Journal of Encapsulation and Adsorption Sciences
Vol.06 No.01(2016), Article ID:64986,12 pages
10.4236/jeas.2016.61004
Precision Synthesis of a Long-Chain Silane Coupling Agent Using Micro Flow Reactors and Its Application in Dentistry
Kiyomi Fuchigami*, Hidefumi Fujimura, Mitsuji Teramae, Toshiyuki Nakatsuka
Research and Development Department, Shofu Inc., Kyoto, Japan

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 26 January 2016; accepted 22 March 2016; published 25 March 2016
ABSTRACT
In dentistry, a wide range of materials is available for restorative treatment; a typical product of such restorative materials mainly consists of radically polymerizable monomer(s) and inorganic filler(s) (for added physical strength), as well as a surface modifier (e.g. silane coupling agent) for improved affinity between monomer and filler. It is favorable to use an optimal surface modifier depending on the respective restorative materials. However, commercially available surface modifiers, which are synthesized by the ton, are not always suited for what is required for properties of the many different dental restorative materials. As a potential solution to such a problem, we focused on the latest technology, “micro flow reactors” that enabled an on-demand low-volume synthesis of many types of surface modifiers. Using micro reaction fields of such flow reactors, we synthesized a novel long-chain silane coupling agent. Compared to the control system synthesized using a conventional reaction flask, the novel system enabled significant reduction in reaction time without inducing any major side reactions. A dental composite resin that was treated with the novel coupling agent exhibited higher toughness, suggesting that such a silane coupling agent was an effective surface modifier.
Keywords:
Micro Flow Reactor, Precision Synthesis, Composite Resins, Molecular Design, Urethanization, Hydrosilylation

1. Introduction
For industrial application, fillers used in composite resins are commonly surface-treated with silane coupling agents that contain radically polymerizable groups [1] . Such a surface treatment improves affinity of filler for radically polymerizable monomers to increase filler content, and thus enhancing physical properties including compressive strength and flexural strength of a resulting composite resin. It is for this reason that, for dental composite resins, 3-Methacryloxypropyltrimethoxysilane (γ-MPS) has been conventionally used as a silane coupling agent [2] - [7] . However, γ-MPS has some drawbacks: due to its short alkylene chains, it cannot sufficiently hydrophobize the filler surface, and also cannot give sufficient fracture toughness to a cured composite resin [8] ; and has a low affinity for radically polymerizable monomers, such as urethane-based or glycidyl methacrylate-based binder resins, because γ-MPS has only one linking group, namely an ester group, in a molecule.
Many researchers have been tackling these problems caused by the short alkylene chains. For example, Japan Patent No. H2-134307 describes a method for synthesizing a long-chain (alkylene) silane coupling agent containing an ester group as an only linking group using reaction flasks. Another example is Japan Patent No. H6-33290, which decribes a method for distilling a synthesized long-chain silane coupling agent. Since both of the silane coupling agents claimed in the said patents are synthesized by means of batch-type reaction flasks, the synthesized molecules are exposed to heat for a long period of time. Consequently, a large amount of polymerization inhibitor needs to be added to avoid unnecessary polymerization, in addition to a post-synthesis distillation procedure for purification to eliminate the residual polymerization inhibitor.
In order to solve all these problems, we design a molecule that contains a urethane group and sufficiently long alkylene chains to synthesize a novel long-chain (alkylene) silane coupling agent (L-SCA). The concept of its effect is illustrated in Figure 1 and Figure 2. With a urethane linkage in a molecule, L-SCA synthesized in this study forms hydrogen bonding between urethane bonds of urethane-based binder resin and hydroxyl groups of glycidyl methacrylate-based binder resin as shown in Figure 2. This contributes to an increase in fracture toughness of a resultant cured composite resin [8] .
Since the above-mentioned conventional batch-type reaction method requires continuous heating until all the reactive substrates disappear, the occurrence of side reactions is unavoidable. However, owing to the latest microprocessing technology, reaction fields with the size of only a few tens of microliters have enabled a continuous precision synthesis of a uniform compound with almost no occurrence of so-called “hot spots”. Therefore, for the synthesis of L-SCA in the present study, we used micro flow reactors, as shown in Figure 3, to perform urethanization and hydrosilylation in a continuous manner in each micro reaction field of 32 µL, which substantially reduced side reactions as well as environmental burdens.
2. Experimental
2.1. Reagents
2-isocyanatoethyl methacrylate (MOI; Tokyo Chemical Industry, Japan), 10-undecene-1-ol (Tokyo Chemical Industry, Japan), Dibutyl tin(IV) dilaurate (Wako Pure Chemical Industries, Japan), p-methoxyphenol (Tokyo Chemical Industry, Japan), Triethoxysilane (Tokyo Chemical Industry, Japan), and Platinum(0)-1,3-divinyl- 1,1,3,3-tetramethyl disiloxane complex solution (2 wt% in xylene; Sigma-Aldrich, USA) were used without

Figure 1. Molecular state of composite resin before and after curing (conventional silane coupling agent used).

Figure 2. Molecular state of composite resin before and after curing (novel silane coupling agent used).
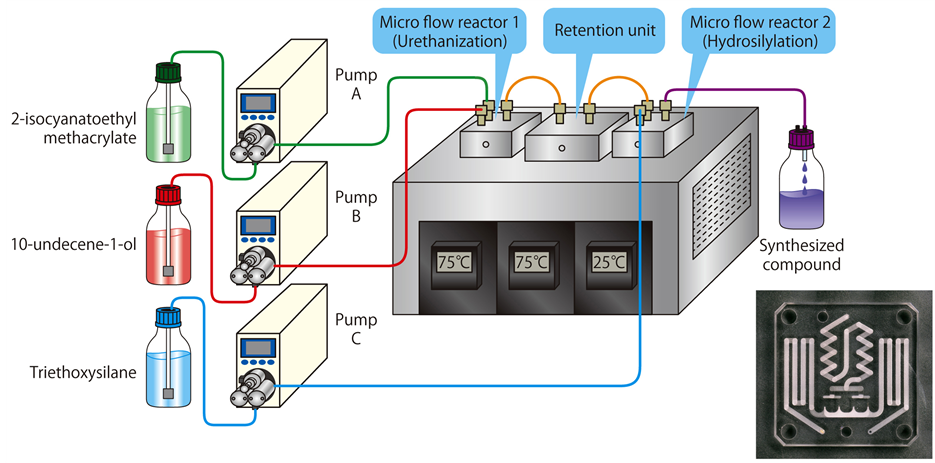
Figure 3. Schematic diagram of micro flow reactors.
further purification.
2.2. Analytical Instruments
Analytical instruments used in this study were: a Fourier transform infrared spectrometer (FT/IR-6300; JASCO, Japan) for infrared/near-infrared spectral measurement; a barrier discharge ionization detector/gas chromatograph (BID-GC-2010 Plus; Shimadzu, Japan) for gas chromatography measurement; an NMR spectrometer (JEOL ESC-400; JEOL, Japan) for 1H-NMR analysis; a rheometer (MCR301; Anton Paar, Austria) for dynamic viscoelasticity measurement; and a universal testing machine (Instron 5567; Instron, USA) for mechanical strength measurement.
Settings for BID-GC analysis: GC column: Agilent J&W GC Columns DB-1 (length: 30 M, ID: 0.25 mm, film thickness: 0.25 µm); Vaporizing chamber temperature: 270˚C; Linear velocity of He carrier gas: 30.0 cm/sec; Purge flow rate: 3.0 mL/min; Split ratio: 1:50.0; Temperature programming: 40˚C-5 min hold, 40˚C - 270˚C, heating rate: 10˚C/min, 270˚C-60 min hold; BID temperature: 270˚C.
Settings for FT-IR analysis: Transmission method using KBr plates; Resolution: 4 cm−1; Measuring range: 400 - 4500 cm−1.
Settings for 1H-NMR: 400 MHz; Chloroform-d (containing 0.05% TMS; Sigma-Aldrich, USA); Number of scans: 8.
2.3. Synthesis of L-SCA and Its Precursor
Figure 4 shows the reagents used and chemical schemes of the reactions for synthesis of L-SCA and its precursor. Synthesis reactions to determine the optimal reaction temperature and reaction time were performed as follows: reagent 1 (containing a reaction catalyst) and reagent 2 were placed in a separate gas tight glass syringe and delivered into micro flow reactor 1 (for urethanization) with a syringe pump at the flow rate of 1.75 - 5.25 mmol/h at the reaction temperature of 70˚C - 80˚C with the heating time (retention time in the retention unit) adjusted by the flow rate, and then a precursor obtained was introduced to micro flow reactor 2, while reagent 3 (containing a reaction catalyst) placed in another gas tight glass syringe was also delivered into micro flow reactor 2 (for hydrosilylation) at the flow rate of 1.75 - 5.25 mmol/h at the reaction temperature of 25˚C. The precursor and a target compound obtained were analyzed and identified by gas chromatography (BID-GC), FT-IR and 1H-NMR.
2.4. Preparation of Dental Flowable Composite Resins (Low Viscosity Composite Resins) and Evaluation of Their Physical Properties
1) Silane treatment of 0.4 µm filler
Table 1 shows chemical structural formulae of the experimental and two commercially-available control silane coupling agents (KBM-5803 and KBM-503; Shin-Etsu Chemical, Japan). Using these silane coupling agents, silane treatment liquid (see Table 2) was prepared, and uniformly added to 100 g of 0.4 µm filler (GM32087 UF-0.4; Schott, Germany) sieved through an 80-mesh sieve, so that the content of silane coupling agent was 5 wt% in the mixture. The filler with uniformly-dispersed silane treatment liquid was subjected to a coupling reaction for 3 h at 115˚C, dried and then sieved again through the 80-mesh sieve. Each of the resulting

Figure 4. Chemical schemes of reactions (1. Urethanization; 2. hydrosilylation).

Table 1. Chemical structural formulae of the control and experimental silane coupling agents.
Table 2. Composition of silane treatment liquid.
fillers was used as silane-treated filler.
2) Preparation of dental flowable composite resins (low viscosity composite resins)
Using the respective silane-treated fillers, UDMA-based and Bis-phenol A-GMA (Bis-GMA)-based flowable composite resins (filler content: 50 wt%) were prepared (Table 3 and Table 4), and their physical properties (viscosity, flow value, flexural strength and compressive strength) were evaluated.
3) Test methods for evaluating physical properties of flowable composite resins
2.4.1. Viscosity
The viscosity of each flowable composite resin prepared was measured using a rheometer. The measurement conditions are shown in Table 5.
2.4.2. Flow Value (Flowability)
Each uncured paste sample of the prepared flowable composite resins (0.2 g each) was placed on a glass plate, covered by another glass plate, and then 400 g of load was applied on the specimen. The load was removed after 1min, and vertical and horizontal distances on the spread of the specimen were measured. The above procedure was performed twice in total, and the obtained values were averaged to represent the flowability of each composite resin.
2.4.3. Three-Point Flexural Strength
Each test composite resin paste was filled in a mold (2 × 2 × 25 mm) used for preparing flexural test specimens, with a cover glass placed with pressure on the surface of the paste, and light-cured from top and bottom sides through each cover glass for 3 min/side using a dental curing unit (Solidilite V; Shofu, Japan). The cured test specimens were stored in 37˚C ion-exchanged water for 24 h, and subjected to three-point flexural testing (at a crosshead speed of 1mm/min using Instron 5567).
2.4.4. Compressive Strength
Each test composite resin paste was filled in a mold (4 mm diameter, 4 mm height) used for preparing compressive test specimens, with a cover glass placed with pressure on the surface of the paste, and light-cured from the top and bottom sides through each cover glass for 3 min/side using a dental curing unit (Solidilite V; Shofu,
Table 3. Composition of UDMA-based flowable composite resin.
Table 4. Composition of Bis-GMA-based flowable composite resin.
Table 5. Conditions for viscosity measurement using a rheometer.
Japan). The cured test specimens were stored in 37˚C ion-exchanged water for 24 h, and subjected to compressive strength testing (at a crosshead speed of 1 mm/min using Instron 5567).
3. Results and Discussion
Before the results of analysis using BID-GC are shown in Figure 5. The urethanization reaction using the micro flow reactor 1 was completed in approx. 1.5 min at 75˚C, whereas by the conventional batch method with a reaction flask and a dropping funnel the reaction was not completed, with the raw materials left unreacted in the flask even at 8 h after completion of adding 2-isocyanatoethyl methacrylate. This suggested that molecules synthesized by the conventional batch method were exposed to heat for such an unnecessarily long period of time that urethane bonds formed were decomposed, which leads to high risks of side reactions and consequent yellowing of a precursor. With the experimental device, the subsequent reaction of hydrosilylation successfully occurred without any side reactions in the precisely temperature-controlled micro reaction field (32 µL) of the micro flow reactor 2 in a manner that silane was correctly added to a terminal alkene. These facts were also evident from the FT-IR (Figure 6) and 1H-NMR (Figure 7(a) and Figure 7(b)) results.
Figures 8(a)-(b) show diffuse reflectance FT-IR analysis results of the respective glass fillers that were surface-treated with L-SCA (synthesized using the micro flow reactors) and the two control silane coupling agents KBM-5803 and KBM-503. It was confirmed that the absorption (at 3740 cm−1) of the isolated silanol groups on the filler surface disappeared by their condensation with the silane coupling agents. All the silane-treated glass fillers exhibited the absorption of the functional groups (alkylene: 2800 - 2900 cm−1, carbonyl: 1650 - 1700 cm−1) derived from the respective silane coupling agents used; particularly the intensity of the alkylene absorption became more notable with the increase of the number of alkylene. These results confirmed that the glass fillers used in this study were adequately treated with the respective silane coupling agents.
Figure 9(a) and Figure 9(b) show the results of viscosity testing (using a rheometer) on the experimental flowable composite resins prepared using the fillers treated with the respective silane coupling agents. For both the UDMA/TEGDMA-based and the Bis-GMA/TEGDMA-based composite resins, the viscosity values of the respective groups (formed according to which silane coupling agent was used) were in the following order: L- SCA < KBM-5803 < KBM-503; the L-SCA group exhibited significantly lower viscosity than the control groups. Table 6 shows their flow values, which were largely dependent on their viscosity values (that affect actual

Figure 5. Gas chromatograms.

Figure 6. Raw materials and synthesized compound characterized by FT-IR.
 (a)
(a)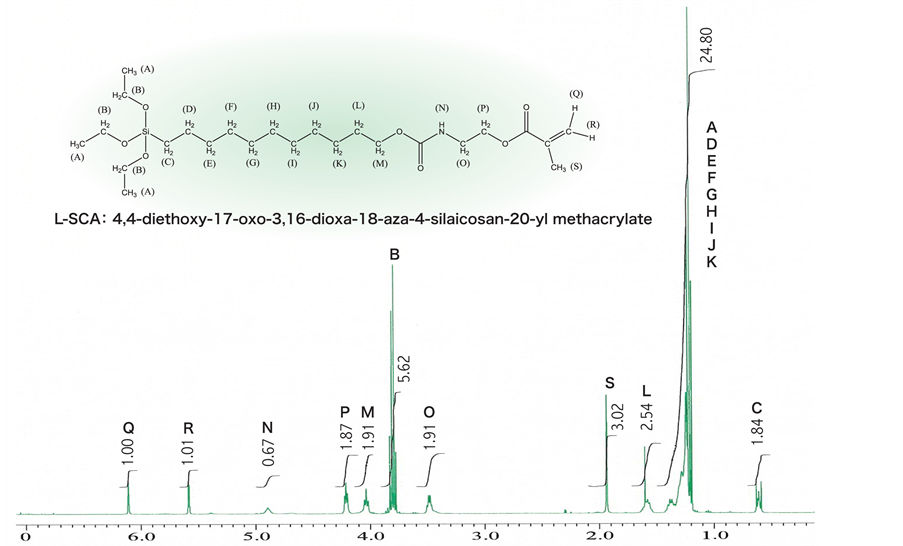 (b)
(b)
Figure 7. (a) Precursor characterized by 1H-NMR; (b) L-SCA characterized by 1H-NMR.
handling properties of the pastes). Again, for both the UDMA/TEGDMA-based and the Bis-GMA/TEGDMA- based composite resins, the flow values were in the following order: L-SCA > KBM-5803 > KBM-503; the L-SCA group had the highest flowability. For these rheological properties, the L-SCA group of flowable composite resin was thus clinically most favorable of all groups accessed in this study.
 (a)
(a) (b)
(b) (c)
(c)
Figure 8. (a) FT-IR analysis results (for filler treated with L-SCA); (b) FT-IR analysis results (for filler treated with KBM-503); (c) FT-IR analysis results (for filler treated with KBM-5803).
Tables 7(a)-(b) show the results of flexural strength testing on the experimental flowable composite resins. Irrespective of whether they are UDMA/TEGDMA-based or Bis-GMA/TEGDMA-based, the KBM-503 group showed the highest flexural modulus and the lowest fracture energy, indicating its hard but brittle characteristics. In contrast, the groups treated with the long-chain silane coupling agents (L-SCA and KBM-5803) had slightly lower flexural modulus values, but higher flexural strength and higher fracture energy values than the KBM-503 group, suggesting that the long-chain (alkylene) silane coupling agents provided fracture toughness to the resultant

 (a) (b)
(a) (b)
Figure 9. (a) Viscosity of UDMA/TEGDMA-based CR, compared according to silane coupling agent used; (b) Viscosity of Bis-GMA/TEGDMA-based CR, compared according to silane coupling agent used.
Table 6. Results of flow value measurement.
(a) (b) (c)
Table 7. (a) Three-point flexural strength; (b) Three-point flexural modulus; (c) Three-point flexural fracture energy.
*Significant difference between values marked with different letters (p < 0.05).
*Significant difference between values marked with different letters (p < 0.05).
*Significant difference between values marked with different letters (p < 0.05).
flowable composite resins. In other words, L-SCA and KBM-5803, which have the larger number of alkylene (C11 and C8, respectively) than KBM-503 (C3), have more distance between the filler and the rigid binder resin network; the carbon chains, which create the distance, bend flexibly to absorb stress, resulting in high fracture toughness. Meanwhile, although there was no marked difference in the number of alkylene between L-SCA (C11) and KBM-5803 (C8), L-SCA showed significantly higher physical properties when the highly polar binder resin, i.e. Bis-GMA, was present. This was clearly due to an interaction between the polar group (hydroxyl) of Bis-GMA and the urethane group within a molecule, which achieved our intended effects.
Finally, the results of compressive strength testing on the experimental flowable composite resins are shown in Table 8(a) and Table 8(b). There was no significant difference in compressive strength among all the groups of UDMA/TEGDMA-based flowable composite resins, whereas for the Bis-GMA/TEGDMA-based flowable composite resins, the L-SCA group had a significantly higher compressive strength than the control groups. This was likely to be due to the effect of the long alxylene chains of L-SCA, in combination with the effect that L-SCA, which was bound to the inorganic filler, formed a hydrogen bond with the binder resin (radically polymerizable monomer) as shown in Figure 2; in particular, the hydroxyl group of Bis-GMA is highly polar, as mentioned above, to form a strong hydrogen bond with the urethane linkage portion.
4. Conclusion
In this study, we conducted a continuous precision synthesis of a long-chain (alxylene) silane coupling agent using micro flow reactors. The conventional batch reaction method using a reaction flask required the reaction time of more than 8 h at 75˚C, while our experimental synthesis method took 1.5 min to complete a urethanization reaction at 75˚C. Our method has enabled the production of the pure compound, causing no side reactions, as is evident from the analyses using BID-GC (Figure 5), FT-IR (Figure 6) and 1H NMR (Figure 7(a) and Figure 7(b)). With a urethane linkage and long-chain alkylene groups in a molecule, the novel silane coupling agent synthesized in this study exhibited a high affinity for urethane-based binder resin as well as glycidyl methacrylate-based binder resin. Furthermore, our method using the micro flow reactors facilitated the synthesis of the compound with less environmental burden, because after the reaction procedure, cleaning of the flow reactor channels could be done with only a small amount of organic solvent such as acetone. It was confirmed that the
(a) (b)
Table 8. (a) Compressive strength; (b) Compressive elasticity modulus.
*Significant difference between values marked with different letters (p < 0.05).
*Significant difference between values marked with different letters (p < 0.05).
*Corresponding author.
Cite this paper
KiyomiFuchigami,HidefumiFujimura,MitsujiTeramae,ToshiyukiNakatsuka, (2016) Precision Synthesis of a Long-Chain Silane Coupling Agent Using Micro Flow Reactors and Its Application in Dentistry. Journal of Encapsulation and Adsorption Sciences,06,35-46. doi: 10.4236/jeas.2016.61004
References
- 1. Masuhara, E. and Fujisawa, S. (1979) Shika Juutenyou Rejin (Dental Composite Resins). Ishiyaku Publishing, Inc., Tokyo.
- 2. Nishiyama, N. and Hayakawa, T. (1987) Silane Coupling Agents. Journal of Adhesive Dentistry, 5, 129-133.
- 3. Arai, K., Yagi, K., Sakamoto, I., Asada, M. and Hashimoto, H. (1973) Characteristics of the Precipitations from Commercial Composite Resins during Storage in Water. JDMA, 30, 22-28.
- 4. Soderholm, K.J. (1981) Degradation of Glass Filler in Experimental Composites. Journal of Dental Research, 60, 1867-1875.
http://dx.doi.org/10.1177/00220345810600110701 - 5. Nishiyama, N., Katsuki, H., Hayakawa, T., Horie, K., Tanioka, Y., Teshima, H. and Akaiwa, Y. (1985) Study on Surface Treatment of Silica Filler, Part 4. Japanese Journal of Dental Materials, 4, 60-61.
- 6. Nagae, T. (1979) Shiran Kappuringu Zai (Silane Coupling Agents). Kogyo Zairyo, 27, 34-38.
- 7. Sakanashi, K. and Nishiyama, N. (1984) Studies on the Treatment of Glass Surface. Japanese Journal of Dental Materials, 3, 284-294.
- 8. Tanaka, J., Takahashi, H., Takeda, S., Nakamura, M., Fukase, Y., Nishiyama, M. and Suzuki, K. (2005) Development of High Strength, High Modulus, High Toughness Dental Matrix Resin without Environmental Endocrine Disruptor. Japanese Association for Dental Science, 24, 51-60.
NOTES

*Corresponding author.