, 193-197. doi:10.1038/35055104
- Forrester, M.T., Foster, M.W. and Stamler, J.S. (2007) Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. Journal of Biological Chemistry, 282, 13977-13983. doi:10.1074/jbc.M609684200
- Singh, K., Chaturvedi, R., Asim, M., Barry, D.P., Lewis, N.D., Vitek, M.P. and Wilson, K.T. (2008) The apolipoprotein e-mimetic peptide COG112 inhibits the inflamematory response to Citrobacter rodentium in colonic epithelial cells by preventing NF-κB activation. Journal of Biological Chemistry, 283, 16752-16761. doi:10.1074/jbc.M710530200
- Tanaka, H., Fujita, N. and Tsuruo, T. (2005) 3-Phosphoinositide-dependent protein kinase-1-mediated IκΒ Kinase β (IKKβ) phosphorylation activates NF-κΒ signaling. Journal of Biological Chemistry, 280, 40965- 40973. doi:10.1074/jbc.M506235200
- Kang, J.L., Lee, H.W., Kim, H.J., Lee, H.S., Castranova, V., Lim, C.M. and Koh, Y. (2005) Inhibition of src tyrosine kinase suppresses activation of nuclear factor-κB, and serine and tyrosine phosphorylation of IκΒ-α in lipopolysaccharide-stimulated Raw 264.7 macrophages. Journal of Toxicology and Environmental Health, Part A, 68, 1643-1662. doi:10.1080/15287390500192114
- Gupta, S.C., Prasad, S. Reuter, S., et al. (2010) Modification of cysteine 179 of IκBa kinase by nimbolide leads to down-regulation of NF-κB-regulated cell survival and proliferative proteins and sensitization of tumor cells to chemotherapeutic agents. Journal of Biological Chemistry, 285, 35406-35417. doi:10.1074/jbc.M110.161984
- Brandt, S., Kwok, T., Harting, R., Konig, W. and Backert, S. (2005) NF-κΒ activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proceedings of the National Academy of Sciences of the USA, 102, 9300-9305. doi:10.1073/pnas.0409873102
- Backert, S. and Neumann, M. (2010) What a disorder: Proinflammatory signaling pathways induced by Helicobacter pylori. Trends in Microbiology, 18, 479-486. doi:10.1016/j.tim.2010.08.003
- Rieke, C., Papendieck, A., Sokolova, O. and Naumann, M. (2011) Helicobacterpylori-induced tyrosine phosphorylation of IKkβ contributes to NF-κΒ activation. Biological Chemistry, 392, 387-393. doi:10.1515/bc.2011.029
- Haynes, M.P., Li, L., Sinha, D., et al. (2003) Src kinase mediates phosphatidylinositol 3-kinsae/Akt-dependent rapid endothelial nitric oxide synthase activation by ghrelin. Journal of Biological Chemistry, 278, 2118-2123. doi:10.1074/jbc.M210828200
- Lodeiro, P., Theodoropoulou, M., Pardo, M., Casanueva, F.F. and Camina, J.P. (2009) c-Src regulates Akt signaling in response to ghrelin via b-arrestin signaling-independent and-dependent mechanism. PLoS ONE, 4, e4686.
- Yu, S.M., Wu, J.F., Lin, T.L. and Kuo, S.C. (1997) Inhibition of nitric oxide synthase expression by PPM-18, a novel anti-inflammatory agent, in vitro and in vivo. Biochemical Journal, 328, 363-369.
- Mori, N., Yamada, Y., Ikeda, S., et al. (2002) Bay 11- 7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood, 100, 1828-1834. doi:10.1182/blood-2002-01-0151
- Slomiany, B.L. and Slomiany, A. (2011) Ghrelin suppression of Helicobacter pylori-induced gastric mucosal expression of iNOS is mediated through the inhibition of KKK-β activation by cNOS-dependent S-nitrosylation. Open Journal of Cell Biology, 1, 1-10. doi:10.4236/ojcb.2011.11001
- Reynaert, N.L., Ckless, K., Korn, S.H., et al. (2004) Nitric oxide represses inhibitory κΒ kinase through S-nitrosylation. Proceedings National Academy of Sciences of the USA, 101, 8945-8950. doi:10.1073/pnas.0400588101
- Marshall, H.E., Hess, D.T. and Stamler, J.S. (2004) S-nitrosylation: Physiological regulation of NF-κΒ. Proceedings of the National Academy of Sciences of the USA, 101, 8841-8842. doi:10.1073/pnas.0403034101
- Perkins, N.D. (2007) Integrating cell-signalling pathways with NF-κΒ and IKK function. Nature Review Molecular Cell Biology, 8, 49-62. doi:10.1038/nrm2083