 Journal of Cancer Therapy, 2013, 4, 1411-1425 Published Online November 2013 (http://www.scirp.org/journal/jct) http://dx.doi.org/10.4236/jct.2013.49168 Open Access JCT 1411 Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal* Nandini Dey1,2,#, Yuliang Sun1, Brian Leyland-Jones1,2, Pradip De1,2,# 1Edith Sanford Breast Cancer, Sanford Research, Sioux Falls, USA; 2Dept. of Internal Medicine, University of South Dakota, Ver- million, USA. Email: #pradip.de@sanfordhealth.org, #nandini.dey@sanfordhealth.org Received October 15th, 2013; revised November 5th, 2013; accepted November 13th, 2013 Copyright © 2013 Nandini Dey et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT Patient derived xenograft (PDX) is defined as a growth of patients’ tumor in the xenograft setting. The evolution of cancer model in animal has a century old history. The most single reason that exerted the pressure on the traditional animal model of cancer to evolve to PDX is that the traditional models have not delivered as expected and traditional models have not predicted clinical success. In spite of well above 50 drugs developed and approved for oncology over the last several decades, there remains a nirking paucity of clinical success as a reminder that this war on cancer riding on the animal model is far from won. In a backbreaking attempt to analyze the failure, the limitation of the “model” system appeared to be the most rational cause of this shortcoming. It was more of a failure to test a drug rather than a failure to make a drug that stunted our collective growth and success in cancer research. PDX is the product of this age-old failure and its fitness is currently tested in virtually all organ-type solid tumors. This review will present and appraise PDX model in the context of its evolution, its future promise, its limitations and more specifically, the current content of PDX in different solid tumors including breast, lung, colorectal, prostrate, GBM, pancreatic, hepatocellular carcinoma and melanoma. Keywords: Patient Derived Xenograft Model; Solid Tumors; Breast Cancer; Lung Cancer; Colorectal Cancer; Prostrate Cancer; GBM; Pancreatic Cancer, Melanoma; Hepatocellular Cancer 1. Introduction After more than forty years of the National Cancer Act of 1971, the nation’s declaration of the “War on Cancer”, to- day, two-thirds of patients survived at least five years after being diagnosed with cancer compared with just half of all diagnosed patients surviving five years after diagnosis in 1975 [1]. The bill fueled major investments in cancer research and led to significant increases in can- cer survival. The last decade achieved significant pro- gress in cancer understanding and therapy as the cardinal genetic drivers of individual tumors can be identified, and different tailor drugs have evolved to specifically in- tercept these driver mutations/pathways [2]. The empha- sis in cancer drug development in the course of last few decades has shifted from cytotoxic, non-specific chemo- therapies to molecularly targeted, rationally designed drugs promising greater efficacy and fewer side effects [3]. Clearly, personalized oncology, an evidence-based and individualized medicine that delivers the right care to the right cancer patient at the right time, is the precious outcome of the effort of the last 50 years [4]. 2. Why We Need “the Tumor Copycat” in Mouse? In spite of significant resource expended on cancer re- search over the last half century, the contribution of newly developed therapeutics to cure the disease or to improve patient survival has been limited [3]. The major contributors in the improvements to overall survival have been either technological (genetic testing e.g., character- izing BRCA1 and BRCA2 mutations; biomarker detec- tion e.g., PSA; Tissue monitoring e.g., colonoscopy or mammography) or medical awareness (incidence of smo- king] which immensely improved survival/reduced deaths through early detection of the disease, reduced preva- lence or increased the prevention of the respective dis- *Conflict of interest: None. #Corresponding author.  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1412 eases. In contrast, only a handful of options have been made open for the treatment in solid tumors in addition to age-old treatment with untargeted chemotherapeutic drugs, surgical resection and radiotherapy as primary and often secondary courses of action in the course of last 30 years [5]. There are two major reasons for this failure. The first one is in the built-in nature and the origin of the disease, the tumor heterogeneity. The accumulated weal- th of information during the last decades in the field of molecular markers, gene expression profiling, and the more recent implementation of next-generation DNA sequencing technologies have helped disclose a broader spectrum of heterogeneity among patients presenting different tumors [6-11]. To the tumor heterogeneity add- ed more complexity is the paradigm of the cancer stem cell [12-14]. The second one is the limitation to have the perfect model for the experiment. Traditional models those evolved with the advent of genetically engineered mice (GEM) or xenograft using athymic mice appears as not fit for the survival as they have failed to predict clinical success [5]. More than 68 drugs have been de- veloped and approved for oncology over the last several decades [15]. Based on the data from the US Food and Drug Administration (FDA), company surveys, and pub- licly available commercial business intelligence data- bases on new oncology drugs approved in the United States and on investigational oncology drugs to estimate average development and regulatory approval times, cli- nical approval success rates, first-in-class status, and glo- bal market diffusion, DiMasi and Grabowski have de- termined that the market success of oncology drugs has induced a substantial amount of investment in oncology drug development in the last decade [15]. The major road block therefore in developing cancer targeted therapies is not the lack of knowledge about specific molecular driv- ers in these diseases, but is the inability to test the target- based drugs in predictable preclinical models. Even a collective effort of the scientific community failed to mend the gap between bench and bedside due to a lack of preclinical models capable of reliably predicting clinical activity of novel compounds in cancer patients [16]. This explains why only 5% of cancer therapies targeted to specific molecular drivers tested in the clinic proved ef- ficacious [17], indicating an unmet need for marked im- provement in predictive preclinical research. In an old study to assess trends in the process of global commer- cial development of cancer treatments, Reichert JM, and Wenger JB analyzed data for 1111 candidates that en- tered clinical study during 1990-2006. Their results show that although the average number of therapeutic candi- dates entering clinical study per year more than doubled, the US approval success rate was as low as 8% during the period [18]. The paucity of success stories in clinics as therapies fail far too many patients remains as a fact; the lesson learned from the use of far-from-perfect and proven to be a clinically irrelevant model [19]. From the failures or limitations of these xenograft or GEM models evolved patient derived xenograft (PDX; patient-derived xenografts; tumor graft models ), “the tu- mor copycat” in mouse, a 21st century preclinical model [5,19-22] with a promise to deliver clinically relevant data. PDX models are generated using freshly resected patient tumors immediately transplanted into immune- compromised murine hosts without an intermediate in vitro culture step [5]. Continuous passages of tumors through consecutive generations of murine hosts without in vitro cell culture permits ongoing propagation of tu- mor lines (Figure 1). The establishment of tumorgrafts constitutes a long-term process consisting of various steps, with the final objective to show that the validated model accurately reproduces human cancer, with a high predictive value of therapeutic efficacy (regardless of the type of treatment), and closely mimics clinical situations frequently observed in patients with cancer, such as resis- tance to standard treatments, metastases, and relapse after initial therapies (involving residual tumor-initiating cells) [23]. Currently subcutaneous (hindquarters) or in the mammary fat pad are more common primary xenografts sites. Transplanting under the kidney capsule or or- thotopically is also practiced as the latter of which may better replicate the tumor microenvironment than subcu- taneous models [5]. Growth of PDX tumors faithfully maintain, [1] the cellular complexity and architecture of the original tumor in its natural state complete with in- vading vasculature and supporting stromal cells and [2] the chromosomal, transcriptomic, genomic and proteo- mic architectural landscape of the original tumor [24]. In an older study using 5 human malignant tumors, trans- planted to athymic nude mice, effect of long-term serial Figure 1. Schematic representation of PDX and PD ex vivo culture; a human mouse interface at the preclinical transla- tional level. Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1413 transplantation were studied by comparing their growth patterns and chromosomal constitutions with their early appearance. After 27 - 56 passages over 3 1/2 to 5 1/2 years, all of the tumors (2 adenocarcinomas of the colon, 2 malignant melanomas, and 1 Burkitt’s lymphoma) were found to retained the cytological and histological ap- pearance. All the mitoses observed in the chromosome studies were of human karyotype. No total chromosomal species shift, no interspecies hybridization, and no changes in biological properties were observed [25]. A number of tumor-specific PDX models have been estab- lished in melanoma, breast, pancreatic, ovarian, lung, colorectal, and brain-derived tumors those exhibit bio- logical stability when passaged in mice in terms of global gene-expression patterns, molecular diversity, cellular heterogeneity, mutational status, metastatic potential, che- moresponsiveness to anti-neoplastic agents, histopathol- ogy, and tumor architecture [16,26]. Mathew Ellis group has characterized patient-derived xenografts (PDXs) for functional studies to demonstrate that the originating tu- mor genome provides a benchmark for assessing genetic drift and clonal representation after transplantation. They published whole-genome comparisons with originating breast cancers representating major intrinsic subtypes. They observed that structural and copy number aberra- tions were retained with high fidelitywhile, at the sin- gle-nucleotide level, variable numbers of PDX-specific somatic events were documented, although they were only rarely functionally significant. It is reported that va- riant allele frequencies were often preserved in the PDXs, demonstrating that clonal representation can be trans- plantable. Estrogen-receptor-positive PDXs were associ- ated with ESR1 ligand-binding-domain mutations, gene amplification, or an ESR1/YAP1 translocation. These events produced different endocrine-therapy-response phenotypes in human, cell line, and PDX endocrine-re- sponse studies. Their study concluded that deeply se- quenced PDX models are an important resource for the search for genome-forward treatment options and capture endocrine-drug-resistance etiologies that are not found in standard cell lines [27]. The success of PDX model in future will depend on its ability to reflect human disease in the experimental settings. Outcome driven predictive preclinical research in future expects PDX to model can- cer as closely as possible so that the evolution of this gene-based disease in human can be forecasted in the laboratory. Considering the complex nature of the evolu- tion of cancer with respect to its diverse molecular eti- ologies, it can be argued that morepredictably we mimic the disease in experimental settings before it happens in a patient’s body, more chance we have to clinically en- counter it. Today’s medicine in oncology is essentially genomically informed science towards selection of tar- geted therapies to treat individual patients, precision cancer medicine [28]. The preclinical component of the precision medicine demands right model to test targeted drugs directed towards genomically informed pathways, and is banking heavily on PDX. 3. Evolution to PDX The proof of concept that it is possible to successfully xenograft fragments of a patient’s tumor into nude mice way back in 1969, paved the path to find to answer a number of questions regarding the cause, prevention, drug-screening, and targeted therapy in cancer [29]. Di- rect transfer xenografts of tumor surgical specimens con- serve the inter-individual diversity and the genetic het- erogeneity typical of the tumors of origin, and yet give a unique and flexible opportunity for drug-combination for the preclinical analysis with a high extrapolative value in clinics [30]. Although the challenges like limited avail- ability of the tumor source and technical difficulties were encountered, human tumor models established in serial passages proved their superiority in predicting drug-re- sponse as well as predicting drug-resistance in clinics. Despite of technical and procedural difficulties, the use of PDX as a tool has evolved greatly from testing a more broader aspect of untargeted chemotherapeutic drug- response to addressing specific and focused challenges including a development of endocrine resistance in ER+ (estrogen receptors) subset of breast cancer (BC). As an example, Kabosetal. in their study have recently de- scribed patient-derived ER+ luminal breast tumor models for the study of intra-tumor hormone and receptor action to evoke the importance of mapping both conserved and tumor-unique ER programs in breast cancers [31]. Their study highlights the importance of modeling in widely diverse patient-derived ER+ breast cancers in vivo to advance our understanding towards improving the treat- ment of this disease. The outcome of their study demon- strates that patient-derived ER+ tumor xenografts display estrogen-dependent growth and have unique ER tran- scriptomes showing that although multiple other factors such as genetic and epigenetic signatures, and co-regu- lator expression patterns, collectively influence tumor behavior, ER remains the common denominator driving tumor growth and survival, at least initially. Since ER is retained in most drug-resistant tumors, their study pointed out the importance of determining the “switch” in acti- vated/deactivated genes and signaling networks under treatment pressure in individual tumors as the cornerstone to overcome persistent ER+ breast disease. Patient-de- rived xenografts thus provided a unique opportunity to dissect the contributions of steroid hormones and their receptors in the context of development of resistance in this subset of breast cancer. Using patient-derived xeno- grafts from metastatic colorectal carcinomas which relia- bly mimicked disease response in humans, prospectively Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1414 recapitulated biomarker-based case stratification, and identified HER2 as a predictor of resistance to anti-epi- dermal growth factor receptor antibodies Bertotti et al., studied the response to combination therapies against HER2 and epidermal growth factor receptor. This proof- of-concept, multi-arm study in HER2-amplified “xenopa- tients” revealed that the combined inhibition of HER2 and EGFR induced overt, long-lasting tumor regression and suggested a promising therapeutic opportunity in cetuxi- mab-resistant patients with metastatic colorectal cancer [30]. Another example of a recent development in the oncology research is the use of PDX models as a renew- able tissue resource of phenotypically stable, biologically and ethnically diverse breast cancers which serve as a renewable, quality-controlled tissue resource for precli- nical studies investigating treatment response and metas- tasis [32]. PDX would more accurately recapitulate the phase I/II or phase III clinical trial situation in which treat- ment is initiated on patients with advanced, high-volume metastatic disease [33]. Recently, Li et al. showed that the analysis of genetic changes in patient-derived xeno- grafts can reveal crucial details of tumor evolution, in- cluding the emergence of functional estrogen receptor mutations in endocrine-resistant breast cancer [34]. 4. PDX as Model for Drug Discovery in Different Organ Sites Solid Tumors The preservation of the patient’s tumor genomic profile and tumor microenvironment in PDX gives the opportu- nity to use primary patient tumorgrafts as a relevant mo- del to support the translation of new drug-based thera- peutic strategies in oncology [35]. PDXs maintain at least certain aspects of the human microenvironment for initial weeks with the complete substitution with host (murine) stroma occurring after 2 - 3 passages in mouse. Hence this model provided more appropriate window for studies of tumor-microenvironment interaction [36]. This is an unique property of PDX model which provides a rare insight to patients’ tumor-stromal interactions (at least under controlled conditions that can be exploited for drug discovery). In the following sections, an overview of the state of art of the use of PDX models as main experi- mental platforms to understand the biology of tumor cells, their response to drugs, and their mechanism of resis- tance to drugs will be presented in the context of differ- ent solid tumors. 4.1. PDX in Breast Cancer As early as 2007, study by Marangoni et al., established a panel of human breast cancer xenografts in immune- deficient mice suitable for pharmacologic preclinical assays. The panel consisted of breast cancer xenografts of 15 triple-negative, one ER positive and 2 ERBB2 positive tumors. Data showed that almost all patient tumors es- tablished as xenografts displayed an aggressive phenotype, i.e., high-grade, triple-negative status. The histology of the xenografts recapitulated the features of the original tumors. Mutation of p53 and inactivation of Rb and PTEN proteins were found in 83%, 30%, and 42% of PDX, re- spectively [37]. This work provided preliminary results to demonstrate the concordance between clinical outcome and response of xenografts supporting the use of human tumor xenografts for the preclinical evaluation of new compounds and predicting drug response in breast cancer. In the subsequent years a number of studies using PDX model opened different avenues in different subtypes of breast cancer. Report by de Plater’s group described es- tablishment and characterization of a breast cancer xeno- graft obtained from a woman carrying a germline BRCA2 mutation [38]. A transplantable xenograft was obtained by grafting the sample into nude mice and the biological and genetic profiles of the xenograft were compared with that of the patient's tumor in terms of histology, immunohis- tochemistry (IHC), BRCA2 sequencing, comparative genomic hybridisation (CGH), and qRT-PCR. Tumor responses to standard chemotherapies including sensitiv- ity to anthracyclin-based chemotherapy, radiotherapy and cisplatin-based treatments as well as resistance to do- cetaxel were also evaluated. Since PDX preserves the genomic landscape of the tumor, the aggressive triple negative breast tumors were more frequently modeled in PDX compared to the other subtypes of breast cancer. Romanelli et al., used PDX model to demonstrate that in- hibition of aurora kinases reduces tumor growth and suppresses tumor recurrence after chemotherapy in triple negative breast cancer [39]. Although statistically more PDX models were established in the triple negative sub- sets of breast cancers, the retention of hormone receptor heterogeneity has been reported in luminal PDX [31]. In this study, five transplantable patient-derived ER+ breast cancer xenografts established from tumors derived from both primary and metastatic cases were assessed for es- trogen dependency, steroid receptor expression, cancer stem cell content, and endocrine therapy response. Gene expression patterns were determined in select tumors ± estrogen and ±endocrine therapy. Xenografts morpho- logically resembled the patient tumors of origin, and ex- pressed similar levels of ER (5% - 99%), and progester- one and androgen receptors, over multiple passages. Analysis of the ER transcriptome in select tumors re- vealed notable differences in ER mechanism of action, and downstream activated signaling networks, in addition to identifying a small set of common estrogen-regulated genes. These results evoked the importance of mapping both conserved and tumor-unique ER programs in breast cancers and helped to define unique estrogen-dependent gene signatures. In another study, response to hormone Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1415 therapy was evaluated in 6 luminal PDX models. The result showed different sensitivities, thus exhibiting het- erogeneity similar to what is encountered in the clinic. The data demonstrated that the primary human luminal breast cancer xenografts, recapitulates the biological and clinical behaviors of patient tumors, and therefore can be suitable for preclinical experiments [40]. DNA copy num- ber analysis and gene expression analysis were carried out by Reyal et al., using Affymetrix Microarrays comparing PDX and patient’s original tumors for the molecular characterization. Comparison analysis showed that 14/18 pairs of tumors shared more than 56% of copy number alterations (CNA). Unsupervised hierarchical clustering analysis showed that 16/18 pairs segregated together, confirming the similarity between tumor pairs. Analysis of recurrent CNA changes between patient tumors and xenografts showed losses in 176 chromosomal regions and gains in 202 chromosomal regions. Interestingly, it was demonstrated that less than 5% of genes had recurrent variations between patient tumors and their respective xenografts; these genes largely corresponded to human stromal compartment genes. Different passages of the same tumor showed that sequential mouse-to-mouse tu- mor grafts did not affect genomic rearrangements or gene expression profiles, suggesting genetic stability of these models over time [24]. Based on these studies and others [41], patient-derived tumors were used to assess efficacy of GDC-0941 (pan PI3K inhibitor developed by Genen- tech Inc., CA) and docetaxel in vivo to show that GDC- 0941 augments the efficacy of docetaxel by increasing drug-induced apoptosis in breast cancer models [42]. Breast cancer like other solid tumors possesses a rare population of cells capable of extensive self-renewal that contributes to metastasis and treatment resistance. PDX model has been used to test the contribution of cancer stem cells. Three primary human breast cancer xenografts generated from 3 different patients. This study by Gi- nestier et al., demonstrated that CXCR1 blockade selec- tively targets human breast cancer stem cells in vitro and in xenografts and suggested that combination therapy using conventional chemotherapy and drugs against can- cer stem cells specific targets can lead to better therapeu- tic results, both in terms of tumor growth as well as in terms of tumor relapse [43]. 4.2. PDX in Lung Cancer PDX models of non small cell lung cancer (NSCLC), established following direct implants of lung cancer tissue fragments in immune-compromised mice has been used for targeted therapies and new drug development [36]. In non-small cell lung cancer, the ability to form primary tumor xenografts is itself demonstrated to have a predic- tive value of increased risk of disease recurrence in early- stage. Thus, there is a correlation between the ability of the tumor to form PDXs and the risk of disease recurrence in early stage. In this study, xenografts were established and passaged successfully from 63 of 157 (40%) im- planted tumor fragments from non-small cell lung cancer patients undergoing curative surgery into NOD-SCID (nonobese diabetic-severely combined immunodeficient) mice [44]. Tumor factors associated with engraftment included squamous histology, poor differentiation, and larger tumor size. Interestingly, there was a correlation between the success of PDX model establishment and mutation status of the tumor. Significantly fewer EGFR (epidermal growth factor receptor)-mutated tumors were engrafted (P = 0.03) than KRAS-mutated tumors (P = 0.05). In an earlier study, similar results have been ob- tained in early passages of the non-small cell lung cancer xenografts which revealed a high degree of similarity with the original clinical tumor sample with regard to histology, immunohistochemistry, as well as mutation status [45]. Even the chemotherapeutic responsiveness of the xenografts resembled the clinical responses as the shrinkage of tumor was obtained with paclitaxel (4 of 25), gemcitabine (3 of 25), and carboplatin (3 of 25) with a lower effectiveness of etoposide (1 of 25) and vinorel- bine (0 of 11). Although, the response to the anti-EGFR therapies did not correlate with mutations in the EGFR or p53, but there appeared a correlation of K-Ras mutations and erlotinib resistance. In a recent study, PDX models were established based on first generation non-small cell lung cancer subrenal capsule xenografts, which provided suitable platform for quick assessment (6 - 8 weeks) of the chemosensitivity of patients’ cancers and selection of the most effective regimens [46]. In this study, xenografts were established at a very high engraftment rate (90%) with the retention of major biological characteristics of the original cancers. PDX provided a model to test the drug sensitiveness in human tumor-stromal settings. Thus it provided a tool to find out the drug resistance cells with tumor whose contribution can be extrapolated in terms of tumor recurrence as it frequently occurs in pa- tients after partial or even complete response. This is important in the context of the cancer stem cell hypothe- sis of the presence of small subpopulations of tumor ini- tiating cells within the tumor which is proposed to ex- plain tumor heterogeneity and the carcinogenesis process, pre and post drug treatment. In fact, in a study by Krum- bach et al., primary resistance to cetuximab in a panel of patient-derived tumourxenograft models was observed via activation of MET(receptor for hepatocyte growth factor) as one mechanism for drug resistance indicating that at the preclinical level, a combined treatments of a MET inhibitor and cetuximabare additive [47]. The study was undertaken with the background fact that despite wide expression of EGFR, only a subgroup of cancer patients responds to cetuximab therapy. They assessed the Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1416 cetuximab response in vivo of 79 human patient-derived xenografts originating from five tumour histotypes. A cetuximab response score including positive and negative factors affecting therapeutic response is proposed in the study. In cetuximab resistant non small cell lung adeno- carcinoma, overexpression due to gene amplification and strong activation of MET was identified. Recently, ten passable patient-derived non small cell lung carcinoma xenograft models were established containing a variety of genetic aberrations including EGFR activating mutation, KRAS mutation, and FGFR1 and cMET amplification [48]. Anti-tumor efficacy of gefitinib in this study dem- onstrated that the EGFR activating mutation model had superior sensitivity and that the KRAS mutation models were resistant to gefitinib, which were consistent with the results reported from clinical trials. Also, models with FGFR1 gene amplification were found insensitive to ge- fitinib treatment. In the case of overcoming resistance, report by Yang et al., demonstrated a novel practical ap- proaches to overcome the two most common resistances to EGFR-TKIs seen in the clinic using marketed target therapies with the help of PDX model [49]. A tailored treatment regimen was tested using patient-derived xeno- grafts from naïve Asian non small cell lung cancer pa- tients including those containing “classic” EGFR activate- ing mutations towards overcoming erlotinib resistance. Using different combination of drugs and their response rate in the PDX model the study was able to identify the main drivers of resistance in patients. 4.3. PDX in Colorectal Cancer A step forward towards personalized (evidence-based) medicine for patients with colorectal cancer (CRC) was achieved following the work of Bertotti et al., who re- ported a molecularly annotated platform of PDX (“xeno- patients”) which identified HER2 as an effective thera- peutic target in cetuximab-resistant colorectal cancer [50]. It is known that only a fraction of patients with metastatic colorectal cancer receive clinical benefit from therapy with anti-epidermal growth factor receptor (EGFR) anti- bodies, indicating the urgent need to identify novel bio- markers for better personalized medicine. Bertotti et al., produced xenograft cohorts from 85 patient-derived, ge- netically characterized metastatic colorectal cancer sam- ples (“xenopatients”) to identify novel determinants of therapeutic response and new oncoprotein targets. Serially passaged tumors retained the morphologic and genomic features of their original counterparts. Xenopatients re- sponded to the anti-EGFR antibody cetuximab with rates and extents analogous to those observed in the clinic which were prospectively stratified as responders or non- responders on the basis of several predictive biomarkers. Genotype-response correlations indicated HER2 ampli- fication specifically in a subset of cetuximab-resistant, KRAS/NRAS/BRAF/PIK3CA wild-type cases. Impor- tantly, HER2 amplification was also enriched in clinically nonresponsive KRAS wild-type patients. Their study indicated that PDX models could be utilized successfully to determine the biomarkers in the drug-resistance condi- tions with different genetic backgrounds. Julien et al., comprehensively characterized a large panel of patient- derived tumor xenografts representing the clinical het- erogeneity of human colorectal cancer [51]showing that their collection recapitulates the clinical situation about the histopathology and molecular diversity of CRC. It was observed that patient tumors and corresponding models are clustering together allowing comparison studies be- tween clinical and preclinical data. Based on this result, they also conducted pharmacologic mono-therapy studies with standard of care for CRC (5-fluorouracil, oxaliplatin, irinotecan, and cetuximab). Through an experimental cetuximab phase II trial, we confirmed the key role of KRAS mutation in cetuximab resistance. They reported the loss of human stromal cells after engraftment, ob- served a metastatic phenotype in some models, and finally compared the molecular profile with the drug sensitivity of each tumor model. In colorectal cancer, PDX model has also been used to report a suppression of the growth of tumors by an adenovector expressing small hairpin RNA targeting Bcl-XL[52]. The result showed that Ad/Bcl-XL shRNA with or without 5-Fu has effective anti-tumor effects on the patient tumor-derived rectal cancer xeno- grafts. 4.4. PDX in Prostate Cancer Toivanen et al., reviewed the current status of xenograft- ing human primary prostate cancer, and their potential application to translational research [53]. Earlier, pa- tient-derived intra-femoral xenograft model of bone me- tastatic prostate cancer that recapitulates mixed osteolytic and osteoblastic lesions has been reported by Raheem et al., [54]. In their study, xenograft tumors were developed from a femoral bone metastasis of prostate cancer (re- moved during hemiarthroplastywhich was transplanted into Rag2(-/-); γc(-/-) mice either intra-femorally or sub cutaneously) were analyzed and validated for prostate cancer biomarker expression. Similarly, Aparicio et al., reported derivatization of neuroendocrine prostate cancer xenografts with large-cell and small-cell features from a single patient’s tumor [55].In another study, an andro- gen-dependent prostate cancer xenograft model was de- rived from a metastatic skin lesion of a Japanese hormone- refractory prostate cancer (HRPC) patient with poorly differentiated prostatic adenocarcinoma. The model ex- pressed wild-type AR and PSA and showed androgen dependence [56]. PDX model had been proved useful for the development of new therapies for androgen abla- tion-resistant prostate cancers. Previously, Yosida et al., Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1417 established a serially transplantable human prostate can- cer xenograft model from liver metastatic tissue of a pa- tient treated with antiandrogen bicalutamide which ex- pressed the AR with a point mutation at amino acid 741 (tryptophan to cysteine; W741C) in the ligand-binding domain[57]. This mutation was also present in cancerous tissue used for generation of xenograft. Data showed that agonistic effect of bicalutamide to a xenograft with clinically induced AR mutation. Although the growth of KUCaP in male mice was androgen dependent, bicalu- tamide aberrantly promoted the growth and prostate- specific antigen production of KUCaP. Thus the bicalu- tamide-responsive mutant AR may serve in the develop- ment of new therapies for androgen ablation-resistant prostate cancers. PDX model provided experimental model to test the effect of kava root extract (Piper me- thysticum Forst is a perennial plant indigenous to the Pa- cific Islands) and flavokawain B on the tumor growth. The kava root extract and flavokawain B reduced tumor growth, AR expression in tumor tissues and levels of serum PSA in the patient-derived prostate cancer xeno- graft models suggesting a potential usefulness of a safe kava product or its active components for prevention and treatment of advanced prostate cancer by targeting AR [58]. 4.5. PDX in GBM Recently, high-resolution mutational profiling suggested the genetic validity of glioblastoma patient-derived pre- clinical models. In a comprehensive study, Yost et al., identified somatic coding mutations and copy number aberrations in four glioblastoma (GBM) primary tumors and their matched pre-clinical models: serum-free neu- rospheres, adherent cell cultures, and mouse xenografts. Their analysis identified known GBM mutations altering PTEN and TP53 genes, and new actionable mutations such as the loss of PIK3R1, and revealed clear patient- to-patient differences. They observed approximately 96% primary-to-model concordance in copy number calls in the high-cellularity samples [59]. Jarzabek et al., had shown that a xenograft model based on serial xenotrans- plantation of human biopsy spheroids in immune-defi- cient rodents maintains the genotype and phenotype of the original patient tumor [60]. Based on their study they later reported an in vivo bioluminescence imaging vali- dation of a human biopsy-derived orthotopic mouse mo- del of glioblastomamultiforme. PDX model has been utilized to identify the role of Wnt/β-catenin signaling as a key downstream effector of MET signaling and con- tributor of GBM malignancy and the maintenance of glioblastoma stem cells [61]. Kim et al., have identified Wnt/β-catenin signaling pathway as one of the pathways that is enriched in MET(high/+) cells populations com- pared with bulk tumor cells in the established a series of GSCs and xenograft tumors derived from freshly disso- ciated specimens from patients with GBM. 4.6. PDX in Hepatocellular Carcinoma Xenografts of human hepatocellular carcinoma SCID mice have been reported by Tran’s group [62] and by other groups [63]. These established patient-derived hepato- cellular carcinoma xenografts were extensively used in later years for the testing of different drugs including AZD6244 (MEK Inhibitor), RAD001 (everolimus), Suni- tinib (a potent inhibitor of two receptors involved in an- giogenesis—vascular endothelial growth factor receptor and platelet-derived growth factor receptor PDGFR), and Brivanibalaninate (a dual inhibitor of vascular endothe- lial growth factor receptor and fibroblast growth factor receptor tyrosine kinases) at the preclinical setting [64- 67]. Huynh et al., treated patient-derived HCC xeno- grafts with 1) Sorafenib (a small molecule inhibitor of several receptor tyrosine kinases including VEGFR, PDGFR, c-Kit and RAF-kinase), 2) AZD6244 (ARRY- 142886), and 3) Sorafenib plus AZD6244. The study showed that the pharmacological inhibition of the MEK/ ERK pathway by AZD6244 enhanced the anti-tumor effect of Sorafenib in both orthotopic and ectopic models of hepatocellular carcinoma underscoring the potential of a combined therapeutic approach with Sorafenib and MEK inhibitors in the treatment of hepatocellular carci- noma [64]. 4.7. PDX in Melanoma As early as 2004, Krepler et al., evaluated the in vivo anti-tumoral effects of CpG oligonucleotides against hu- man malignant melanomaxenografts in NOD/SCID mice and demonstrated the antitumor activity of oligonucleo- tides containing immune-stimulatory CpG motifs in a xeno-transplantation model with absent B, T cells and a lack of natural killer (NK) cell function [68]. CpG oli- gonucleotides administered in single peri-tumoral subcu- taneous injections three times per week resulted in ele- vated plasma levels of interleukin-12 and significant in- hibition of the growth of established tumor xenografts by 60% (p < 0.016) compared to the placebo control. Pa- tient-derived tumor xenografts engrafted in immune- compromised mice have been proposed as valuable pre- clinical models in melanoma that can predict clinical response to treatments[69]. Patient-derived tumor xeno- graft model was utilized to guide the use of BRAF in- hibitors in metastatic melanoma. Recently, Guerreschi et al., established a PDX model of BRAF V600E mela- noma useful for testing the efficacy of Vemurafenib (B-RAF enzyme inhibitor developed by Plexxicon and Genentech Inc.) [70]. They validated the stability of the model that was similar to the original tumor with respect Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1418 to histology, immunohistochemistry, mutational status, and fluorine-18 fluorodeoxyglucose ([F]FDG)-PET/com- puted tomography (CT). The sensitivity of the xenografts to Vemurafenib was determined by tumor growth inhibi- tion and decreased in standardized uptake value on [F] FDG-PET/CT. Their result allowed successful rechal- lenge with Vemurafenib in a patient who was adminis- tered a lower dose of Vemurafenib because of the onset of adverse events. Authors claimed that their study found that PDX provides “real-time” results in an animal that phenocopies the biology and expected Vemurafenib re- sponses of the tumor in a patient with BRAF V600E melanoma and this “coclinical” trial using PDX appears to guide Vemurafenib treatment for metastatic melanoma. PDX has been also established in uveal melanoma. The uveal melanoma xenografts in immunodeficient mice have been reported to accurately recapitulate the genetic features of primary human uveal melanoma and they exhibited genetic stability over the course of their in vivo maintenance [71]. These models constitute valuable pre- clinical tool for drug screening in uveal melanoma. Es- tablishment and characterization of a panel of human uveal melanoma xenografts derived from metastatic tu- mors has also been reported [72]. It has been observed that the establishment rate of human uveal melanoma in their study was 28% (25 of 90) which was independent of size, histologic parameters, or chromosome 3 mono- omy but was significantly higher in metastatic tumors as compared to the primary tumors. In vivo tumor growth was found prognostic for a lower metastasis-free survival in patients with primary tumors. There was a high con- cordance between the patients’ tumors and their corre- sponding xenografts for all parameters tested (histology, genetic profile, and tumor antigen expression). Interest- ingly, the four xenografts studied displayed different response profiles to chemotherapeutic agents. 4.8. PDX in Pancreatic Cancer Recently, PDX model has been used to isolate and cul- ture rare cancer stem cells CSC) from pancreatic ductal adenocarcinoma [73]. Pancreatic tumor cells from pa- tient-derived xenografts were screened for the presence of surface markers of pancreatic CSCs, CD24, CD44, and CD326. Following cell isolation and culture, 35% of sorted human xenograft cells formed tumor spheroids retaining high expression levels of CD24, CD44, and CD326. In another study, the efficacy of inhibition of EGFR/HER2 receptors and the downstream KRAS ef- fector, mitogen-activated protein kinase/extracellular- signal regulated kinase (ERK) kinase 1 and 2 (MEK1/2), on pancreatic cancer proliferation was tested following a combination of MEK inhibitor trametinib and lapatinib using pancreatic PDX model [74]. An inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is shown to be aug- mented by combined treatment with the epidermal grow- th factor receptor/HER2 inhibitor lapatinib. Data indi- cated that inhibition of the EGFR family receptor signal- ing may contribute to the effectiveness of MEK1/2 inhi- bition of tumor growth possibly through the inhibition of feedback activation of receptor tyrosine kinases in re- sponse to inhibition of the RAS-RAF-MEK-ERK path- way. In an earlier study, Rajeshkumar et al., reported a collection of freshly generated patient-derived pancreatic ductal adenocarcinoma xenografts were used to test the effect of gemcitabine, the first-line chemotherapeutic agent for pancreatic ductal adenocarcinoma, which ini- tially proved effective in reducing tumor size. However gemcitabine was largely ineffective in diminishing the CSC populations, and eventually culminated in tumor relapse. Since death receptor 5 (DR5) was found to be enriched in pancreatic CSCs compared with the bulk of the tumor cells, a combination of tigatuzumab, a fully hu- manized DR5 agonist monoclonal antibody, with gem- citabine was more efficacious by providing a double hit to kill both CSCs and bulk tumor cells. This combination therapy produced remarkable reduction in pancreatic CSCs, tumor remissions, and significant improvements in time to tumor progression in a model that is consid- ered more difficult to treat. Thus data provided the ra- tionale to explore the DR5-directed therapies in combi- nation with chemotherapy as a therapeutic option to im- prove the current standard of care for pancreatic cancer patients [75]. A molecular profiling of direct xenograft tumors established from human pancreatic adenocarci- noma. Engraftment of human pancreatic tumors into im- munodeficient mice prior to and following neoadjuvant therapy was demonstrated by Kim et al., which provided an in vivo platform for comparison of global gene ex- pression patterns [76]. Recently, a prospective and ran- domized testing was reported in a set of almost 200 sub- cutaneous and orthotopic implanted whole-tissue primary human tumor xenografts in pancreatic ductal adenocar- cinoma [77]. The most pronounced therapeutic effects were observed in gemcitabine-resistant patient-derived tumors. Intriguingly, the proposed triple therapy ap- proach could be further enhanced by using a PEGylated formulation of gemcitabine, which significantly increas- ed its bioavailability and tissue penetration, resulting in a further improved overall outcome. The study demonstrat- ed that a multimodal (combining chemotherapy, hedge- hog pathway inhibition, and mTOR inhibition) treatment can eliminate cancer stem cells and leads to long-term survival in primary human pancreatic cancer tissue xeno- grafts. PDXs as a tool for cancer stem cell studies have been well developed for pancreatic cancer. A direct pan- creatic cancer xenograft model has been used as a plat- Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal Open Access JCT 1419 form for cancer stem cell therapeutic development and to bridge the enormous gap between the anti-proliferative and in vivo antitumor efficacy of gemcitabine in cell line-based models and its clinical efficacy [78]. Jimeno et al. demonstrated that the chemotherapeutic treatment of pancreatic PDXs caused an increase of cancer stem cell markers including ALDH and CD24 in the residual tu- mor population, proving the hypothesis of an obviously enhanced chemo-resistance of the cancer stem cells sub- population. Combined treatment with gemcitabine and cyclopamine induced tumor regression and decrease in cancer stem cell markers and hedgehog signaling. Hed- gehog inhibitors were able to further reduce tumor grow- th and decreased both static and dynamic markers of can- cer stem cell. Their study proved that direct tumor xeno- grafts are a valid platform to test multi-compartment therapeutic approaches in pancreatic cancer. Similar ef- fects were reported with PDXs derived from other can- certypes, including colorectal cancer [79]. 5. Limitations of PDX Model Like any other model system, PDX has limitations. The pros and cons of the PDX model have been schematically presented in Figure 2. First, PDX “do not” and “cannot” represent human immune response neither from the tu- mor side nor from the host side. The researcher and phy- sicians scientists will have to either use a next generation sophistication to extrapolate/simulate the immune com- ponent or they have to rationally couple the inference drawn from the cancer immunologists in circumventing this built-in limitation of the model. Second, the estab- lishment of PDX is neither cost effective nor human re- source friendly. The burden of cost, infrastructure and trained human resource is certainly a primary hold back factor in the development, growth and the future evolu- tion of PDX model. However, considering 1) the limita- tions of different existing models (Figure 3), 2) the un- met need to treatment different challenging aspects of cancer including metastasis, stem cell involvement, tu- mor dormancy and drug resistance, PDX model remains our best hope in the preclinical translational research. 6. PDX Is Here to Stay: Future Perspectives PDX has a promise to fulfill the primary goal of cancer biologists to better understand tumorigenesis and cancer progression over time which is the most formidable challenge in cancer research [5]. PDX provides, more realistic, gemonically stable, hispopathologically faithfull platform of study than any other animal model of cancer with a unique flexilibily for drug testing for various pur- poses, 1) tumor shrinkage, 2) development of drug re- sistance 3) metastasis and stem cell studies (Figure 4). The preclinical setting in which PDXs can be relevant is represented by the evaluation of the potential of new drugs in cancer treatment as PDX faithfully represents the heterogeneity of a cancer type as well as representing their various subcategories on one hand and provides consistent genomic landscape on the other hand. For the same reason the PDX model represents an improved model to study the evolution of a tumor and development of drug resistance. The uniqueness of the PDX is its ca- pability to retain the genetic stability of the tumor as well as capability to evolve in the experimental settings as closely as the tumor evolves and respond/resists to drugs Figure 2. Promise of PDX model. 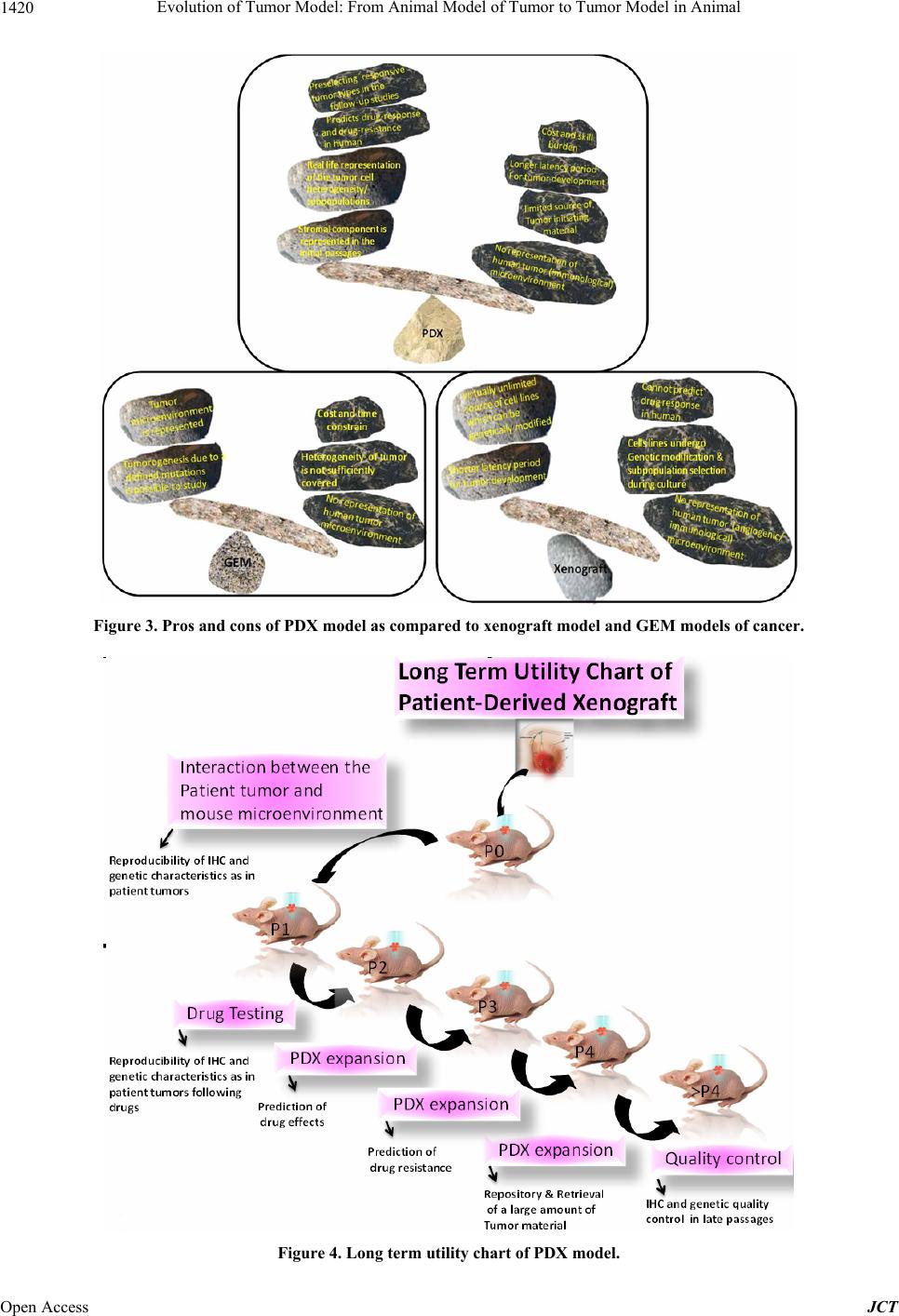 Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1420 Figure 3. Pros and cons of PDX model as compared to xenograft model and GEM models of cancer. Figure 4. Long term utility chart of PDX model. Open Access JCT  Current Distortion Evaluation in Traction 4Q Constant Switching Frequency Converters 1421 in patients. Today’s precision medicine will have to de- pend on the power and promise of PDX model. 7. Conclusion Mouse models are invaluable tools for preclinical eva- luation of new therapeutic strategies in cancer. The enor- mous burden of cost and a high failure rate of cancer drug development frankly highlight the need for new preclinical strategies and resources. PDX models from patient-derived tumor tissue at low passage have proven to conserve original tumor characteristics such as het- erogeneous histopathology, clinical bio-molecular signa- ture, malignant phenotypes and genotypes, tumor archi- tecture, gene expression profiles and tumor vasculature. Based on this hypothesis, primary tumor xenografts have shown to provide relevant predictive insights into clinical outcomes when evaluating the worth of new cancer therapies. 8. Acknowledgements Authors acknowledge Edith Sanford Breast Cancer Re- search, Sanford Research, Sioux Falls, SD. 9. Review Criteria The information for this review is compiled in part by searching the PubMed database for full length research articles and review articles those were published before October, 1st 2013. Electronic early-release publications listed in these databases are also included. Only articles published in English are considered. The search terms used included “patient derived xenograft” and “PDX model” in association with the following search terms: “Breast cancer”, “Colorectal cancer”, “Lung cancer”, “GBM”, “Tumor growth”, “Hepatocellular carcinoma”, “Melanoma” “Pancreatic Cancer”, “Prostrate Cancer”, “Angiogenesis”, and “Therapeutics”. REFERENCES [1] N. J. Vogelzang, et al., “Clinical Cancer Advances 2011: Annual Report on Progress against Cancer from the American Society of Clinical Oncology,” Journal of Cli- nical Oncology: Official Journal of the American Society of Clinical Oncology, Vol. 30, No. 1, 2012, pp. 88-109. [2] Y. Yarden and C. Caldes, “Basic Cancer Research Is Es- sential for the Success of Personalised Medicine,” Euro- pean Journal of Cancer, Vol. 49, No. 12, 2013, pp. 2619- 2620. http://dx.doi.org/10.1016/j.ejca.2013.04.020 [3] J. S. de Bono and A. Ashworth, “Translating Cancer Re- search into Targeted Therapeutics,” Nature, Vol. 467, No. 7315, 2010, pp. 543-549. http://dx.doi.org/10.1038/nature09339 [4] M. Kalia, “Personalized Oncology: Recent Advances and Future Challenges,” Metabolism: Clinical and Experi- mental, Vol. 62, No. Suppl 1, 2013, pp. S11-S14. http://dx.doi.org/10.1016/j.metabol.2012.08.016 [5] S. A. Williams, W. C. Anderson, M. T. Santaguida and S. J. Dylla, “Patient-Derived Xenografts, the Cancer Stem Cell Paradigm, and Cancer Pathobiology in the 21st Cen- tury,” Laboratory Investigation; A Journal of Technical Methods and Pathology, Vol. 93, No. 9, 2013, pp. 970- 982. [6] E. R. Mardis, “Genome Sequencing and Cancer,” Current Opinion in Genetics & Development, Vol. 22, No. 3, 2012, pp. 245-250. http://dx.doi.org/10.1016/j.gde.2012.03.005 [7] J. A. Baron, “Screening for Cancer with Molecular Mark- ers: Progress Comes with Potential Problems,” Nature Reviews. Cancer, Vol. 12, No. 5, 2012, pp. 368-371. http://dx.doi.org/10.1038/nrc3260 [8] A. Prat and C. M. Perou, “Deconstructing the Molecular Portraits of Breast Cancer,” Molecular Oncology, Vol. 5, No. 1, 2011, pp. 5-23. http://dx.doi.org/10.1016/j.molonc.2010.11.003 [9] R. Fisher, L. Pusztai and C. Swanton, “Cancer Heteroge- neity: Implications for Targeted Therapeutics,” British Journal of Cancer, Vol. 108, No. 3, 2013, pp. 479-485. http://dx.doi.org/10.1038/bjc.2012.581 [10] M. J. Ellis and C. M. Perou, “The Genomic Landscape of Breast Cancer as a Therapeutic Roadmap,” Cancer Dis- covery, Vol. 3, No. 1, 2013, pp. 27-34. [11] Cancer Genome Atlas Network, “Comprehensive Mole- cular Portraits of Human Breast Tumours,” Nature, Vol. 490, No. 7418, 2012, pp. 61-70. http://dx.doi.org/10.1038/nature11412 [12] M. F. Clarke, et al., “Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells,” Cancer Research, Vol. 66, No. 19, 2006, pp. 9339-9344. http://dx.doi.org/10.1158/0008-5472.CAN-06-3126 [13] C. A. O’Brien, A. Kreso and C. H. Jamieson, “Cancer Stem Cells and Self-Renewal,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 16, No. 12, 2010, pp. 3113-3120. [14] P. Valent, et al., “Cancer Stem Cell Definitions and Ter- minology: The Devil Is in the Details,” Nature Reviews. Cancer, Vol. 12, No. 11, 2012, pp. 767-775. http://dx.doi.org/10.1038/nrc3368 [15] J. A. DiMasi and H. G. Grabowski, “Economics of New Oncology Drug Development,” Journal of Clinical Onco- logy: Official Journal of the American Society of Clinical Oncology, Vol. 25, No. 2, 2007, pp. 209-216. [16] J. J. Tentler, et al., “Patient-Derived Tumour Xenografts as Models for Oncology Drug Development,” Nature Re- views. Clinical Oncology, Vol. 9, No. 6, 2012, pp. 338- 350. [17] T. Van Dyke, “Finding the Tumor Copycat: Approximat- ing a Human Cancer,” Nature Medicine, Vol. 16, No. 9, 2010, pp. 976-977. http://dx.doi.org/10.1038/nm0910-976 [18] J. M. Reichert and J. B. Wenger “Development Trends Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1422 for New Cancer Therapeutics and Vaccines,” Drug Dis- covery Today, Vol. 13, No. 1-2, 2008, pp. 30-37. http://dx.doi.org/10.1016/j.drudis.2007.09.003 [19] L. M. Ellis and I. J. Fidler “Finding the Tumor Copycat. Therapy Fails, Patients Don’t,” Nature Medicine, Vol. 16, No. 9, 2010, pp. 974-975. http://dx.doi.org/10.1038/nm0910-974 [20] D. Siolas and G. J. Hannon “Patient-Derived Tumor Xeno- grafts: Transforming Clinical Samples into Mouse Mod- els,” Cancer Research, Vol. 73, No. 17, 2013, pp. 5315- 5319. http://dx.doi.org/10.1158/0008-5472.CAN-13-1069 [21] K. Jin, et al., “Patient-Derived Human Tumour Tissue Xenografts in Immunodeficient Mice: A Systematic Re- view,” Clinical & Translational Oncology: Official Pub- lication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico, Vol. 12, No. 7, 2010, pp. 473-480. [22] C. L. Morton and P. J. Houghton, “Establishment of Hu- man Tumor Xenografts in Immunodeficient Mice,” Na- ture Protocols, Vol. 2, No. 2, 2007, pp. 247-250. http://dx.doi.org/10.1038/nprot.2007.25 [23] D. Decaudin, “Primary Human Tumor Xenografted Mod- els (Tumorgrafts) for Good Management of Patients with Cancer,” Anticancer Drugs, Vol. 22, No. 9, 2011, pp. 827-841. http://dx.doi.org/10.1097/CAD.0b013e3283475f70 [24] F. Reyal, et al., “Molecular Profiling of Patient-Derived Breast Cancer Xenografts,” Breast Cancer Research: BCR, Vol. 14, No. 1, 2012, p. R11. http://dx.doi.org/10.1186/bcr3095 [25] C. O. Povlsen, J. Visfeldt, J. Rygaard and G. Jensen, “Growth Patterns and Chromosome Constitutions of Hu- man Malignant Tumours after Long-Term Serial Trans- plantation in Nude Mice,” Acta Pathologica et Microbi- ologica Scandinavica. Section A, Pathology, Vol. 83, No. 6, 1975, pp. 709-716. [26] S. Kopetz, R. Lemos and G. Powis, “The Promise of Pa- tient-Derived Xenografts: The Best Laid Plans of Mice and Men,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 18, No. 19, 2012, pp. 5160-5162. [27] S. Li, et al., “Endocrine-Therapy-Resistant ESR1 Vari- ants Revealed by Genomic Characterization of Breast- Cancer-Derived Xenografts,” Cell Reports, Vol. 4, No. 6, 2013, pp. 1116-1130. [28] J. Mendelsohn, “Personalizing Oncology: Perspectives and Prospects,” Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, Vol. 31, No. 15, 2013, pp. 1904-1911. [29] Fiebig H-H & Burger AM (Human Tumor Xenografts and Explants, Springer, 2002. [30] A. Bertotti, et al., “A Molecularly Annotated Platform of Patient-Derived Xenografts (Xenopatients) Identifies HER2 as an Effective Therapeutic Target in Cetuximab- Resistant Colorectal Cancer,” Cancer Discovery, Vol. 1, No. 6, 2011, pp. 508-523. [31] P. Kabos, et al., “Patient-Derived Luminal Breast Cancer Xenografts Retain Hormone Receptor Heterogeneity and Help Define Unique Estrogen-Dependent Gene Signa- tures,” Breast Cancer Research and Treatment, Vol. 135, No. 2, 2012, pp. 415-432. http://dx.doi.org/10.1007/s10549-012-2164-8 [32] X. Zhang, et al., “A Renewable Tissue Resource of Phe- notypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Mod- els,” Cancer Research, Vol. 73, No. 15, 2013, pp. 4885- 4897. http://dx.doi.org/10.1158/0008-5472.CAN-12-4081 [33] R. S. Kerbel, “Human Tumor Xenografts as Predictive Preclinical Models for Anticancer Drug Activity in Hu- mans: Better than Commonly Perceived, But They Can Be Improved,” Cancer Biology & Therapy, Vol. 2, No. 4, 2003, pp. S134-S139. [34] S. Oesterreich, A. M. Brufsky and N. E. Davidson, “Us- ing Mice to Treat (Wo)men: Mining Genetic Changes in Patient Xenografts to Attack Breast Cancer,” Cell Reports, Vol. 4, No. 6, 2013, pp. 1061-1062. [35] D. J. Monsma, et al., “Genomic Characterization of Ex- plant Tumorgraft Models Derived from Fresh Patient Tu- mor Tissue,” Journal of Translational Medicine, Vol. 10, No. 1, 2012, p. 125. http://dx.doi.org/10.1186/1479-5876-10-125 [36] M. Moro, G. Bertolini, M. Tortoreto, U. Pastorino, G. Sozzi and L. Roz, “Patient-Derived Xenografts of Non Small Cell Lung Cancer: Resurgence of an Old Model for Investigation of Modern Concepts of Tailored Therapy and Cancer Stem Cells,” Journal of Biomedicine & Bio- Technology, Vol. 2012, No. 2012, 2012, Article ID: 568567. http://dx.doi.org/10.1155/2012/568567 [37] E. Marangoni, A. Vincent-Salomon, N. Auger, A. De- georges, F. Assayag, P. de Cremoux, L. de Plater, C. Guyader, G. De Pinieux, J. G. Judde, M. Rebucci, C. Tran-Perennou, X. Sastre-Garau, B. Sigal-Zafrani, O. Delattre, V. Diéras and M. F. Poupon, “A New Model of Patient Tumor-Derived Breast Cancer Xenografts for Pre- clinical Assays,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 13, No. 13, 2007, pp. 3989-3998. [38] L. de Plater, A. Laugé, C. Guyader, M.-F. Poupon, F. As- sayag, P. de Cremoux, A. Vincent-Salomon, D. Stoppa-Ly- onnet, B. Sigal-Zafrani, J.-J. Fontaine, R. Brough, C. J. Lord, A. Ashworth, P. Cottu, D. Decaudin and E. Marangoni, “Establishment and Characterisation of a New Breast Can- cer Xenograft Obtained from a Woman Carrying a Germ- line BRCA2 Mutation,” British Journal of Cancer, Vol. 103, No. 8, 2010, pp. 1192-1200. http://dx.doi.org/10.1038/sj.bjc.6605900 [39] A. Romanelli, A. Clark, F. Assayag, S. Chateau-Joubert, M.-F. Poupon, J.-L. Servely, J.-J. Fontaine, X. H. Liu, E. Spooner, S. Goodstal, P. de Cremoux, I. Bièche, D. De- caudin and E. Marangon, “Inhibiting Aurora Kinases Re- duces Tumor Growth and Suppresses Tumor Recurrence after Chemotherapy in Patient-Derived Triple-Negative Breast Cancer Xenografts,” Molecular Cancer Therapeu- tics, Vol. 11, No. 12, 2012, pp. 2693-2703. http://dx.doi.org/10.1158/1535-7163.MCT-12-0441-T [40] P. Cottu, E. Marangoni, F. Assayag, P. de Cremoux, A. Vin- cent-Salomon, Ch. Guyader, L. de Plater, C. Elbaz, N. Kar- boul, J. J. Fontaine, S. Chateau-Joubert, P. Boudou-Rou- Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1423 quette, S. Alran, V. Dangles-Marie, D. Gentien, M.-F. Pou- pon and D. Decaudin, “Modeling of Response to Endo- crine Therapy in a Panel of Human Luminal Breast Can- cer Xenografts,” Breast Cancer Research and Treatment, Vol. 133, No. 2, 2012, pp. 595-606. http://dx.doi.org/10.1007/s10549-011-1815-5 [41] Y. S. DeRose, K. M. Gligorich, G. Wang, A. Georgelas, P. Bowman, S. J. Courdy, A. L. Welm, B. E. Welm, “Pa- tient-Derived Models of Human Breast Cancer: Protocols for in Vitro and in Vivo Applications in Tumor Biology and Translational Medicine,” Current Protocols in Phar- macology, 2013, Chapter 14: Unit 14 23. [42] J. J. Wallin, J. Guan, W. W. Prior, L. B. Lee, L. Berry, L. D. Belmont, H. Koeppen, M. Belvin, L. S. Friedman, D. Sampath, “GDC-0941, a Novel Class I Selective PI3K Inhibitor, Enhances the Efficacy of Docetaxel in Human Breast Cancer Models by Increasing Cell Death in Vitro and in Vivo,” Clinical Cancer Research: An Official Jour- nal of the American Association for Cancer Research, Vol. 18, No. 14, 2012, pp. 3901-3911. [43] C. Ginestier, S. L. Liu, M. E. Diebel, H. Korkaya, M. Luo, M. Brown, J. Wicinski, O. Cabaud, E. Charafe-Jauffret, D. Birnbaum, J.-L. Guan, G. Dontu and M. S. Wicha, “CXCR1 Blockade Selectively Targets Human Breast Cancer Stem Cells in Vitro and in Xenografts,” The Journal of Clinical Investigation, Vol. 120, No. 2, 2010, pp. 485-497. http://dx.doi.org/10.1172/JCI39397 [44] T. John, D. Kohler, M. Pintilie, N. Yanagawa, N. A. Pham, M. Li, D. Panchal, F. Hui, F. Meng, F. A. Shep- herd and M. S. Tsao, “The Ability to Form Primary Tu- mor Xenografts Is Predictive of Increased Risk of Disease Recurrence in Early-Stage Non-Small Cell Lung Cancer,” Clinical Cancer Research: An Official Journal of the Ameri- can Association for Cancer Research, Vol. 17, No. 1, 2011, pp. 134-141. [45] Fichtner I, J. Rolff, R. Soong, J. Hoffmann, S. Hammer, A. Sommer, M. Becker and J. Merk, “Establishment of Patient-Derived Non-Small Cell Lung Cancer Xenografts as Models for the Identification of Predictive Biomark- ers,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 14, No. 20, 2008, pp. 6456-6468. [46] X. Dong, J. Guan, J. C. English, J. Flint, J. Yee, K. Evans, N. Murray, C. Macaulay, R. T. Ng, P. W. Gout, W. L. Lam, J. Laskin, V. Ling, S. Lam and Y. Wang, “Patient-Derived First Generation Xenografts of Non-Small Cell Lung Cancers: Promising Tools for Predicting Drug Responses for Personalized Chemotherapy,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 16, No. 5 , 2010, pp. 1442-1451. [47] R. Krumbach, J. Schüler, M. Hofmann, T. Giesemann, H.-H. Fiebig and T. Beckers, “Primary Resistance to Cetu- ximab in a Panel of Patient-Derived Tumour Xenograft Models: Activation of MET as One Mechanism for Drug Resistance,” European Journal of Cancer, Vol. 47, No. 8, 2011, pp. 1231-1243. http://dx.doi.org/10.1016/j.ejca.2010.12.019 [48] X. C. Zhang, J. C. Zhang, M. Li, X.-S. Huang, X.-N. Yang, W.-Z. Zhong, L. Xie, L. Zhang, M. H. Zhou, P. Gavine, X. Y. Su, L. Zheng, G. S. Zhu, P. Zhan, Q. S. Ji and Y.-L. Wu, “Establishment of Patient-Derived Non-Small Cell Lung Cancer Xenograft Models with Genetic Aberrations within EGFR, KRAS and FGFR1: Useful Tools for Preclinical Studies of Targeted Therapies,” Journal of Translational Medicine, Vol. 11, 2013, p. 168. http://dx.doi.org/10.1186/1479-5876-11-168 [49] M. Yang, B. Shan, Q. Li, X. Song, J. Cai, J. Deng, L. Zhang, Z. Du, J. Lu, T. Chen, J. P. Wery, Y. Chen and Q. Li, “Overcoming Erlotinib Resistance with Tailored Treat- ment Regimen in Patient-Derived Xenografts from Naive Asian NSCLC Patients,” International Journal of Cancer, Vol. 132, No. 2, 2013, pp. E74-E84. [50] A. Bertotti, et al., “A Molecularly Annotated Platform of Pa- tient-Derived Xenografts (‘Xenopatients’) Identifies HER2 as an Effective Therapeutic Target in Cetuxi- mab-Resistant Colorectal Cancer,” Cancer Discovery, Vol. 1, 2011, pp. 508-523. http://dx.doi.org/10.1158/2159-8290.CD-11-0109 [51] S. Julien, et al., “Characterization of a Large Panel of Pa- tient-Derived Tumor Xenografts Representing the Clini- cal Heterogeneity of Human Colorectal Cancer,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 18, No. 19, 2012, pp. 5314-5328. [52] H. B. Zhu, W. Zhou, J. Z. Hu, Z. T. Huang, W. F. Lao, X. F. Huang and C. He, “Suppressing the Growth of Rectal Cancer Xenografts Derived from Patient Tumors by an Adenovector Expressing Small Hairpin RNA Targeting Bcl-XL,” The Journal of Gene Medicine, Vol. 14, No. 12, 2012. pp. 761-768. http://dx.doi.org/10.1002/jgm.2681 [53] R. Toivanen, R. A. Taylor, D. W. Pook, S. J. Ellem and G. P. Risbridger, “Breaking through a Roadblock in Prostate Cancer Research: An Update on Human Model Systems,” The Journal of Steroid Biochemistry and Molecular Bi- ology, Vol. 131, No. 3-5, 2012, pp. 122-131. http://dx.doi.org/10.1016/j.jsbmb.2012.01.005 [54] O. Raheem, A. A. Kulidjian, C. Wu, Y. B. Jeong, T. Ya- maguchi, K. M. Smith, D. Goff, H. Leu, S. R. Morris, N. A Cacalano, K. Masuda, C. H. M. Jamieson, C. J. Kane and C. A. M. Jamieson, “A Novel Patient-Derived Intra-Femoral Xenograft Model of Bone Metastatic Prostate Cancer that Recapitulates Mixed Osteolytic and Osteoblastic Lesions,” Journal of Translational Medicine, Vol. 9, 2011, p. 185. http://dx.doi.org/10.1186/1479-5876-9-185 [55] A. Aparicio, V. Tzelepi, J. C. Araujo, C. C. Guo, S. D. Liang, P. Troncoso, C. J. Logothetis, N. M. Navone and S. N. Maity, “Neuroendocrine Prostate Cancer Xenografts with Large-Cell and Small-Cell Features Derived from a Single Patient’s Tumor: Morphological, Immunohistochemical, and Gene Expression Profiles,” The Prostate, Vol. 71, No. 8, 2011, pp. 846-856. http://dx.doi.org/10.1002/pros.21301 [56] T. Kimura, H. Kiyota1, D. Nakata, T. Masaki, Masami Kusaka and S. Egawa, “A Novel Androgen-Dependent Prostate Cancer Xenograft Model Derived from Skin Metastasis of a Japanese Patient,” The Prostate, Vol. 69, No. 15, 2009, pp. 1660-1667. http://dx.doi.org/10.1002/pros.21016 [57] T. Yoshida, H. Kinoshita, T. Segawa, E. Nakamura, T. Inoue, Y. Shimizu, T. Kamoto and O. Ogawa, “Antian- Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal 1424 drogen Bicalutamide Promotes Tumor Growth in a Novel Androgen-Dependent Prostate Cancer Xenograft Model Derived from a Bicalutamide-Treated Patient,” Cancer Research, Vol. 65, No. 21, 2005, pp. 9611-9616. http://dx.doi.org/10.1158/0008-5472.CAN-05-0817 [58] X. S. Li, Z. B. Liu, X. Xu, C. A. Blair, Z. Sun, J. Xie, M. B. lilly and X. L. Zi, “Kava Components Down-Regulate Expression of AR and AR Splice Variants and Reduce Growth in Patient-Derived Prostate Cancer Xenografts in Mice,” PLoS ONE, Vol. 7, No. 2, 2012, Article ID: e31213. http://dx.doi.org/10.1371/journal.pone.0031213 [59] S. E. Yost, S. Pastorino, S. Rozenzhak, E. N. Smith, Y. S. Chao, P. F. Jiang, S. Kesari, K. A. Frazer and O. Haris- mendy, “High-Resolution Mutational Profiling Suggests the Genetic Validity of Glioblastoma Patient-Derived Pre-Clinical Models,” PLoS ONE, Vol. 8, No. 2, 2013, Article ID: e56185. http://dx.doi.org/10.1371/journal.pone.0056185 [60] M. A. Jarzabek, P. C. Huszthy, K. O. Skaftnesmo, E. McCormack, P. Dicker, J. H.M. Prehn, R. Bjerkvig and A. T. Byrne, “In Vivo Bioluminescence Imaging Validation of a Human Biopsy-Derived Orthotopic Mouse Model of Glioblastoma Multiforme,” Molecular Imaging, Vol. 12, No. 3, 2013, pp. 161-172. [61] K. H. Kim, H. J. Seol, E. H. Kim, J. Rheey, H. J. Jin, Y. Lee, K. M. Joo, J. Lee and D.-H. Nam, “Wnt/Beta-Catenin Signaling Is a Key Downstream Mediator of MET Sig- naling in Glioblastoma Stem Cells,” Neuro-Oncology, Vol. 15, No. 2, 2013, pp. 161-171. http://dx.doi.org/10.1093/neuonc/nos299 [62] H. Huynh, K. C. Soo, P. K. Chow, L. Panasci and E. Tran, “Xenografts of Human Hepatocellular Carcinoma: A Use- ful Model for Testing Drugs,” Clinical Cancer Research, Vol. 12, 2006, pp. 4306-4314. http://dx.doi.org/10.1158/1078-0432.CCR-05-2568 [63] M. Yan, H. Li, F. Y. Zhao, L. X. Zhang, C. Ge, M. Yao, and J. J. Li, “Establishment of NOD/SCID Mouse Models of Human Hepatocellular Carcinoma via Subcutaneous Transplantation of Histologically Intact Tumor Tissue,” Chinese Journal of Cancer Research, Vol. 25, No. 3, 2013, pp. 289-298. [64] H. Huynh, V. C. Ngo, H. N. Koong, D. Poon, S. P. Choo, H. C. Toh, C. H. Thng, P. Chow, H. S. Ong, A. Chung, B. C. Goh, P. D. Smith and K. C. Soo, “AZD6244 Enhances the Anti-Tumor Activity of Sorafenib in Ectopic and Or- thotopic Models of Human Hepatocellular Carcinoma (HCC),” Journal of Hepatology, Vol. 52, No. 1, 2010, pp. 79-87. http://dx.doi.org/10.1016/j.jhep.2009.10.008 [65] H. Huynh, V. C. Ngo, J. Fargnoli, M. Ayers, K. C. Soo, H. N. Koong, C. H. Thng, H. S. Ong, A. Chung, P. Chow, P. Pollock, S. Byron and E. Tran, “Brivanib Alaninate, a Dual Inhibitor of Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Tyro- sine Kinases, Induces Growth Inhibition in Mouse Mod- els of Human Hepatocellular Carcinoma,” Clinical Can- cer Research, Vol. 14, 2008, pp. 6146-6153. http://dx.doi.org/10.1158/1078-0432.CCR-08-0509 [66] H. Huynh, V. C. Ngo, S. P. Choo, D. Poon, H. N. Koong, C. H. Thng, H. C. Toh, L. Zheng, L. C. Ong, Y. Jin, I. C. Song, A. P.C. Chang, H. S. Ong, A. Y.F. Chung, P. K.H. Chow and K. C. Soo, “Sunitinib (SUTENT, SU11248) Suppresses Tumor Growth and Induces Apoptosis in Xenograft Models of Human Hepatocellular Carcinoma,” Current Cancer Drug Targets, Vol. 9, No. 6, 2009, pp. 738-747. http://dx.doi.org/10.2174/156800909789271530 [67] H. Huynh, K. H. P. Chow, K. C. Soo, H. C. Toh, S. P. Choo, K. F. Foo, D. Poon, V, C. Ngo and E. Tran, “RAD001 (Everolimus) Inhibits Tumour Growth in Xeno- graft Models of Human Hepatocellular Carcinoma,” Journal of Cellular and Molecular Medicine, Vol. 13, No. 7, 2009, pp. 1371-1380. http://dx.doi.org/10.1111/j.1582-4934.2008.00364.x [68] C. Krepler, V. Wacheck, S. Strommer, G. Hartmann, P. Polterauer, K. Wolff, H. Pehamberger and B. Jansen, “CpG Oligonucleotides Elicit Antitumor Responses in a Human Melanoma NOD/SCID Xenotransplantation Mo- del,” The Journal of Investigative Dermatology, Vol. 122, No. 2, 2004, pp. 387-391. http://dx.doi.org/10.1046/j.0022-202X.2004.22202.x [69] J. M. Pimiento, E. M. Larkin, K. S. M. Smalley, G. L. Wiersma, N. R. Monks, I. V. Fedorenko, C. A. Peterson and B. J. Nickoloff, “Melanoma Genotypes and Pheno- types Get Personal,” Laboratory Investigation, Vol. 93, No. 8, 2013, pp. 858-867. [70] P. Guerreschi, C. Scalbert, A. Qassemyar, J. Kluza, L. Ravasi, D. Huglo, V. Martinot-Duquennoy, P. Formstecher, P. Marchetti and L. Mortier, “Patient-Derived Tumor Xe- nograft Model to Guide the Use of BRAF Inhibitors in Metastatic Melanoma,” Melanoma Research, Vol. 23, No. 5, 2013, pp. 373-380. http://dx.doi.org/10.1097/CMR.0b013e328363ed92 [71] C. Laurent, D. Gentien, S. Piperno-Neumann, F. Némati, A. Nicolas, B. Tesson, L. Desjardins, P. Mariani, A. Rapinat, X. Sastre-Garau, J. Couturier, P. Hupé, L. de Koning, T. Dubois, S. Roman-Roman, M. H. Stern, E. Barillot, J. W. Harbour, S. Saule and D. Decaudin, “Patient-Derived Xeno- grafts Recapitulate Molecular Features of Human Uveal Melanomas,” Molecular Oncology, Vol. 7, No. 3, 2013, pp. 625-636. http://dx.doi.org/10.1016/j.molonc.2013.02.004 [72] F. Nemati, X. Sastre-Garau, C. Laurent, J. Couturier, P. Mariani, L. Desjardins, S. Piperno-Neumann, O. Lantz, B. Asselain, C. Plancher, D. Robert, I. Péguillet, M. H. Don- nadieu, A. Dahmani, M. A. Bessard, D. Gentien, C. Reyes, S. Saule, E. Barillot, S. Roman-Roman and D. Decaudin, “Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors,” Clinical Cancer Re- search: An Official Journal of the American Association for Cancer Research, Vol. 16, No. 8, 2010, pp. 2352- 2362. [73] P. C. Gach, P. J. Attayek, G. Herrera, J. J. Yeh and N. L. Allbritton, “Isolation and in Vitro Culture of Rare Cancer Stem Cells from Patient-Derived Xenografts of Pancreatic Ductal Adenocarcinoma,” Analytical Chemistry, Vol. 85, No. 15, 2013, pp. 7271-7278. http://dx.doi.org/10.1021/ac401165s [74] D. M. Walters, J. M. Lindberg, S. J. Adair, T. E. New- hook, C. R. Cowan, J. B. Stokes, C. A. Borgman, E. B. Stelow, B. T. Lowrey, M. E. Chopivsky, T. M. Gilmer, J. Open Access JCT  Evolution of Tumor Model: From Animal Model of Tumor to Tumor Model in Animal Open Access JCT 1425 T. Parsons and T. W. Bauer, “Inhibition of the Growth of Patient-Derived Pancreatic Cancer Xenografts with the MEK Inhibitor Trametinib Is Augmented by Combined Treatment with the Epidermal Growth Factor Receptor/ HER2 Inhibitor Lapatinib,” Neoplasia, Vol. 15, No. 2, 2013, pp. 143-155. [75] N. V. Rajeshkumar, Z. A. Rasheed, E. García-García, F. López-Ríos, K. Fujiwara, W. H. Matsui1 and M. Hidalgo, “A Combination of DR5 Agonistic Monoclonal Antibody with Gemcitabine Targets Pancreatic Cancer Stem Cells and Results in Long-Term Disease Control in Human Pancreatic Cancer Model,” Molecular Cancer Therapeu- tics, Vol. 9, No. 9, 2010, pp. 2582-2592. http://dx.doi.org/10.1158/1535-7163.MCT-10-0370 [76] M. P. Kim, M. J. Truty, W. Choi, Y. Kang, X. Chopin- Lally, G. E. Gallick, H. Wang, D. J. McConkey, R. Hwang, C. Logsdon, J. Abbruzzesse and J. B. Fleming, “Molecular Profiling of Direct Xenograft Tumors Estab- lished from Human Pancreatic Adenocarcinoma after Neoadjuvant Therapy,” Annals of Surgical Oncology, Vol. 19, No. Suppl. 3, 2012, pp. 395-403. http://dx.doi.org/10.1245/s10434-011-1839-4 [77] P. C. Hermann, et al., “Multimodal Treatment Eliminates Cancer Stem Cells and Leads to Long-Term Survival in Primary Human Pancreatic Cancer Tissue Xenografts,” PLoS ONE, Vol. 8, No. 6, 2013, Article ID: e66371. http://dx.doi.org/10.1371/journal.pone.0066371 [78] A. Jimeno, G. Feldmann, A Suárez-Gauthier, Z. Rasheed, A. Solomon1, G.-M. Zou, B. Rubio-Viqueira, E. García- García, F. López-Ríos, W. Matsui, A. Maitra1 and M. Hidalgo, “A Direct Pancreatic Cancer Xenograft Model as a Platform for Cancer Stem Cell Therapeutic Devel- opment,” Molecular Cancer Therapeutics, Vol. 8, No. 2, 2009, pp. 310-314. http://dx.doi.org/10.1158/1535-7163.MCT-08-0924 [79] T. Hoey, W.-C. Yen, F. Axelrod, J. Basi, L. Donigian, S. Dylla, M. Fitch-Bruhns, S. Lazetic, I.-K. Park, A. Sato, S. Satyal, X. H. Wang, M. F. Clarke, J. Lewicki and A. Gurney, “DLL4 Blockade Inhibits Tumor Growth and Reduces Tumor-Initiating Cell Frequency,” Cell Stem Cell, Vol. 5, No. 2, 2009, pp. 168-177. http://dx.doi.org/10.1016/j.stem.2009.05.019
|