International Journal of Organic Chemistry
Vol. 2 No. 4 (2012) , Article ID: 25881 , 11 pages DOI:10.4236/ijoc.2012.24047
β-Oxoanilides in Heterocyclic Synthesis: Synthesis and Antimicrobial Activity of Pyridines, Pyrans, Pyrimidines and Azolo, Azinopyrimidines Incorporating Antipyrine Moiety
Department of Chemistry, Faculty of Science, Al-Azhar University, Assiut, Egypt
Email: *a.khames@yahoo.com
Received July 25, 2012; revised August 26, 2012; accepted September 17, 2012
Keywords: β-Oxoanilides; Pyridines; Pyrans; Pyrimidines; Azolo; Azinopyrimidines
ABSTRACT
Condensation of β-Oxoanilide 1 with active methylene derivatives 2a,b afforded the pyridine derivative 5, and with crotononitrile afforded the pyridine 8. Compounds 9 and 11a-c were obtained by reaction of 1 with malononitrile dimer and arylidinemalononitrile 10a-10c. In contrast, when compound 1 reacted with ethoxymethylen malononitrile afforded the pyridine derivative 13. On the other hand, treatment of 1 with anthranilic acid gave the quinoline derivative 14. Also, reactions of 1 with isothiocyanate derivatives afforded compounds 16-18. The reaction of 1 with chalcone derivative afforded the pyridine derivative 22. Treatment of compound 1 with thiourea produced pyrimidine derivative 23. Furthermore, compound 1 converted into pyrimidinethione 24a and pyrimidinone 24b on treatment with a mixture of aromatic aldehydes and thiourea or urea respectively. Reaction of 24a with hydrazonyl halide, thiosemicarbazide and arylidinecyanothioacetamide afforded compounds 26, 28 and 29. Compound 29 was treated with chloroacetonitrile to afford compound 30. Six compounds from the newly synthesized were screened for antibacterial and antifungal activity against bacteria Staphylococcus aureus, Bacillus cereus and Klebsiella pneumonia and fungi Aspergillus flavus and Aspergillus ochraceous, respectively. Some of the tested compounds showed significant antimicrobial activity. IR, 1H NMR, mass spectral data, and elemental analysis elucidated the structures of all the newly synthesized compounds.
1. Introduction
β-Oxoanilides are valuable intermediates in synthetic organic chemistry [1-6]. In the last few years we and others reported a variety of synthesis of heteroaromatics that have been developed utilizing β-oxoanilides as readily obtainable compounds [7-9]. Due to the recent reported biological activities of the heterocyclic moieties mentioned here such as pyridine derivatives which posses antimicrobial [10], and fungicidal [11] activities. As well as, pyrimidine derivatives were reported to be showed antimicrobial [12], activity. On the other hand, pyrazolopyrimidines are widely used as antimicrobial [13], activity. In addition, antipyrine has attracted a great deal of interest due to its wide applications in the field of pharmaceuticals, so many of the heterocyclic compounds incorporating antipyrine moiety exhibited antimicrobial [14], activity. Incorporating antipyrine moiety with the mentioned newly synthesized heterocyclic derivative may be enhancement the biological activities of the synthesized compounds. In continuation to this work we report here the reactions of β-oxoanilide with some electrophilic and nucleophilic reagents to produce some new substituted azines moiety.
2. Results and Discussion
It has been found that condensation of β-oxoanilide derivative 1 [15] with malononitrile 2a in ethanol containing a catalytic amount of piperidine furnished the novel pyridone derivative 5 in good yield. The structure of 5 was confirmed based on spectral data (IR, 1H NMR and MS). So, the mass spectrum of compound 5 showed the molecular ion peak at m/z = 336 (22%, M + 1) and the base peak was found at m/z = 93 (100%) corresponding to C5H3NO ion. Formation of compound 5 was interpreted via intermediacy of condensation product 3 which cyclized to 4 followed by aromatized to the final product 5 (Scheme 1).
Similarly, reaction of 1 with cyanothioacetamide 2b in ethanolic piperidine solution yielded the pyridinone derivative 5 not the expected pyridinethione derivative 7 under the same condition as literature [16]. Structure 5 formed via this route was established based on its elemental analysis and compatible spectral data (IR, 1H NMR and MS). The product 5 formed via this route was assumed to formed by condensation of 1 with cyanothioacetamide to form the condensation product 6 which cyclized via loss of H2S (detected by lead acetate paper) to give the adduct 4. Rearrangement of compound 4 to the final product 5 (Scheme 1).
Also, the reaction of 1 with 3-aminocrotonitrile afforded the 6-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro- 1H-pyrazol-4-yl-amino)-2,4-dimethylnicotinonitrile 8(Scheme 1). Compound 8 was confirmed based on the elemental analysis and spectral data.
The behavior of 1 towards electrophilic reagents under an alkaline condition was investigated. Thus, the reaction of 1 with malononitrile dimer afforded the pyran derivative 9 which was established by spectral data (IR, 1H NMR and 13C NMR) (Scheme 2).
Also, the reaction of 1 with arylidinemalononitrile 10a-10c afforded the pyran moiety 11a-11c or the hydroxy pyridines 12a-12c. Structure 11 was confirmed for the reaction product on the basis of spectroscopic data. The 1H NMR spectrum showed a singlet signal at δ 4.36 ppm for 4H-pyran, whereas structure 12 would be expected to show OH proton at down field.
Further, β-oxoanilide 1 reacted with ethoxymethylene malononitrile to yield the pyridine derivative 13.
On the other hand, treatment of 1 with anthranilic acid gave the quinoline derivative 14. Establishing of the structure 14 by its elemental analysis and spectroscopic data (Scheme 2).
Reaction of β-oxoanilide 1 with ethoxycarbonylisothiocyanate 15a (prepared in situ) in dry acetone afforded the adduct 16a. Compound 16a was established based on its elemental analysis and spectral data. The 1H NMR spectrum of compound 16a revealed the signals of OCH2CH3 protons at δ = 1.22 ppm as triplet, J = 7.2 Hz, and 4.15 as quartet, J = 7.2 Hz Compound 16a was cyclized to the corresponding pyrimidinethione derivative
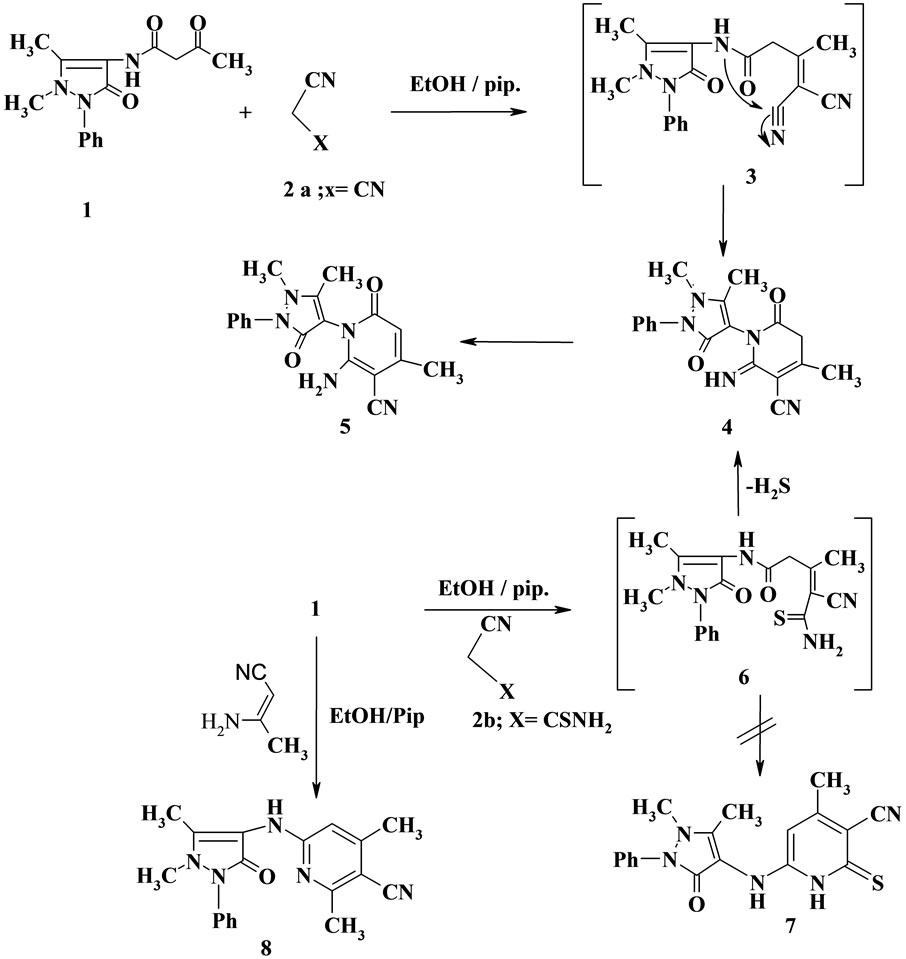
Scheme 1. Synthesis of compounds 5-8.
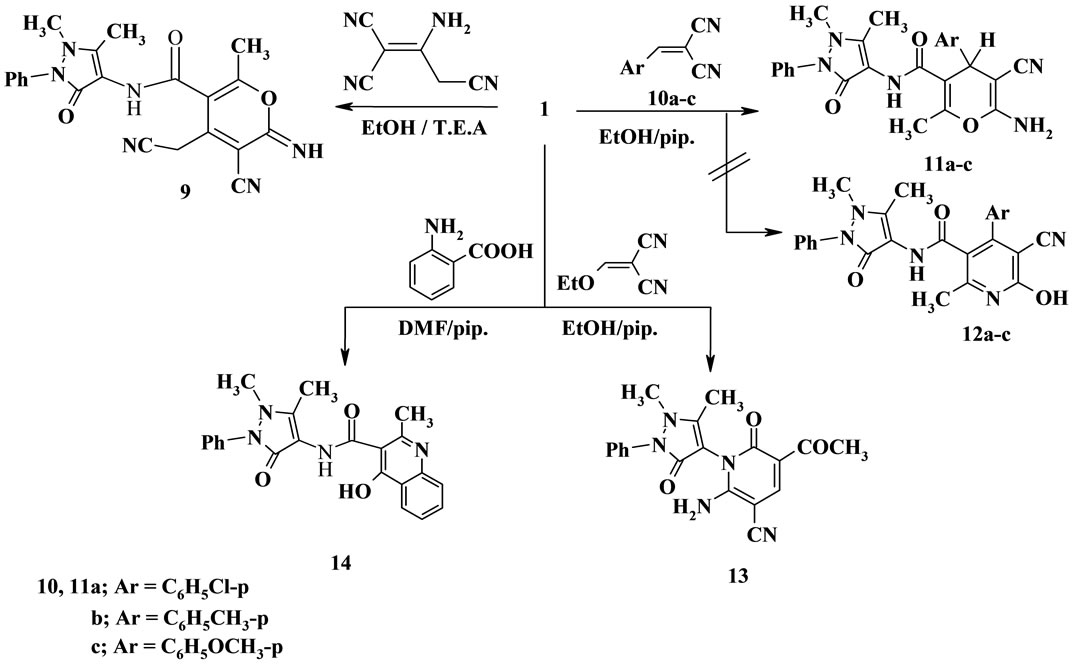
Scheme 2. Synthesis of compounds 9-14.
17 upon boiling in ethanolic sodium ethoxide solution. The 1H NMR spectrum of compound 17 revealed the absence of any signals may be attributed to OCH2CH3 protons, and appearance only the signals assigned to 3CH3, NH, OH and aromatic protons.
On the other hand, reaction of 1 with p-chlorobenzoyl isothiocyanate 15b in dry acetone furnished the pyrimidinethione derivative 18. Establishing the pyrimidinethione derivative 18 based on its elemental analysis and spectral data (IR and 1H NMR). (Scheme 3). The 1H NMR spectrum of compound 18 revealed the absence of any signals may be attributed to OCH2CH3 protons. The formation of compound 18 in this reaction was assumed to proceed via addition of the methylene group in compound 1 to the isothiocyanate group in 15b to give the nun-isolable adduct 16b, which underwent cyclization with the loss of water molecule to give the final product pyrimidinethione derivative 18.
The reaction of β-oxoanilide 1 with the chalcone derivative 19 in ethanolic piperidine solution under reflux afforded a reaction product that could be formulated as the cyclized benzene derivative 21 as literature [17] or the pyridine derivative 22. The structure 21 was ruled out according to the un-identical spectroscopic data for the structure. But the pyridine derivative 22 is considered the sole reaction product of the reaction based on its compatible spectroscopic data (IR, 1H NMR, MS and 13C NMR). Thus, 13C NMR of the reaction product revealed a signal at 167.15, 188.33, 199.23 ppm assigned to three carbonyl function groups and five sp3 signals at 10.91; 14.95; 20.60; 21.06; 36.00 ppm assigned to five CH3 groups. We found that the two carbonyl group at 199.23, 188.33 ppm has high downfield and this high downfield is identical to acetyl carbonyl and carbonyl of pyridine ring not identified to the amidic carbonyl in structure 21 which may be assigned at rang 162.00 - 166.00 ppm. Also, the 1H NMR spectrum of the reaction product revealed the presence of five singlet signals at δ = 1.63, 1.66, 2.26, 2.32, 2. 38, assigned for five CH3 groups. Moreover, the IR spectrum of the reaction product indicated the presence of three absorption bands at ν = 1697, 1655, 1645 cm−1 which attributed to three carbonyl function groups. Also, the mass spectrum of the reaction product indicated the presence of the pyridinone ion peak at m/z = 91 (44%). From the above data we sure that the sole product is the pyridine derivative 22 (Scheme 4).
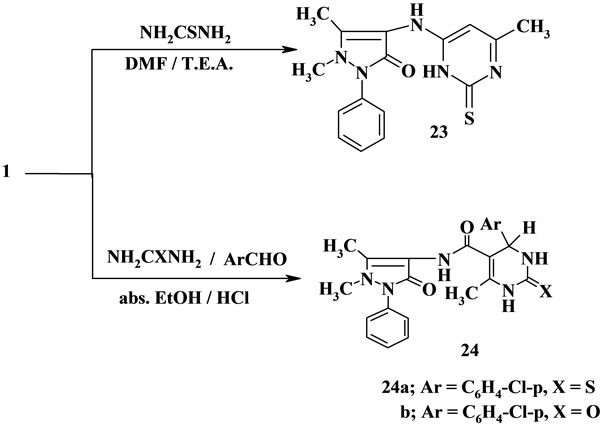
Treatment of 1 with thiourea afforded the pyrimidine derivative 23. Establishing of compound 23 based on its elemental analysis and spectral data (IR and 1H NMR), (Scheme 5).
The pyrimidinethione 24a and pyrimidinone 24b were synthesized by reacting of β-oxoanilide 1 with a mixture of aromatic aldehydes and thiourea or urea as one-pot three components. Thus, β-oxanilide 1 was reacted with a mixture of p-chlorobenzaldehyde and thiourea to form the pyrimidinethione derivative 24a and with urea to form the pyrimidinone derivative 24b. Establishing of structures 24a, 24b based on its spectroscopic data. The mass spectrum of pyrimidinethione 24a showed a molecular ion peak at m/z = 467 (M+) corresponding to the molecular formula C23H22ClN5O2S. Moreover, its 1H NMR spectrum revealed the signals at δ = 5.44 (s, 1H,
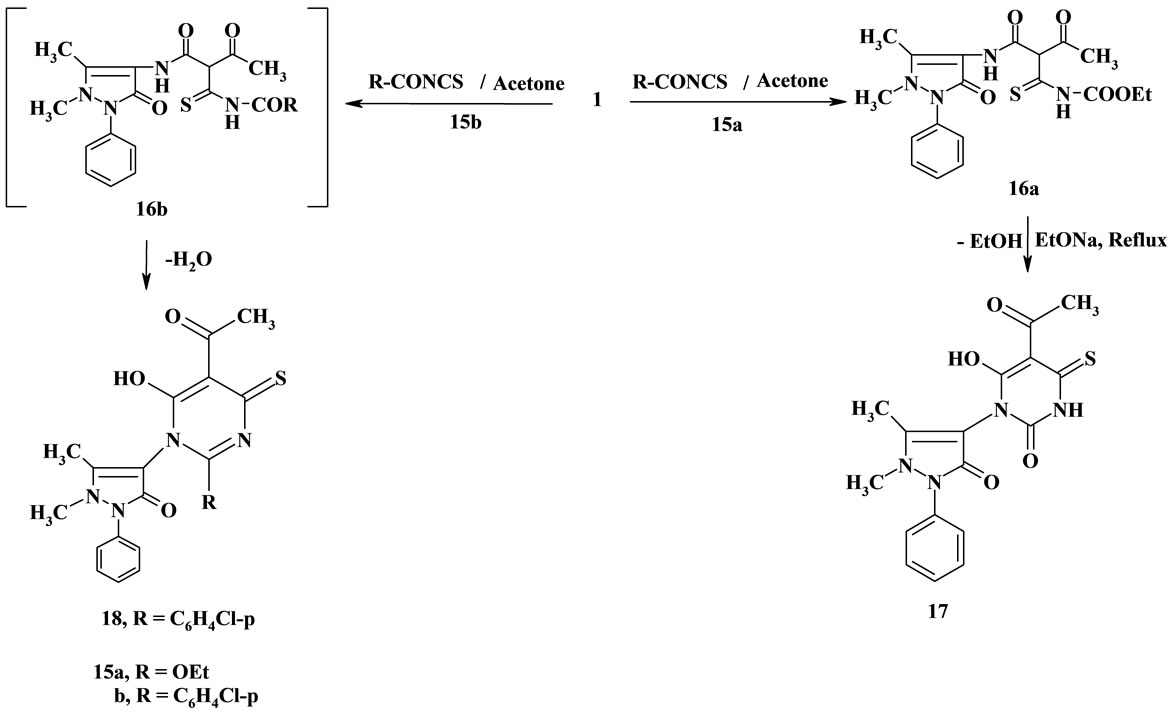
Scheme 3. Synthesis of compounds 16a-18.
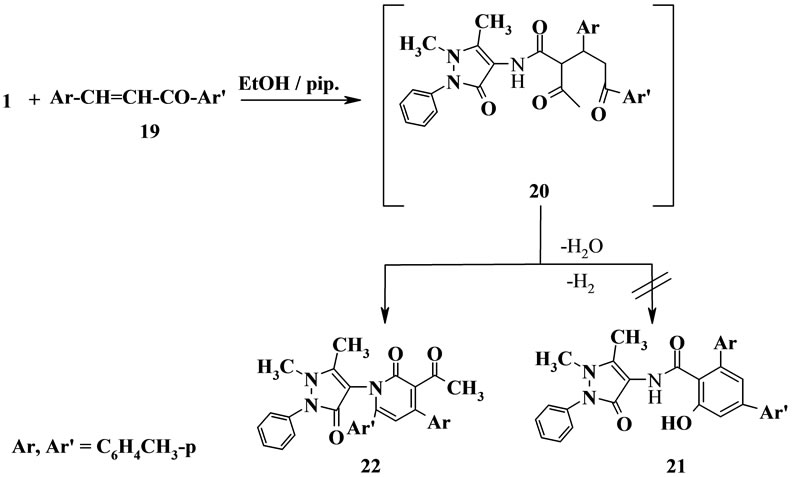
Scheme 4. Synthesis of compound 22.
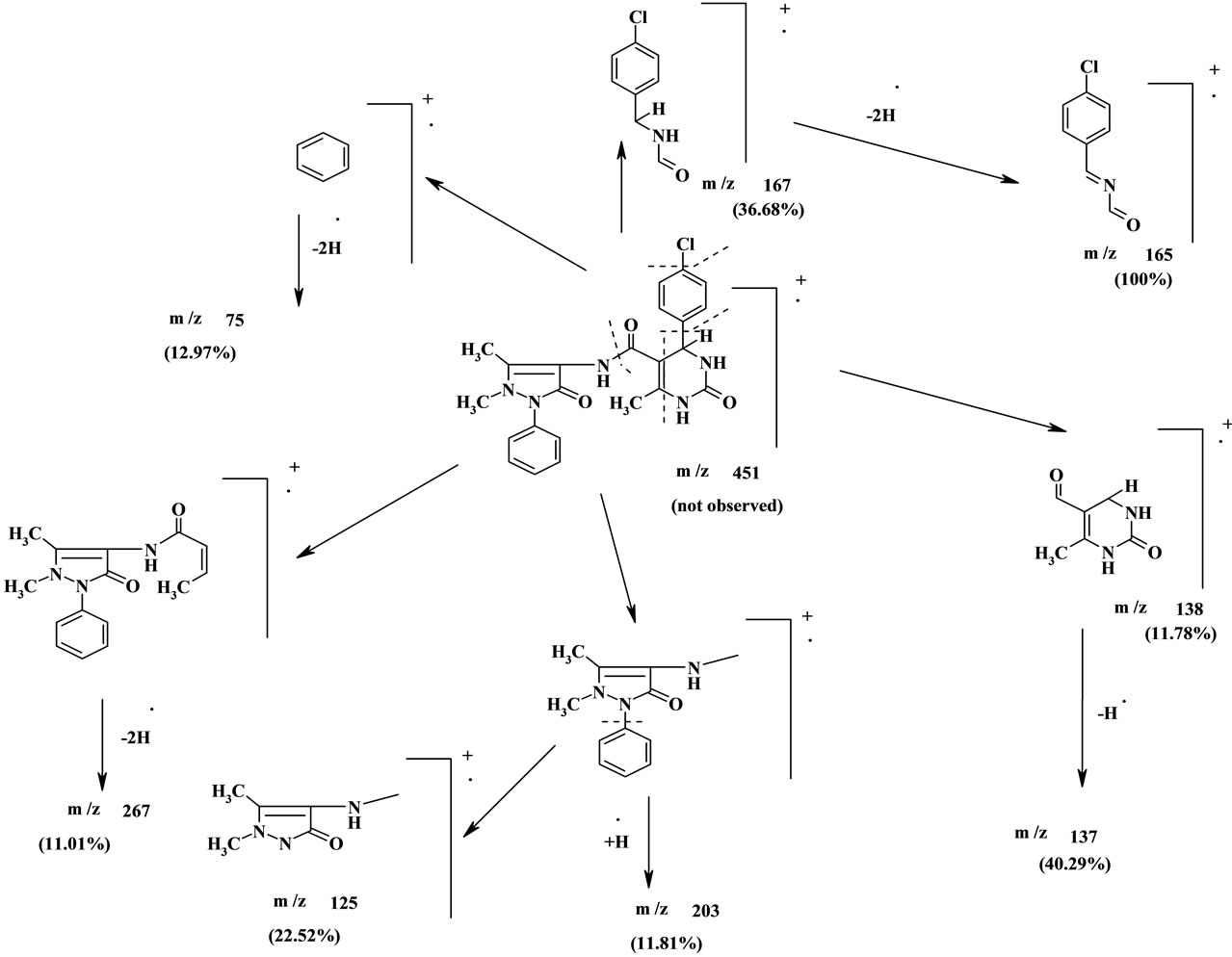
4H-pyrimidine), 9.26 (s, 1H, NH), 9.55 (s, 1H, NH), 9.84 (s, 1H, NH). On the other hand, the molecular ion peak of the structure 24b was not observed [18,19] in its mass spectrum data due to the highly unstable M+. The base peak was found at m/z = 165 (100%) corresponding to M+ (451)-296 (Sheet 1). From the above data the structures 24a, 24b are correct structures for the reaction product (Scheme 5).
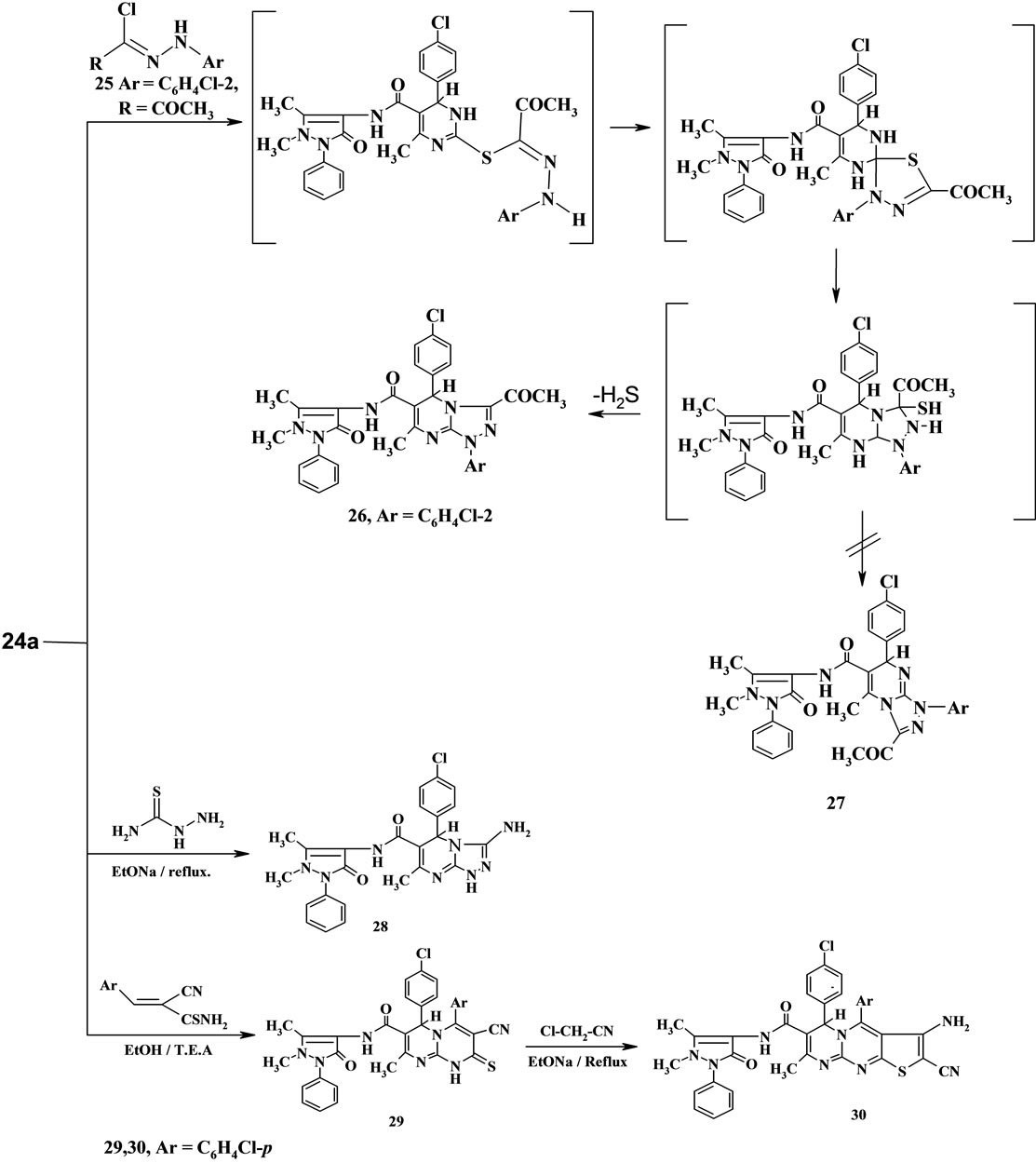
The pyrimidinethione derivative 24a was used as starting material to obtain some fused azines [20]. Thus, it reacted with hydrazonylhalide derivative 25 in ethanol containing little amount of triethylamine to afforded the triazolopyrimidine derivative 26 or the isomeric structure 27. We believe that the structure 26 is the correct one because it has the plane of symmetry than the structure 27 which has not this symmetry [21]. The structure 26 was established based on the compatible elemental analysis and spectroscopic data. The formation of 26 was assumed to proceed according to the mechanism described in scheme 6 as reported in literature [21].
Treatment of compound 24a with thiosemicarbazide as binucleophlic reagent in ethanolic sodium ethoxide solution afforded the amino triazolopyrimidine derivative 28 by loss of H2S.
Scheme 5. Synthesis of compounds 23, 24a, 24b.
Sheet 1. Fragmentation pattern of compound 24b.
On the other hand, the pyrimidinethione 24a was reacted with p-chlorobenzylidinecyanothioacetamide as example to the electrophilic reagents in ethanol containing a little amount of triethylamine under reflux to give the expected pyrimidopyrimidinthione derivative 29, which reacted with one molecule of chloroacetonitrile in refluxing ethanolic sodium ethoxide solution to furnish the fused tricyclic pyrimidothienopyrimidine derivative 30. Establishing the structure 30 based on its spectral data (Scheme 6).
Scheme 6. Synthesis of compounds 26-30.
3. Biological Activity
Seven compounds from the newly synthesized were screened in vitro for their antibacterial activities against Gram positive bacteria; Staphylococcus aureus and Bacillus cereus (G + ve), Gram negative bacteria; Klebsiella pneumonia (G − ve) and for their Antifungal activities against Aspergillus flavus and Aspergillus ochraceous by the agar diffusion technique [22]. 1 mg/ml solution in dimethylformamide (DMF) was used. The bacteria are maintained on nutrient agar. DMF showed no inhibition zones. The agar media were inoculated with different microorganism’s culture tested after 24 hours of inoculated at 37˚C for bacteria and for antifungal tested after 72 hours of inoculated at 28˚C. The diameter of inhibittion zone (mm) was measured. The data obtained is summarized in Table 1.
4. Experimental
All melting points are uncorrected. IR spectra (KBr) were recorded on a FTIR 5300 spectrometer (n, cm−1). The 1H NMR and 13C NMR spectra were recorded in DMSO-d6 at 200, 300 MHz on a Varian Gemini NMR spectrometer (d, ppm) using TMS as an internal standard. Mass spectra were obtained on GC Ms-QP 1000 EX
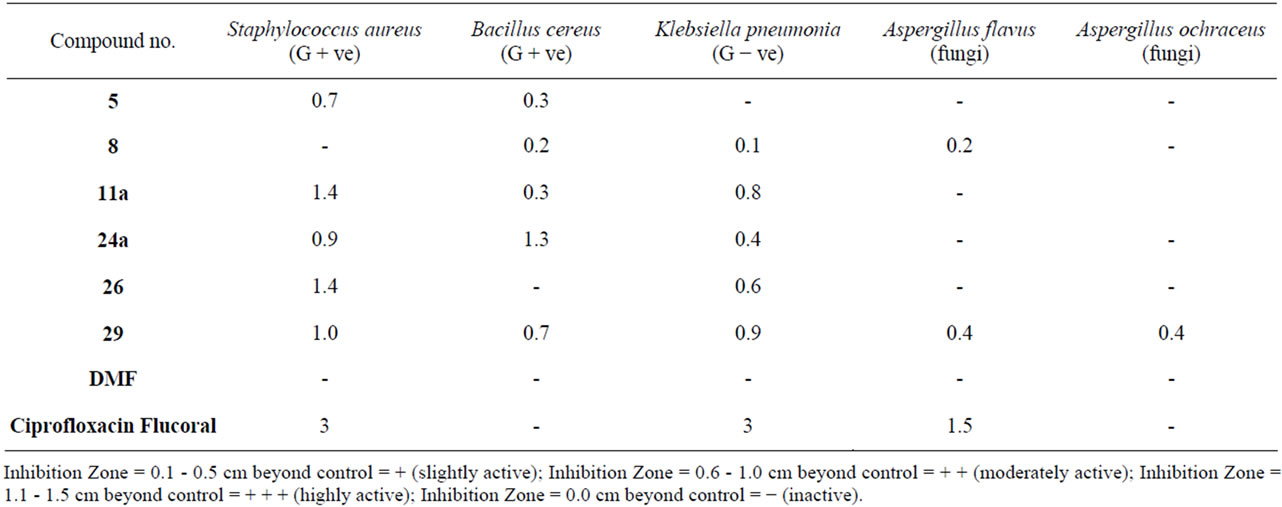
Table 1. Antimicrobial activity of some of the newly synthesized compounds.
mass spectrometer at 70 ev. Elemental analysis were carried out by the Micro analytical Research Center, Faculty of Science, Cairo University, Assiut University and Alazhar University, Faculty of Science, Department of Chemistry, Assiut Branch.
2-Amino-1-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro- 1H-pyrazol-4-yl)-4-methyl-6-oxo-1,6-dihydropyridine-3-carbonitrile (5):
General procedure:
Method A: A mixture of 1 (0.01 mole), malononitrile (2a) (0.01 mole) in ethanol (30 mL) was treated with a few drops of piperidine and refluxed for 12 hrs. The solid precipitate produced on cool was collected by filtration and recrystallized from the proper solvent to give 5.
Method B: A mixture of 1 (0.01 mole), cyanothioacetamide (2b) (0.01 mole) and a few drops of piperidine, was refluxed in ethanol (30 mL) for 12 hrs. The obtained solid on cooling recrystallized from ethanol to give 5.
It was obtained as green crystals from ethanol; yield 74%; mp. 193˚C; IR (KBr) ν cm−1 3327, 3267 (NH2), 3065 (CH-arom), 2934 (CH-aliph), 2193 (CN), 1654, 1635 (2C=O); 1H NMR (DMSO-d6) δ = 2.32 (s, 3H, CH3), 2.71 (s, 3H, CH3), 3.04 (s, 3H, NCH3), 5.61 (s, 2H, NH2), 7.24 - 7.87 (m, 6H, Ar-H + CH-pyridine), MS: m/z = 66 (32%), 93 (100%), 116 (43%), 148 (23%), 168 (31%), 183 (27%), 271 (26%), 299 (20%), 335 (30%) and 336 (22%) [M + 1]. Anal. Calc. For C18H17N5O2 (335.37): C, 64.47; H, 5.11; N, 20.88. Found: C, 64.73; H, 5.38; N, 20.63.
6-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyra-zol-4-ylamino)-2,4-dimethylnicotinonitrile (8):
To a solution of compound 1 (0.01 mole), 3-aminocrotonitrile (0.01 mole) in ethanol (30 mL) treated with a few drops of piperidine was heated under reflux for 12 hrs. Then cool, the solid product so formed was collected by filtration and recrystallized from dioxane as brown crystals; yield 70 %; mp. 187˚C; IR (KBr) ν cm−1 3333 (NH), 3057 (CH-arom), 2923 (CH-aliph), 2193 (CN), 1655 (C=O); 1H NMR (DMSO-d6) δ = 2.17 (s, 3H, CH3), 2.33 (s, 3H, CH3), 2.39 (s, 3H, CH3), 3.04 (s, 3H, NCH3), 7.09 - 7.78 (m, 6H, Ar-H + pyridine-H), 10.56 (s, 1H, NH), MS: m/z = 56 (47%), 77 (16%), 98 (100%), 120 (12%), 131 (3%), 161 (2%), 201 (5%), 213(4.9%), 280 (6%), 318 (3%) and 334 (3.62%) [M+1]. Anal. Calc. For C19H19N5O (333.40): C, 68.45; H, 5.74; N, 21.01. Found: C, 68.72; H, 5.95; N, 21.22.
3-Cyano-4-(cyanomethyl)-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-imino-6-methyl-2H-pyran-5-carboxamide (9):
A mixture of compound 1 (0.01 mole), 2-aminoprop-1-ene-1,1,3-tricarbonitrile (0.01 mole) in ethanol (30 mL) was treated with a few drops of piperidine and heated under reflux for 18 hrs. Then cool, poured into crushed ice and acidified with dil. HCl. The product formed was collected by filtration and recrystallized from DMF/ ethanol as green crystal; yield 73%; mp. > 300˚C; IR (KBr) ν cm−1 3373, 3181 (2NH), 3045 (CH-arom.), 2925 (CH-aliph), 2205, 2194 (2CN), 1666, 1635 (2CO), 1H NMR (DMSO-d6) δ = 2.14 (s, 3H, CH3), 2.19 (s, 3H, CH3), 3.02 (s, 3H, NCH3), 3.97 (s, 2H, CH2), 7.30 - 7.52 (m, 5H, Ar-H), 8.20 (s, 1H, NH), 10.01 (s, 1H, NH),13C NMR δ C = 17.70; 20.63; 24.84; 52.89; 112.38 (CN); 117.25 (CN); 122.42; 126.53; 126.53; 129.16; 129.16; 134.32; 134.32; 138.76; 146.0; 147.25; 147.25; 157.21; 157.21; 161.16 (CO); 180.45 (CO). Anal. Calc. For C21H18N6O3 (402.47): C, 62.68; H, 4.51; N, 20.88. Found: C, 62.89; H, 4.74; N, 20.65.
Preparation of compounds 11a-c:
General procedure:
A mixture of 1 (0.01 mole), arylidinemalononitriles 10a-c (0.01 mole) in ethanol (30 mL) was treated with a few drops of piperidine and heated under reflux for 8 hrs. The reaction mixture allowed to cool, poured into crushed ice and acidified with HCl. The solid product was filtered off and recrystallized from the proper solvent to give 11a-c.
6-Amino-4-(4-chlorophenyl)-5-cyano-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-methyl-4H-pyran-3-carboxamide (11a):
It was obtained as yellow crystals from ethanol; yield 88%; mp. 202˚C; IR (KBr) ν cm−1 3423, 3333 (NH2), 3212 (NH), 3053 (CH-arom.), 2926 (CH-aliph.), 2198 (CN), 1690, 1645 (2CO); 1H NMR (DMSO-d6) δ = 2.10 (s, 3H, CH3), 2.13 (s, 3H, CH3), 3.04 (s, 3H, NCH3), 4.36 (s, 1H, 4H-pyran), 6.16 (s, 2H, NH2), 7.25 - 7.53 (m, 9H, Ar-H), 11.15 (s, 1H, NH). MS: m/z = 68 (100%), 70 (89%), 84 (81%), 115 (83%), 178 (99.69%) and 472 (84%) [M−3]. Anal. Calc. For C25H22ClN5O3 (475.94): Calcd: C, 63.09; H, 4.66; N, 14.71. Found: C, 63.29; H, 4.89; N, 14.97.
6-Amino-5-cyano-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-methyl-4-(4-methylphenyl)-4H-pyran-3-carboxamide (11b).
It was obtained as yellow crystals from ethanol; yield 85%; mp. 192˚C; IR (KBr) ν cm−1 3318, 3283 (NH2), 3161 (NH), 3050 (CH-arom), 2926 (CH-aliph), 2202 (CN), 1665, 1634 (2CO); 1H NMR (DMSO-d6) δ = 2.05 (s, 3H, CH3), 2.13 (s, 3H, CH3), 2.25 (s, 3H, CH3), 3.16 (s, 3H, NCH3), 4.16 (s, 1H, 4H-pyran), 6.66 (s, 2H, NH2), 7.21 - 8.13 (m, 9H, Ar-H), 11.01 (s, 1H, NH). Anal. Calc. For C26H25N5O3 (455.52): C, 68.56; H, 5.53; N, 15.37. Found: C, 68.81; H, 5.76; N, 15.63.
6-Amino-5-cyano-N-(1,5-dimethyl-3-oxo-2-phenyl-2, 3-dihydro-1H-pyrazol-4-yl)-4-(4-methoxy-phenyl)-2-methyl-4H-pyran-3-carboxamide (11c).
It was obtained as yellow crystals from ethanol; yield 87%; mp. 196˚C; IR (KBr) ν cm−1 3313, 3275 (NH2), 3161 (NH), 3049 (CH-arom), 2924 (CH-aliph), 2201 (CN), 1668, 1635 (2CO); 1H NMR (DMSO-d6) δ = 2.25 (s, 3H, CH3), 2.28 (s, 3H, CH3), 3.16 (s, 3H, NCH3), 3.78 (s, 3H, OCH3), 4.52 (s, 1H, 4H-pyran), 6.57 (s, 2H, NH2), 7. 05 - 8.24 (m, 9H, Ar-H), 10.35 (s, 1H, NH). Anal. Calc. For C26H25N5O4 (471.52): C, 66.23; H, 5.34; N, 14.85. Found: C, 66.48; H, 5.61; N, 14.59.
5-Acetyl-2-amino-1-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-6-oxo-1,6-dihydropyridine-3-carbonitrile (13):
To a solution of 1 (0.01 mole) in ethanol (30 mL)/ piperdine (0.5 mL), ethoxymethylene malononitrile (0.01 mole) was added. The reaction mixture was refluxed for 15 hrs. Then left to stand, poured into cold water and acidified with HCl. The product formed was collected by filtration and recrystallized from ethanol to give (13; 66%) as brown crystals; mp. > 300˚C. IR (KBr) ν cm−1 3421, 3400 (NH2), 2923 (CH-aliph.), 2194 (CN), 1721, 1660, 1630 (3CO); 1H NMR (DMSO-d6) δ = 2.12 (s, 3H, CH3), 2.14 (s, 3H, COCH3), 3.02 (s, 3H, NCH3), 5.59 (s, 2H, NH2), 6.67 (s, 1H, CH-pyridine), 7.02 - 7.48 (m, 5H, Ar-H). Anal. Calc. For C19H17N5O3 (363.38): C, 62.80; H, 4.72; N, 19.27. Found: C, 62.55; H, 4.95; N, 19.54.
N-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-4-hydroxy-2-methylquinoline-3-carboxamide(14):
To a solution of acetoacetanilide 1 (0.01 mole) in DMF (30 mL) containing a few drops of triethylamine, anthranilic acid (0.01 mole) was added. The reaction mixture was heated under reflux for 8 hrs. Then cool, poured into crushed ice and acidified with HCl. The solid product was collected by filtration and recrystallized from dioxane to give (14; 64%) as green crystals; mp. 196˚C; IR (KBr) ν cm−1 3425 (OH), 3347 (NH), 3050 (CHarom), 2924 (CH-aliph), 1670, 1648 (2C=O); 1H NMR (DMSO-d6) δ = 2.16 (s, 3H, CH3), 2.24 (s, 3H, CH3), 2.72 (s, 1H, NCH3), 7.37 - 7.76 (m, 10H, Ar-H + OH), 10.23 (br, 1H, NH), MS: m/z = 56 (100%), 77 (78%), 102 (17%), 119(36%), 146 (24%), 166 (15%) and 387 (4.3 %) [M+-1]. Anal. Calc. For C22H20N4O3 (388.43): C, 68.03; H, 5.19; N, 14.42. Found: C, 68.28; H, 5.45; N, 14.63.
Ethyl-2-{[(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino]carbonyl}-3-oxobutanethiocarb-amate (16a):
To a solution of compound 1 (0.01 mole) in dry acetone (30 mL), ethoxy carbonyl isothiocyanate 15a (prepared by reflux mixture of ethyl chloroformate (0.01 mol) and ammonium thiocyanate (0.01 mol) in dry acetone for 30 mins) was added. The reaction mixture was heated under reflux for 6 hrs. Then left to cool, the precipitate was collected by filtration and recrystallized from ethanol to give 16a, as white crystals; mp. 197˚C. IR (KBr) ν cm−1 3254, 3212 (2NH), 2980 (CH-aliph), 1784, 1720, 1661, 1630 (4CO), 1H NMR (DMSO-d6), δ = 1.22 (t, J = 7.2 Hz, 3H, CH3, CH3CH2O), 2.08 (s, 3H, CH3), 2.09 (s, 3H, CH3), 2.16 (s, 3H, NCH3), 3.37 (s, 1H, CH), 4.15 (q, J = 7.2 Hz, 2H, CH2, OCH2CH3), 7.30 - 7.55 (m, 5H, Ar-H), 10.55 (s, 1H, NH), 11.37 (s, 1H, NH). Anal. Calc. For C19H22N4O5 S (418.47): C, 54.53; H, 5.30; N, 13.39; S, 7.66. Found: C, 54.81; H, 5.56; N, 13.63; S, 7.50.
5-Acetyl-1-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-6-hydroxy-4-thioxo-3,4-dihydropyrimidin-2(1H)-one (17):
To a solution of 16a (0.01 mole) in ethanol, sodium ethoxide (0.01 mole) was added. The reaction mixture was refluxed for 12 hrs. Then left to stand, poured into cold water and acidified with HCl. The product formed was collected by filtration and recrystallized from ethanol to give (17; 68%) as brown crystals; mp. > 300˚C; IR (KBr) ν cm−1 3485 (OH), 3357 (NH), 3060 (CH-arom), 2927 (CH-aliph), 1653, 1630 (3CO); 1H NMR (DMSOd6) δ = 1.24 (s, 3H, CH3), 2.18 (s, 3H, COCH3), 2.71 (s, 3H, NCH3), 7.07 - 7.50 (m, 5H, Ar-H), 9.62 (s, 1H, NH), 12.57 (s, 1H, OH). Anal. Calc. For C17H16N4O4S (372.41): C, 54.83; H, 4.33; N, 15.04; S, 8.61. Found: C, 54.57; H, 4.61; N, 15.25; S, 8. 87.
4-(5-Acetyl-2-(4-chlorophenyl)-6-hydroxy-4-thioxopyrimidin-1(4H)-yl)-1,5-dimethyl-2-phenyl-1,2-dihydro-3H-pyrazol-3-one (18):
To solution of compound 1 (0.01 mole) in dry acetone (30 mL), p-chlorobenzoyl isothiocyanate (15b) (prepared by reflux a mixture of p-chlorobenzoyl chloride (0.01 mol) and ammonium thiocyanate (0.01 mol) in dry acetone for 30 mins) was added. The reaction mixture was heated under reflux for 6 hrs, then left to cool. The solid product formed was collected by filtration and recrystallized from ethanol; yield 62% as brown crystals; mp. 190˚C; IR (KBr) ν cm−1 3441 (OH), 3060 (CH-arom), 2926 (CH-aliph), 1660, 1634 (2CO); 1H NMR (DMSOd6), δ = 2.16 (s, 3H, CH3), 2.71 (s, 3H, COCH3), 3.11 (s, 3H, NCH3), 7.07 - 7.93 (m, 9H, Ar-H), 12.52 (br, 1H, OH). Anal. Calc. For C23H19ClN4O3S (466.95): C, 59.16; H, 4.10; N, 12.00; S, 6.87. Found: C, 59.43; H, 4.27; N, 12.24; S, 6.62.
3-Acetyl-1-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-4,6-bis(4-methylphenyl)pyridin-2(1H)-one (22).
A mixture of compound 1 (0.01 mole) and 1,3-bis (4-methylphenyl)prop-2-en-1-one (19) in ethanol (30 mL) containing a few drops of piperidine was heated under reflux for 6 hrs. The solid product formed after cooling was collected by filtration and recrystallized from ethanol to give (22; 87%) as white crystals; mp. 120˚C; IR (KBr) ν cm−1 3060 (CH-arom), 2924 (CH-aliph), 1697, 1655, 1645 (3C=O); 1H NMR (DMSO-d6) δ = 1.63 (s, 3H, CH3), 1.66 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.38 (s, 3H, CH3), 7.10 - 8.02 (m, 13H, Ar-H), 9.34 (s, 1H, pyridine-H), MS: m/z = 57 (100%), 91 (44%), 149 (49%), 221 (77%), 306 (11%), 384 (5%) and 503 (4%) [M+]. 13C NMR δ C = 10.91; 14.95; 20.60; 21.06; 36.00; 120.85; 120.85; 120.85; 123.24; 123.24; 126.10; 127.47; 128.50; 128.70; 128.70; 128.70; 128.70; 129.17; 129.17; 129.17; 129.17; 129.41; 129.41; 131.89; 135.37; 140.34; 140.34; 143.65; 158.00; 167.15 (CO); 188.33 (CO); 199.23 (CO). Anal. Calc. For C32H29N3O3 (503.61): C, 76.32; H, 5.80; N, 8.34. Found: C, 76.58; H, 5.58; N, 8.59.
1,5-Dimethyl-4-[(6-methyl-2-thioxo-2,3-dihydropyrimidin-4-yl)amino]-2-phenyl-1,2-dihydro-3H-pyrazol-3-one (23):
To a solution of acetoacetanilide 1 (0.01 mole) in DMF (30 mL) containing a few drops of triethylamine, thiourea (0.01 mole) was added. The reaction mixture was heated under reflux for 8 hrs. Then cool, poured into crushed ice and acidified with HCl. The solid product was collected by filtration and recrystallized from DMF; yield 59 %; mp. > 300˚C; IR (KBr) ν cm−1 3420, 3378 (2NH), 3053 (CH-arom), 2926 (CH-aliph), 1652 (C=O); 1H NMR (DMSO-d6) δ = 2.46 (s, 3H, CH3), 2.97 (s, 3H, CH3), 3.05 (s, 3H, NCH3), 7.11 - 7.67 (m, 6H, Ar-H + pyrimidine-H), 9.67 (s, 1H, NH), 9.85 (s, 1H, NH). Anal. Calc. For C16H17N5OS (327.41): C, 58.70; H, 5.23; N, 21.39; S 9.79. Found: C 58.48; H, 5.51; N, 21.53; S, 9.65.
Preparation of compounds 24a,b:
General procedure:
To a solution of acetoacetanilide 1 (0.01 mole) in absolute ethanol (30 mL) and (1 mL) HCl. A mixture of aromatic aldehyde (0.01 mole) and thiourea or urea (0.01 mole) was added. The reaction mixture was refluxed for 9 hrs. The solid product was produced on hot was collected by filtration and recrystallized from the proper solvent to give 24a,b.
4-(4-Chlorophenyl)-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (24a):
It was obtained as pale yellow crystals from dioxane/ ethanol; yield 79%; mp. 270˚C; IR (KBr) ν cm−1 3388, 3189 (3NH), 3071 (CH-arom), 2925 (CH-aliph), 1646 (2C=O); 1H NMR (DMSO-d6) δ = 1.88 (s, 3H, CH3), 2.49 (s, 3H, CH3), 3.02 (s, 3H, NCH3), 5.44 (s, 1H, 4Hpyrimidine), 7.34-7.58 (m, 9H, Ar-H), 9.26 (s, 1H, NH), 9.55 (s, 1H, NH), 9.84 (s, 1H, NH). MS: m/z = 61 (18%), 81 (100%), 94 (67%), 131 (78%), 158 (50%), 178 (40%), 187 (49%), 268 (38%), 290 (37%), 438 (36%) and 467 (40%) [M+]. Anal. Calc. For C23H22ClN5O2S (467.98): C, 59.03; H, 4.74; N, 14.97; S; 6.85. Found: C, 59.25; H, 4.97; N, 14.71; S; 6.58.
4-(4-Chlorophenyl)-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (24b):
It was obtained as yellow crystals from dioxane/ethanol; yield 82 %; mp. 184˚C; IR (KBr) ν cm−1 3447, 3275 (3NH), 3049 (CH-arom), 2924 (CH-aliph), 1667, 1647, 1630 (3C=O); 1H NMR (DMSO-d6) δ = 1.87 (s, 3H, CH3), 2.09 (s, 3H, CH3), 3.01 (s, 3H, NCH3), 5.45 (s, 1H, 4H-pyrimidine), 7.30 - 7.58 (m, 9H, Ar-H), 9.34 (s, 1H, NH), 9.61 (s, 1H, NH), 10.03 (s, 1H, NH). MS: m/z = 75 (12.97%), 125 (22.52%), 137 (40%), 138 (11.78%), 165 (100%), 267 (11%). Anal. Calc. For C23H22ClN5O3 (451.92): C, 61.13; H, 4.91; N, 15.50. Found: C, 61.35; H, 4.66; N, 15.77.
3-Acetyl-5-(4-chlorophenyl)-1-(2-chlorophenyl)-7- methyl-N-(1,5-dimethyl-2-phenyl-3-oxo-2,3-dihydro-1Hpyrazol-4-yl)-1,5-dihydro-[1,2,4]triazolo[4,3-a]pyrimi-dine-6-carboxamide (26):
A mixture of compound 24a (0.01 mole), N-(2-chlorophenyl)-2-oxopropane hydrazonoyl chloride (25) in ethanol (30 mL) was treated with a few drops of triethylamine and heated under reflux for 8 hrs. Then cool, poured into crushed ice and acidified with HCl. The solid product was collected and recrystallized from ethanol to give (26; 74 %) as green crystals; mp. 183˚C; IR (KBr) ν cm−1 3421 (NH), 3066 (CH-arom), 2927 (CH-aliph), 1721, 1680, 1647 (3C=O); 1H NMR (DMSO-d6) δ = 2.39 (s, 3H, CH3), 2.45 (s, 3H, CH3), 2.48 (s, 3H, COCH3), 3.19 (s, 3H, NCH3), 5.21 (s,1H, 5H-pyrimidine), 7.35 - 7.84 (m, 13H, Ar-H), 9.56 (s, 1H, NH). Anal. Calc. For C32H27Cl2N7O3 (628.52): C, 61.15; H, 4.33; N, 15.60. Found: C, 61.36; H, 4.61; N, 15.82.
3-Amino-5-(4-chlorophenyl)-N-(1,5-dimethyl-3-oxo- 2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-7-methyl-1,5-dihydro[1,2,4]triazolo[4,3-a]pyrimidine-6-carboxamide(28).
A mixture of 24a (0.01 mole), and thiosemicarbazide (0.01 mole) in ethanol containing sodium ethoxide (0.01 mole) was refluxed for 24 hrs. Then left to cool, poured into cold water and acidified with HCl. The product formed was collected by filtration and recrystallized from ethanol to give (28; 67%) as brown crystals; mp. 205˚C. IR (KBr) ν cm−1 3408, 3320 (NH2), 3234, 3198 (2NH), 3085 (CH-arom), 2925 (CH-aliph.), 1660, 1652 (2CO); 1H NMR (DMSO-d6) δ = 1.97 (s, 3H, CH3), 2.29 (s, 3H, CH3), 3.00 (s, 3H, NCH3), 5.32 (s, 1H, 5H-pyrimidine), 7.05 - 8.02 (m, 11H, Ar-H + NH2), 9.60 (s, 1H, NH), 11.44 (s, 1H, NH). Anal. Calc. For C24H23ClN8O2 (490.96): C, 58.72; H, 4.72; N, 22.82. Found: C, 58.94; H, 4.95; N, 22.60.
4,6-Bis-(4-Chlorophenyl)-3-cyano-N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-8-methyl-2- thioxo-1,6-dihydro-2H-pyrimido[1,2-a]pyrimidine-7-carboxamide (29):
A mixture of compound 24a (0.01 mole), p-chloroarylidinecyanothioacetamide 16b (0.01 mole) in ethanol (30 mL) was treated with a few drops of triethylamine and heated under reflux for 8 hrs. Then cool, poured into crushed ice and acidified with HCl. The solid product was collected and recrystallized from ethanol to give (29, 70%) as yellow crystals; mp. 183˚C; IR (KBr) ν cm−1 3389, 3201 (2NH), 3070 (CH-arom.), 2921 (CH-aliph.), 2214 (CN), 1648, 1630 (2C=O); 1H NMR (DMSO-d6) δ = 2.44 (s, 3H, CH3), 2.45 (s, 3H, CH3), 3.02 (s, 3H, NCH3), 5.42 (s, 1H, 6H-pyrimidine), 7.05 - 7.84 (m, 13H, Ar-H), 9.59 (s, 1H, NH), 9.96 (s, 1H, NH). Anal. Calc. For C33H25Cl2N7O2S (654.58): C, 60.55; H, 3.85; N, 14.98; S, 4.90. Found: C, 60.77; H, 3.68; N, 14.76; S, 4.66.
3-Amino-4,6-bis(4-chlorophenyl)-2-cyano-N-(1,5- dimethyl-2-phenyl-3-oxo-2,3-dihydro-1H-pyrazol-4-yl)- 8-methyl-6H-pyrimido[1,2-a]-thieno[2,3-d]pyrimidine-7-carboxamide (30):
To a solution of 29 (0.01 mole) in ethanol, α-chloroacetonitrile (0.01 mole) and sodium ethoxide (0.01 mole) were added. The reaction mixture was refluxed for 24 hrs. Then left to stand, poured into cold water and acidified with HCl. The product formed was collected by filtration and recrystallized from ethanol to give (30; 60%) as green crystals; mp. 295˚C; IR (KBr) ν cm−1 3430, 3355 (NH2), 3200 (NH), 3065 (CH-arom.), 2925 (CH-aliph.), 2200 (CN), 1664, 1640 (2CO); 1H NMR (DMSO-d6), δ = 1.70 (s, 3H, CH3), 2.93 (s, 3H, CH3), 3.03 (s, 3H, NCH3), 5.42 (s, 1H, 6H-pyrimidine), 6.61 (s, 2H, NH2), 7.11 - 8.12 (m, 13H, Ar-H), 9.45 (s, 1H, NH). Anal. Calc. For C35H26Cl2N8O2S (693.62): C, 60.61; H, 3.78; N, 16.15; S, 4.62. Found: C, 60.80; H, 3.57; N, 16.38; S, 4.86.
REFERENCES
- E. Rajanarendar, P. Ramesh, M. Srinivas, K. Ramu and G. Mohan, “Solid-Supported Synthesis of Isoxazole-Substituted 1,4-Dihydropyridines by Modified Hantzsch Method and Their Aromatization,” Synthetic Communications, Vol. 36, No. 5, 2006, pp. 665-671. doi:10.1080/15459620500408884
- A. T. Manvar, R. R. Pissurlenkar, V. R. Virsodia, K. D. Upadhyay, D. R. Manvar, A. K. Mishra, H. D. Acharya, A. R. Parecha, C. D. Dholakia, A. K. Shah and E. C. Coutinho, “Synthesis, in Vitro Antitubercular Activity and 3D-QSAR Study of 1,4-Dihydropyridines,” Molecular Diversity, Vol. 14, No. 2, 2010 pp. 285-305. doi:10.1007/s11030-009-9162-8
- W. W. Wardakhan, G. A. Elmegeed and F. M. Manhi, “Utility of 2-Aminothiophene-3-carboxamide in the Synthesis of Biologically Active, Fused Heterocyclic Derivatives” Phosphorus, Sulfur, Silicon, and Related Elements, Vol. 180, No. 1, 2005, pp. 125-140. doi:10.1080/104265090508028
- A. M. Hussein, A. A. Harb and I. A. Mousa, “β-Oxoanilides in Heterocyclic Synthesis: An Expeditious Synthesis of New Polyfunctionally Substituted Pyridine and Pyrazole Derivatives,” Journal of Heterocyclic Chemistry., Vol. 45, No. 6, 2008, pp. 1819-1823. doi:10.1002/jhet.5570450641
- T. M. Abu Elmaati, “Alkyl Heterocycles in Heterocyclic Synthesis (II): Novel Synthesis of Isoquinoline, Thiazolopyridine, and Thieno[2,3-b]pyridine Derivatives,” Journal of Heterocyclic Chemistry, Vol. 41, No. 6, 2004, pp. 947-950. doi:10.1002/jhet.5570410614
- A. A. Harb, A. M. Hussein and I. A. Mousa, “Acetoacetanilides in Heterocyclic Synthesis, Part 1: An Expeditious Synthesis of Thienopyridines and Other Fused Derivatives,” Phosphours, Sulfur and Silicon, and Related Elements, Vol. 181, No. 10, 2006, pp. 2247-2261.
- A. M. Hussein, A. A. M. El-Adasy, M. A. M. Gad-Elkareem and I, M. Othman, “β-Oxoanilides in Heterocyclic Synthesis: A Convenient Synthetic Route to Polyfunctionally Substituted Pyridazines and Pyridines,” Organic Chemistry an Indian Journal, Vol. 4, No. 3, 2008, pp. 178-186.
- A. M. Hussein, M. S. El-Gaby, F. A. Abu-Shanab and M. A. M. Abdel-Raheim, “β-Oxoanilides in Heterocyclic Synthesis: Synthesis of Triand Tetracyclic Heteroaromatic Containing a Bridgehead Nitrogen Atom,” Organic Communications, Vol. 2, No. 3, 2009, pp. 66-71.
- A. M. Hussein, M. A. M. Gad-Elkareem, A. A. M. ElAdasy and I. M. Othman, “N-1-Naphthyl-3-oxo-butanamide in Heterocyclic Synthesis: A Facile Synthesis of Nicotinamide, Thieno[2,3-b]pyridine, and Bior Tricyclic Annulated Pyridine Derivatives Containing Naphthyl Moiety,” Phosphorus, Sulfur, Silicon, and Related Elements, Vol. 184, No. 9, 2009, pp. 2263-2280. doi:10.1080/10426500802453658
- A. Riahi, M. Wurster, M. Lalk, U. Lindequist and P. Langer, “Synthesis and Antimicrobial Activity of 4-Hydroxy-4-(pyridyl)alk-3-en-2-ones,” Bioorganic & Medicinal Chemistry, Vol. 17, 2009, pp. 4323-4326. doi:10.1016/j.bmc.2009.05.023
- X. Zhu and D. Shi, “Convenient Synthesis and Biological Activities of Pyridine Derivatives of 3,4-Dihydropyrimidin-2(1H)-ones,” Journal of Heterocyclic Chemistry, Vol. 48, No. 3, 2011, pp. 572-576.
- B. Ramesh and C. M. Bhalgat, “Novel Dihydropyrimidines and Its Pyrazole Derivatives: Synthesis and Pharmacological Screening,” European Journal of Medicinal Chemistry, Vol. 46, 2011, pp. 1882-1891. doi:10.1016/j.ejmech.2011.02.052
- H. Behbehani, H. M. Ibrahim, S. Makhseed and H. Mahmoud, “Applications of 2-Arylhydrazononitriles in Synthesis: Preparation of New Indole Containing 1,2,3- Triazole, Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives and Evaluation of Their Antimicrobial Activities,” European Journal of Medicinal Chemistry, Vol. 46, 2011, pp. 1813-1820. doi:10.1016/j.ejmech.2011.02.040
- H. Bayrak, A. Demirbas, N. Demirbas and S. A. Karaoglu, “Cyclization of Some Carbothioamide Derivatives Containing Antipyrine and Triazole Moieties and Investigation of Their Antimicrobial Activities,” European Journal of Medicinal Chemistry, Vol. 45, 2010, pp. 4726-4732. doi:10.1016/j.ejmech.2010.07.018
- A. A. H. Abdel Rahman, A. H. A. Ahmed and M. M. M. Ramiz, “Synthesis and Anti-Hbv Activity of 4-Aminoantipyrine Derivatives,” Chemistry of Heterocyclic Compounds, Vol. 46, No. 1, 2010, pp. 72-78. doi:10.1007/s10593-010-0472-7
- S. Krivokolysko, V. Dyachenko and V. Litvinov, “A Convenient Method for the Synthesis of Substituted 2- Alkylthio-3-cyano-4,6-dimethyl-5-phenylcarbamoyl-1,4-dihydropyridines,” Chemistry of Heterocyclic Compounds, Vol. 36, No. 3, 2000, pp. 284-286. doi:10.1007/BF02256865
- V. L. Gein, E. V. Levandovskaya, N. V. Nosova, N. V. Antonova, E. V. Voronina, M. I. Vakhrin and A. P. Krivenko, “Synthesis and Antibacterial Activity of N,N’-diaryl-2-aryl-6-hydroxy-6-methyl-4-oxocyclohexane-1,3-dicarboxamides,” Pharmaceutical Chemistry Journal, Vol. 41, No. 12, 2007, pp. 21-23. doi:10.1007/s11094-008-0043-8
- M. A. M. Gad-Elkareem, M. A. A. Elneairy and A. M. Taha, “Reactions with 3,6-Diaminothieno[2,3-b]-pyridines: Synthesis and Characterization of Several New Fused Pyridine Heterocycles,” Heteroatom Chemistry, Vol. 18, No. 4, 2007, pp. 405-413. doi:10.1002/hc.20313
- H. M. Madkour, M. R. Mahmoud, M. H. Nassar and M. M. Habashy, “Behaviour of Some Activated Nitriles toward Barbituric Acid, Thiobarbituric Acid and 3-Methyl- 1-phenylpyrazol-5-one,” Molecules, Vol. 5, No. 5, 2000, pp. 746-755. doi:10.3390/50500746
- S. A. Ahmed, A. M. Hussein, W. G. Hozayen, A. H. H. El-Ghandour and A. O. Abdelhamid, “Synthesis of Some Pyrazolopyrimidines as Purine Analogues,” Journal of Heterocyclic Chemistry, Vol. 44, No. 4, 2007, pp. 803- 810. doi:10.1002/jhet.5570440408
- A. B. A. El-Gazzar, H. N. Hafez and E. M. A. Yakout, “One-Pot Synthesis of Pyrido[2,3-d]pyrimidine-2-thiones and a Study of Their Reactivity towards Some Reagents,” Journal of the Chinese Chemical Society, Vol. 54, No. 5, 2007, p. 1303.
- National Committee for Clinical Laboratory Standards, “Performance Standards for Antimicrobial Disk Suscepti bility Tests,” Approved Standard NCCLS. M2-A5, 13, 24, NCCLS, Villanova, 1993.
NOTES
*Corresponding author.