International Journal of Organic Chemistry
Vol.04 No.05(2014), Article ID:52662,4 pages
10.4236/ijoc.2014.45033
The First Synthesis of Sessiline
Viktor Ilkei1*, Kornél Faragó1, Zsuzsanna Sánta2, Miklós Dékány2, László Hazai1, Csaba Szántay Jr.2, Csaba Szántay1, György Kalaus1
1Department of Organic Chemistry and Technology, Budapest University of Technology and Economics, Budapest, Hungary
2Gedeon Richter Plc, Budapest, Hungary
Email: *viktor.ilkei@gmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 9 October 2014; revised 23 November 2014; accepted 9 December 2014
ABSTRACT
Sessiline is an alkaloid which was recently isolated from the fruits of Acanthopanax sessiliflorus. The molecule contains two five-membered heterocyclic units joined together by an acylaminocarbinol-ether type bond. Here, we describe the first, simple synthesis of sessiline from 5-hydrox- ypyrrolidin-2-one and 5-hydroxymethylfurfural, which are prepared from succinimide and furfuryl alcohol, respectively. The coupling reaction takes place on moderate heating under neat conditions.
Keywords:
Sessiline, Acylaminocarbinol, Iminium Ion, Alkaloid Synthesis

1. Introduction
Sessiline (1) (Figure 1) was isolated in 2002 from the fruits of Acanthopanax sessiliflorus, a herbaceous plant, which is distributed in East Asia [1] . Its structure was elucidated by spectroscopic methods. The molecule consists of two heterocyclic units joined together by an ether-bond. The alkaloid is found in the plant as a racemate.

Figure 1. The structure of sessiline (1).
It is known that acylaminocarbinols, as well as the iminium ions that arise from them, are electrophilic reagents, which can be utilized in the synthesis of sessiline (1). Taking the above assumption into consideration, we outlined a simple retrosynthetic scheme for 1 (Scheme 1). By disconnecting the ether bond, we obtain two known compounds: 5-hydroxypyrrolidin-2-one (2) and 5-hydroxymethylfurfural (3).
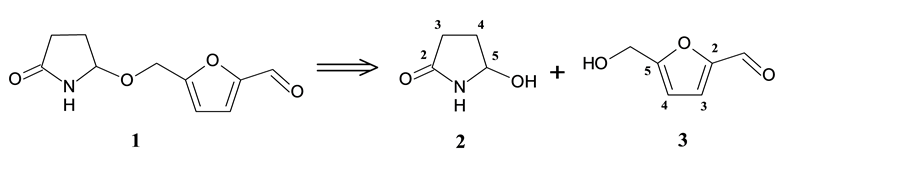
Scheme 1. Retrosynthetic analysis of sessiline (1).
2. Results and Discussion
In order to accomplish our goal, we synthesized 5-hydroxypyrrolidin-2-one (2) and 5-hydroxymethylfurfural (3) (Scheme 2). Compound 2 can be prepared from succinimide (4) in two steps. The partial reduction of 4 yields 5-ethoxypyrrolidin-2-one (5) [2] , which can be hydrolysed in boiling water to give the amidocarbinol 2 [3] .
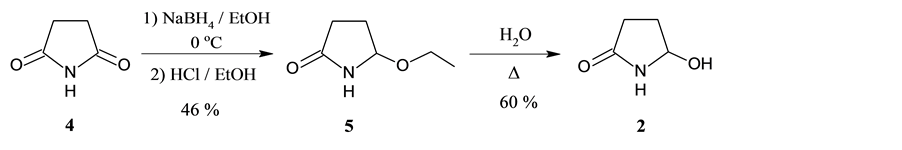
Scheme 2. Synthesis of 5-hydroxypyrrolidin-2-one (2).
Next, we prepared 5-hydroxymethylfurfural (3) from furfuryl alcohol (6) (Scheme 3). First, 6 was protected by acetylation to give furfuryl acetate (7) [4] , which was subjected to Vilsmeier formylation to yield 5-(for- myl)furfuryl acetate (8) [5] . Then, 3 was obtained by deacetylation [6] .
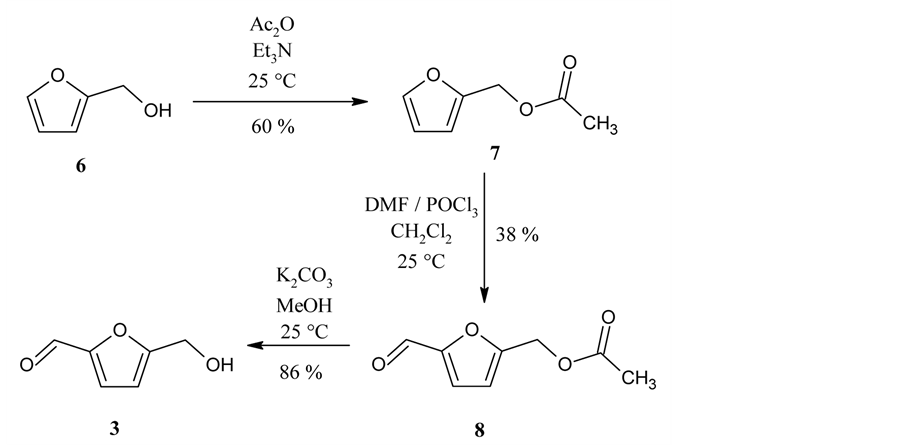
Scheme 3. Synthesis of 5-hydroxymethylfurfural (3).
Next, we needed to synthesize target molecule 1 by coupling 2 and 3. Although the type of the planned reaction is known, relatively few examples can be found in the literature. The majority of these transformations were carried out under mild conditions using acid catalysis [7] [8] .
In the present case, 2 was allowed to react with an excess of 3 at 60˚C under neat conditions. The reaction gave sessiline (1) in an acceptable yield (Scheme 4).
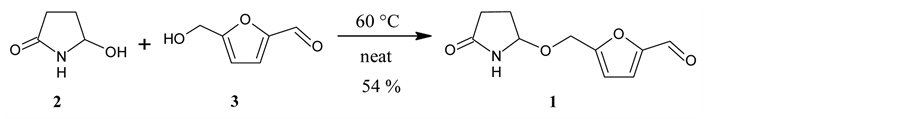
Scheme 4. The synthesis of sessiline (1).
Surprisingly, no acid catalysis or solvent was necessary for the reaction to take place, raising of the temperature proved to be sufficient. In light of our successful synthesis, we proposed a plausible reaction mechanism for the formation of sessiline (1) (Scheme 5). According to our assumption, higher temperatures cause the equilibrium between the amidocarbinol 2 and its ionic form 2/a to shift towards the latter, which results in a higher concentration of acyliminium ions in the reaction mixture, thus enabling the reaction to take place.

Scheme 5. Proposed reaction mechanism for the formation of sessiline (1).
3. Experimental
3.1. General
Melting points were measured on a SANYO Gallenkamp apparatus and are uncorrected. IR spectra were recorded on a Bruker FT-IR instrument. 1H-NMR and 13C-NMR measurements were performed on Varian 400 MHz, Varian 500 MHz and Varian 800 MHz spectrometers. Chemical shifts are given on the delta scale as parts per million (ppm) with tetramethylsilane (TMS) (1H) or dimethylsulfoxide-d6 (13C) as the internal standard (0.00 ppm and 39.5 ppm, respectively). MS spectra were recorded on VG-Trio-2 and Finnigan MAT 95SQ instruments using EI or ESI techniques. HRMS analyses were performed on an LTQ FT Ultra (Thermo Fischer Scientific, Bremen, Germany) system. TLC was carried out using Kieselgel 60 F254 (Merck) coated glass plates. Column chromatography was performed using Geduran Si 60 (Merck) silica.
3.2. Furfuryl Acetate (7)
To a mixture of 56.6 g (577 mmol; 50 ml) furfuryl alcohol (6) and 12.1 g (120 mmol; 16.7 ml) triethylamine was added 67 g (656 mmol; 62.4 ml) of acetic anhydride dropwise over 15 minutes. The reaction mixture was stirred at ambient temperature for 21 hours, then it was extracted with 3 × 30 ml water. The organic phase was dried over magnesium sulphate and evaporated under reduced pressure. The crude product was distilled in vacuo (bp. 76˚C/15 mmHg; lit.: 67˚C/8 mmHg [4] ). 48 g (60%) pure 7 was obtained as a clear liquid. nd: 1.4619 (lit.: 1.4603 [4] ). IR νmax (film, cm−1): 1738, 1502, 1437, 1231, 1150, 1079, 1015, 918, 885, 817, 744.
3.3. 5-(Formyl)Furfuryl Acetate (8)
A mixture of 97 g (1.33 mol; 103 ml) dimethylformamide and 300 ml dichloromethane was cooled to 0˚C. To this mixture 152 g (0.99 mol; 91 ml) phosphoryl chloride was added dropwise at 0˚C over 5 minutes. The re- action mixture was stirred at 0˚C for 1 hour, then 48 g (0.343 mol; 43 ml) furfuryl acetate (7) was added dropwise over 5 minutes. The reaction mixture was stirred at ambient temperature for 22 hours, after which it was neutralised with 15% sodium carbonate solution. The precipitate was filtered and washed with 100 ml dichlo- romethane. The aqueous-organic mixture was extracted with 10 × 100 ml dichloromethane, the organic phase was dried over magnesium sulphate and evaporated under reduced pressure. The crude product was crystallized from ether, after which 22.7 g (39%) of pure 8 was obtained as colourless crystals. Rf 0.81 (ethyl acetate:hexane = 2:1). Mp.: 55˚C - 56˚C (lit.: 55˚C - 56˚C [5] ). IR νmax (KBr, cm−1): 3122, 2943, 2833, 1740, 1675, 1588, 1523, 1437, 1403, 1366, 1273, 1221, 1022. 1H NMR (800 MHz, DMSO-d6) δH 2.08 (s, 3H, Ac); 5.15 (s, 2H, CH2O); 6.81 (d, 1H, J = 3.5 Hz, H-4); 7.53 (d, 1H, J = 3.5 Hz, H-3); 9.60 (s, 1H, CH=O). MS(EI): 168 (C8H8O4). EI-MS (rel. int.%): 168(1); 126(100); 109(22); 97(7); 79(30); 53(9); 44(22); 43(28).
3.4. 5-Hydroxymethylfurfural (3)
To 2.60 g (15.5 mmol) 5-(formyl)furfuryl acetate (8) dissolved in 20 ml methanol, 0.330 g (2.39 mmol) potassium carbonate was added. The reaction mixture was strirred at ambient temperature for 1 hour, after which 15 ml water was added and the mixture was extracted with 10 × 15 ml dichloromethane. The organic phase was dried over magnesium sulphate and evaporated under reduced pressure. The crude product was subjected to column chromatography using a mixture of ethyl acetate:hexane = 2:1 as the eluent. 1.671 g (86%) of pure 3 was obtained as a yellow liquid, which crystallized on cooling. Rf 0.55 (ethyl acetate:hexane = 2:1). Mp.: 28˚C - 32˚C (lit.: 31˚C - 32˚C [6] ). IR νmax (KBr, cm−1): 3405, 3123, 2926, 2851, 1675, 1523, 1397, 1370, 1334, 1280, 1192, 1023. 1H NMR (800 MHz, DMSO-d6) δH 4.51 (d, 2H, J = 6.0 Hz, CHO); 5.59 (t, 1H, J = 6.0 Hz, OH); 6.61 (d, 1H, J = 3.5 Hz, H-4); 7.50 (d, 1H, J = 3.5 Hz, H-3); 9.55 (s, 1H, CH=O). MS(EI): 126 (C6H6O3). EI-MS (rel. int.%): 126(96); 109(10); 97(100); 81(4); 69(24); 53(9); 41(50); 39(22).
3.5. 5-Ethoxypyrrolidin-2-One (5)
A solution of 7.156 g (72.22 mmol) succinimide (4) in 300 ml ethanol was cooled to 0˚C. To this solution 4.00 g (105.74 mmol) sodium borohydride was added in one portion. The solution was stirred at 0˚C for 4 hours, during which time every 15 minutes 5 drops of 2 M ethanolic hydrogen chloride solution were added. Then the reaction mixture was acidified to pH = 3 with 2 M ethanolic hydrogen chloride solution over 30 minutes, after which it was stirred at 5˚C for 45 minutes. Then the reaction mixture was neutralised (pH = 7) with 5% ethanolic potassium hydroxide solution and evaporated to dryness under reduced pressure. The remaining syrupy solid was suspended in 80 ml chloroform, filtered, and the precipitate washed with 3 × 20 ml chloroform. The filtrate was evaporated under reduced pressure, and the remaining colourless oil was dissolved in 80 ml dichloro- methane and washed with 3 × 10 ml water. The aqueous phase was extracted with 6 × 20 ml dichloromethane, then the organic phases were unified, dried over magnesium sulphate and evaporated under reduced pressure. The remaining colourless oil crystallized on standing, after which 4.327 g (46%) pure 5 was obtained as colour- less crystals. Rf 0.67 (acetone). Mp.: 51˚C - 53˚C (lit.: 48˚C - 53˚C [2] ). IR νmax (KBr, cm−1): 3200, 2978, 1707, 1689, 1668, 1457, 1282, 1250, 1067, 986. 1H NMR (400 MHz, DMSO-d6) δH 1.10 (t, 3H, J = 7.0 Hz, CH3); 1.78 - 1.89 (m, 1H, Hx-4); 1.95 - 2.05 (m, 1H, Hx-3); 2.11 - 2.30 (m, 2H, Hy-4, Hy-3); 3.27-3.35 (m, 1H, OCH2x); 3.44 - 3.54 (m, 1H, OCH2y); 4.83 - 4.89 (m, 1H, H-5); 8.62 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC 15.1 (CH3); 27.7 (C-4); 28.1 (C-3); 61.6 (OCH2); 85.0 (C-5); 177.4 (CON). MS(ESI): 130 (C6H12NO2). ESI-MS-MS (cid = 35) (rel. int.%): 84(100).
3.6. 5-Hydroxypyrrolidin-2-One (2)
2.0 g (15.5 mmol) 5-ethoxypyrrolidin-2-one (5) was dissolved in 25 ml water and the solution was refluxed for 3 hours. Then the water was evaporated under reduced pressure. The remaining oil was triturated with ethyl acetate, from which a white solid crystallized on cooling. The solid was filtered and recrystallized from acetone. 0.89 g (60%) pure 2 was obtained as colourless crystals. Rf 0.19 (acetone:hexane = 2:1). Mp.: 94˚C - 96˚C (lit.: 90˚C [3] ). IR νmax (KBr, cm−1): 3254, 2996, 2962, 1668, 1475, 1415, 1323, 1271, 1166, 1101, 1070, 1016. 1H NMR (400 MHz, DMSO-d6) δH 1.66 - 1.75 (m, 1H, Hx-4); 1.94 - 2.03 (m, 1H, Hx-3); 2.14 - 2.24 (m, 1H, Hy-4); 2.23 - 2.33 (m, 1H, Hy-3); 5.06 (m, 1H, H-5); 5.70 (d, 1H, J = 6.9 Hz, OH); 8.18 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δC 28.4 (C-3); 30.3 (C-4); 78.5 (C-5); 176.7 (CON). HRMS: 102.05491 (C4H8NO2; calc. 102.05496). ESI-MS-MS (cid = 55) (rel. int.%): 85(100).
3.7. Sessiline (1)
200 mg (1.98 mmol) 5-hydroxypyrrolidin-2-one (2) and 800 mg (6.34 mmol) 5‑hydroxymethylfurfural (3) were stirred at 60˚C under neat conditions for 1 hour. Then the reaction mixture was diluted with 1 - 2 ml dichloro- methane, filtered, the solid washed with 3 × 2 ml dichloromethane, and air-dried with suction. 225 mg (54%) pure 1 was obtained as colourless crystals. Rf 0.47 (acetone:hexane = 2:1). Mp.: 169˚C - 172˚C (lit.: 171˚C - 172˚C [1] ). IR νmax (KBr, cm−1): 3321, 3176, 3117, 2965, 2896, 2863, 2790, 2763, 1772, 1709, 1666, 1531, 1463, 1412, 1389, 1338, 1284, 1276, 1262, 1250, 1208, 1201, 1177, 1096, 1062, 1030, 1007. 1H NMR (400 MHz, DMSO-d6) δH 1.86 - 1.95 (m, 1H, Hx-4’); 2.01 - 2.10 (m, 1H, Hx-3’); 2.16 - 2.34 (m, 2H, Hy-4’, Hy-3’); 4.49 + 4.58 (AB, 2 × 1H, Jgem = 13.2 Hz, CHO); 5.02 (m, 1H, H-5’); 6.73 (d, 1H, J = 3.4 Hz, H-4); 7.51 (d, 1H, J = 3.4 Hz, H-3); 8.80 (s, 1H, NH); 9.58 (s, 1H, CH=O). 13C NMR (100 MHz, DMSO-d6) δC 27.5 (C-4’); 27.9 (C-3’); 60.3 (CH2O); 85.2 (C-5’); 111.8 (C-4); 123.9 (C-3); 152.2 (C-2); 157.7 (C-5); 177.6 (CON); 178.3 (CH=O). HRMS: 210.07605 (C10H12NO4; calc. 210.07608). ESI-MS-MS (cid=65) (rel. int.%): 192(100); 164(8); 126(2); 109(4).
Acknowledgements
The authors are grateful to Gedeon Richter Plc for the financial support.
References
- Lee, S., Ji, J., Shin, K.H. and Kim, B.-K. (2002) Sessiline, A New Nitrogenous Compound from the Fruits of Acantho- panax sessiliflorus. Planta Medica, 68, 939-941. http://dx.doi.org/10.1055/s-2002-34925
- Hubert, J.C., Wijnberg, J.B.P.A. and Speckamp, W.N. (1975) NaBH4 Reduction of Cyclic Imides. Tetrahedron, 31, 1437-1441. http://dx.doi.org/10.1016/0040-4020(75)87076-1
- Cue Jr., B.W. and Chamberlain, N. (1979) An Improved Method for the Preparation of 5-Hydroxy-2-Pyrrolidone. Organic Preparations and Procedures International, 11, 285‑286. http://dx.doi.org/10.1080/00304947909355413
- Renvall, I. and Mattila, T. (1977) Esterification of Furfuryl Alcohol and Its Derivatives. US Patent No. 4008256.
- Mehner, A., Montero, A.L., Martinez, R. and Spange, S. (2007) Synthesis of 5-Acetoxymethyl- and 5-Hydroxymethyl- 2-Vinylfuran. Molecules, 12, 634-640. http://dx.doi.org/10.3390/12030634
- Schinzer, D., Bourguet, E., Ducki, S. (2004) Synthesis of Furano-Epothilone D. Chemistry―A European Journal, 10, 3217-3224. http://dx.doi.org/10.1002/chem.200400125
- Toja, E., Gorini, C., Zirotti, C., Barzaghi, F. and Galliani, G. (1991) Amnesia-Reversal Activity of a Series of 5-Alkoxy- 1-Arylsulfonyl-2-Pyrrolidinones. European Journal of Medicinal Chemistry, 26, 403-413. http://dx.doi.org/10.1016/0223-5234(91)90101-R
- Toja, E., Gorini, C., Zirotti, C., Barzaghi, F. and Galliani, G. (1991) Amnesia-Reversal Activity of a Series of 5-Alkoxy- 1-Arylcarbonyl-2-Pyrrolidinones and 5-Alkoxy-1-Arylmethyl-2-Pyrrolidinones. European Journal of Medicinal Che- mistry, 26, 415-422. http://dx.doi.org/10.1016/0223-5234(91)90102-S
NOTES

*Corresponding author.